

Abstract
miR-155 is involved in immune and inflammatory diseases and is associated with liver fibrosis and steatohepatitis. However, the mechanisms involved in miR-155 regulation of liver injury are largely unknown. The role of miR-155 in acute liver injury was assessed in wild type (WT), miR-155−/− and miR-155−/− mice transplanted with WT bone marrow. Additionally, miR-155 expression was evaluated in liver tissue and peripheral blood mononuclear cells (PBMC) of patients with autoimmune hepatitis. Concanavalin A (ConA) but not acetaminophen (APAP) treatment increased the expression of miR-155 in liver tissue of WT mice. ConA induced an increase of cell death, liver transaminases and a higher expression of pro-inflammatory cytokines (Cxcl1, 5, 9, 10, 11, Ccl2, 20 and Icam1) in miR-155−/− compared to WT mice. Importantly, these animals showed a significant decrease in CD4+CXCR3+ and Foxp3+ cells recruitment but no changes in other inflammatory cell populations. Mechanistically, miR-155 deficient Tregs showed increased Ship1 expression, a known target of miR-155. Inhibition of Ship1 in miR-155−/− mice restored Foxp3 recruitment and reduced liver cytokine expression. Transplantation of bone marrow from WT animals into miR-155−/− mice partially reverted the effect of ConA on miR-155−/− mice as assessed by proinflammatory cytokines and cell death protein expression. Patients with autoimmune hepatitis showed a marked increase in miR-155 expression in the liver but a reduced expression of miR-155 in PBMC.
Conclusions
miR-155 expression is altered in both liver tissue and circulating inflammatory cells during liver injury, thus regulating inflammatory cell recruitment and liver damage. These results suggest that maintaining miR-155 expression in inflammatory cells might be a potential strategy to modulate liver injury.
Keywords: acute liver injury, liver inflammation, ConA, Tregs, SHIP1
INTRODUCTION
MicroRNAs (miRNAs) are small non-coding RNAs that act as post-transcriptional regulators of gene expression. Each microRNA frequently targets several genes and each gene can be targeted by several miRNAs, making microRNAs important fine tuning regulators of gene expression in physiological or disease conditions (1,2). miRNA expression is context dependent, with a specific miRNA expression profile for every organ, cell type and physiological situation. In the context of liver disease a dysregulation of miRNAs expression has been well-described in liver tissue being of pathophysiological relevance (3–5). However, the expression and role of miRNAs in inflammatory cells in the context of liver damage is still largely unknown.
miR-155 is ubiquitously expressed in several tissues and cell types. miR-155 participates in the correct development of immune cells such as B and T lymphocytes, dendritic cells and macrophages by regulating their function and activation (6–11). In addition, miR-155 regulates oncogenic pathways in solid tumors and hematological malignancies (12,13). Importantly, miR-155 function is context dependent. The interaction of miR-155 with its target gene SOCS1 determines immune responses such as Treg competitive fitness and natural killer (NK) cell expansion. However, when miR-155 functions independently of SOCS1, it regulates CD8+ T cell responses and Th17 differentiation (14,15). Other target genes of miR-155 such as SHIP1 are known to regulate Th17 and Treg development and function (16,17).
In the context of liver diseases, miR-155 is expressed in hepatocytes (18,19), endothelial cells (20) and inflammatory cells such as monocytes, NK cells and macrophages (19,21). Moreover, miR-155 has been described to be up-regulated in liver tissue of patients with hepatitis C virus and alcoholic liver disease (4,21,22) and to mediate cellular growth and transforming growth factor-β-dependent epithelial to mesenchymal transition in liver carcinogenesis (18). However, the role of miR-155 in liver diseases is still not well understood, and may depend on disease context. While miR-155 has been shown to be involved in the progression of liver inflammation, steatosis and fibrosis in experimental models of chronic alcoholic liver disease (22–24), in a non-alcoholic steatohepatitis model, miR-155 played a hepatoprotective role (25).
In the present study, miR-155 deficiency was found to enhance acute liver injury and promote an alteration of inflammatory cells recruitment. Interestingly, by restoring miR-155 expression in inflammatory cells in miR-155 deficient mice (miR-155−/−) the phenotype was partially reverted, thus suggesting that miR-155 deficiency in immune cells may enhance liver injury. Similar to previous studies in mice, the expression of miR-155 in patients with liver disease was found increased in liver tissue, but decreased in circulating inflammatory cells. These results suggest that miR-155 expression in immune cells may play a role in liver injury and disease, and thus restoration of miR-155 expression in inflammatory cells might be a strategy to modulate liver injury.
Experimental Procedures
Patients
Liver biopsies were obtained from a cohort of consecutive patients with clinical, analytical and histological features of autoimmune hepatitis (AIH, n=15), and liver cirrhosis [alcoholic liver disease (n=16) or non-alcoholic steatohepatitis (n=3)]. All patients were admitted to the Liver Unit of the Hospital Clinic of Barcelona from July 2009 to December 2016 and an informed consent was obtained from all patients, according to the ethical guidelines of the 1975 Declaration of Helsinki; the study was approved by the Ethics Committee of the Hospital Clinic of Barcelona. The characteristics of patients with AIH from whom liver biopsies were obtained are shown in Supplementary Table 1. A group of livers samples obtained from optimal cadaveric liver donors or resections of liver metastasis were used as controls. All controls had normal serum transaminases levels and normal histology, as described previously (26).
Peripheral blood mononuclear cells (PBMC) were isolated from patients with AIH (Supplementary Table 2) and from patients with liver cirrhosis (Supplementary Table 3).
Isolation of peripheral blood mononuclear cells
PBMC were isolated from peripheral blood samples using cell preparation tubes with sodium citrate and a density gradient liquid (Ficoll) following the manufacturer’s instructions (BD, NJ, USA).
Mice
miR-155 knockout mice (miR-155−/−) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Congenic CD45.1 mice (B6.SJL-PtprcaPepcb/BoyCrl) were purchased from Charles River Laboratories (l’Arbresle, France). As control wild type (WT) mice we used C57BL/6J inbred strain as suggested by the provider of the miR-155 deficient animals. The wild type mice were housed and bred in the same animal facility and in the same conditions as miR-155−/− animals. Wild type, miR-155−/− and CD45.1 mice share a C57BL/6J genetic background, including their major histocompatibility complex molecules.
Animal Models
For concanavalin A (ConA) treatment, mice were injected intravenously with 10 mg/Kg of ConA. Animals were sacrificed to perform analysis at 8 or 18 hours after the injection. Blood and liver samples were collected.
For acetaminophen (APAP) treatment, 12-weeks-old male mice were fasted overnight with free access to water. Afterwards the mice were intraperitoneally injected with 500 mg/kg of APAP resuspended in warm saline (NaCl 0, 9%). Twenty-four hours after the injection, the animals were sacrificed and blood and liver samples were collected.
To inhibit SHIP1 activity the inhibitor 3AC (Millipore) was used (herein referred to as iSHIP). A solution of iSHIP was prepared in 0, 3% hydroxypropylcellulose in PBS (w/v). Mice received, during 2 days, intraperitoneally injections of iSHIP at 26’5 mg/Kg or vehicle. Twenty four hours after the last iSHIP injection ConA was administered intravenously at 10mg/kg to all animals. After 18 hours animals were sacrificed. Blood and liver samples were collected.
A bone marrow (BM) transplant was performed in female miR-155−/− mice. To this end, miR-155−/− recipient mice were intraperitoneally administered with 25 mg/kg of an antineoplastic drug (Busulfan) during 3 days prior to BM transplantation. The BM was obtained from either WT donors (with a CD45.1 phenotype) or from miR-155−/− donors (with a CD45.2 phenotype). All donor mice were sacrificed by cervical dislocation; afterwards tibias, femurs and iliac crests were collected and used to obtain BM cells. Recipient mice received an intravenous injection of 20 x106 BM cells. Three weeks after BM transplantation, peripheral blood was collected from the tail and used to perform a chimerism analysis. Eight weeks after BM transplantation, the recipient mice were intravenously injected with 10mg/Kg of ConA and sacrificed 18 hours thereafter. Blood and liver samples were then collected.
CD4+ cell isolation from spleen
Female WT and miR-155−/− mice were injected with 6, 5 mg/Kg of ConA, and 18 hours later the animals were sacrificed. Spleens were collected and physically disaggregated using a 70 μm cell strainer and erythrocytes were lysed with Lysing Buffer (BD PhamLyse). CD4+ cells were isolated using the CD4+ T cell Isolation Kit (Miltenyi Biotec, Gladbach, Germany) following the manufacturer’s instructions. Next, CD4+ T cells were cultured and stimulated with anti-CD3e at 0, 5 μg/ml (BD Pharmigen) during 48 hours. Supernatants were collected at 24 and 48 hours. Cells were harvested at 48 hours and RNA was extracted with the Rneasy Mini Kit (Qiagen, Venlo, Netherlands).
Treg isolation from spleen
CD4+CD25+ (Tregs) cells were isolated with the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi) following manufacturer’s instruction. Afterwards isolated Tregs were cultured and treated with IL-2 at 2000 U/ml, CD3e at 1 μg/ml and CD28 at 5 μg/ml during 6 days. RNA was extracted with the Rneasy Mini Kit (Qiagen).
Statistical analysis
The results of the quantitative variables are expressed as mean ± standard deviation. Comparisons between groups were performed using the Student´s t test.
Further experimental procedures are described in the supplementary material.
RESULTS
miR-155 deficiency enhances Concanavalin A-induced acute liver injury
In order to determine the effect of miR-155 expression during liver injury, we evaluated the effect of ConA and APAP intoxication in miR-155−/− mice, two well-described acute liver injury models with different modes of action. ConA treatment induces acute liver injury mediated by T cells, and is considered to be a good experimental model of AIH (27,28). As shown in figure 1A, ConA treatment induced a marked increase of miR-155 expression in liver tissue of WT mice, 20-fold more than vehicle-treated animals. Next, we evaluated the effect of ConA in miR-155−/− mice. ConA treatment induced a significant increase in serum levels of liver transaminases [i.e. aspartate transaminase (AST), alanine transaminase (ALT)] and lactate dehydrogenase (LDH)] in WT mice, which were substantially higher in miR-155 deficient animals at both 18 and 8 hours after ConA injection (Figure 1B and Supplementary Figure 1A). To evaluate liver damage, we performed a TUNEL assay, which stains apoptotic nuclei and necrotic areas. We observed that miR-155 deficient animals had an increase in liver necrosis compared to WT-treated animals (Figure 1C and Supplementary Figure 1B). Representative pictures of haematoxylin-eosin staining are shown in (Supplementary Figure 2A).
Figure 1. miR-155 deficiency aggravates liver injury in a ConA model.
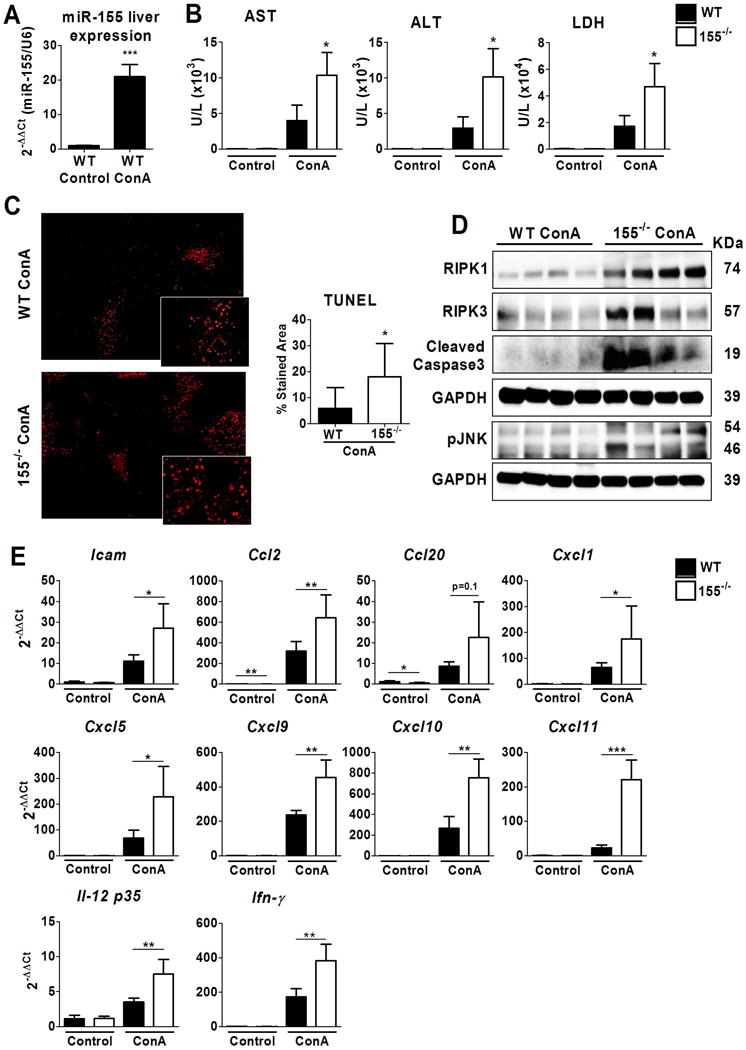
Wild type and miR-155−/− mice were treated with physiological saline (WT, n=6; miR-155−/−, n=6) or with 10 mg/Kg of ConA (WT, n=5; miR-155−/− n=4). Animals were evaluated at 18 hours after ConA treatment. (A) miR-155 expression in livers of WT animals (B) AST, ALT and LDH levels were measured in serum (C) Representative images of livers analysed by TUNEL assay staining at X40 magnification, and quantification of the stained area; (n≥9 *p<0.05) with respect to WT-ConA treated animals (D) Protein levels of RIPK1, RIPK3, cleaved Caspase 3 and pJNK were determined by Western Blot in livers of WT and miR-155−/− ConA-treated animals. GAPDH was used as a loading control. (E) Hepatic gene expression of several inflammatory mediators was measured by qPCR and expressed as 2−ΔΔCt fold change vs. WT vehicle treated animals. Significant p values are shown above the bars: *p<0.05, **p<0.01 with respect to WT vehicle-treated animals.
We next explored whether cell death signaling was thus increased in miR-155−/− mice at 8 and 18 hours after ConA treatment. We observed that RIPK1, RIPK3, cleaved caspase 3 and activation of JNK were overexpressed in miR-155−/− livers compared with WT ConA-treated animals (Figure 1D and Supplementary Figure 1C). These results suggested that lack of miR-155 increases apoptotic cell death. Furthermore, miR-155−/− ConA-treated animals showed an increased gene expression of inflammatory mediators compared to WT animals, which was significant at 18 hours (Figure 1E and Supplementary Figure 1D). Animals deficient in miR-155 had a significant increase in liver expression of inflammatory mediators such as Icam1, Il-12 as well as the CXC and CCL chemokine family members Cxcl1, 5, 9, 10, 11 and Ccl2 compared to WT ConA-treated animals (Figure 1E). No changes were observed in the expression of other inflammatory mediator such as Tnfα, IL1β, Il-6 or Il-10, Nos2, Arg1, Mrc1and Il-4 (Supplementary figure 2B).
On the other hand, APAP intoxication triggers hepatocyte death and release of danger-associated molecular patterns, thus promoting infiltration of macrophages and NKT cells (29,30). As shown in Supplementary figure 3A, APAP did not induce miR-155 expression in WT compared to vehicle-treated animals. APAP administration in miR-155 deficient mice induced a non-significant increase in serum liver transaminases and LDH as compared to APAP-treated WT mice (Supplementary figure 3B). Likewise, no major changes were observed in the liver histology and inflammatory cytokine expression (Supplementary figure 3D).
Overall these results suggest that the effect of miR-155 deficiency on liver injury may depend on the type of injury, being most important in T cell-mediated liver injury.
Deficiency of miR-155 alters inflammatory cell recruitment in liver injury
Expression of miR-155 is important for inflammatory cell differentiation and recruitment (8). Therefore, we next evaluated the inflammatory cell populations recruited to the liver after ConA treatment in WT and miR-155 deficient mice. Staining of the pan-leucocyte marker CD45 and the neutrophil marker myeloperoxidase (MPO) showed a non-significant decrease in infiltrating inflammatory cells and neutrophils respectively in miR-155−/− mice as compared to WT ConA-treated animals (Figure 2A). Furthermore, flow cytometry analysis showed that while recruitment of the total population of CD8+ and CD4+ cells did not change between the two groups after ConA-induced liver injury, CD4+CXCR3+ (tissue-homing T cells) was significantly decreased in miR-155−/− compared to WT ConA-treated mice (Figure 2B and Supplementary Figure 4). Moreover, F4/80lowCD11bmid monocyte-derived macrophages were slightly increased in liver tissue of miR-155−/− as compared to WT mice (Figure 2B). On the other hand, recruitment of NKT cell population (CD3+ NK1.1+) and F4/80highCD11bhigh were not increased in livers of miR-155−/− mice (Figure 2B).
Figure 2. miR-155 deficiency alters liver recruitment in a ConA model.
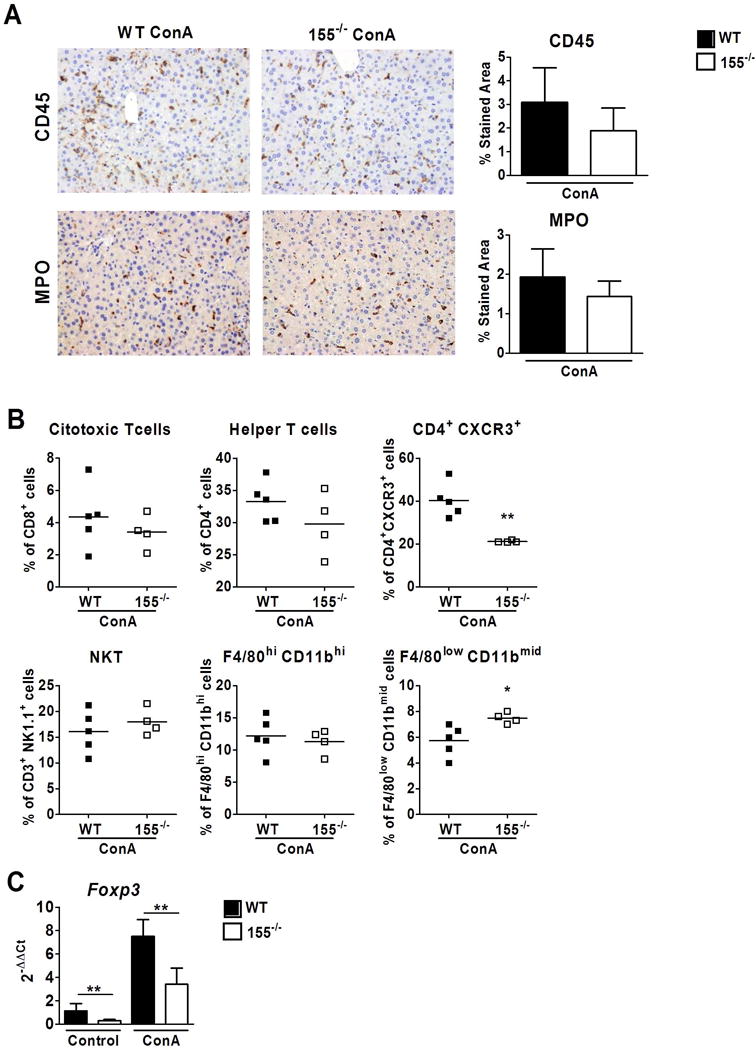
(A) Representatives images and quantification of CD45 and MPO staining of livers of WT and miR-155−/− ConA-treated mice after 18 hours (X200 magnification) (B) Scatter plots representing percentages of different immune cells measured by flow cytometry in livers of WT (n=5) and miR-155−/− (n=4) ConA-treated mice after 18 hours (C) Hepatic expression of Foxp3 measured by qPCR. Significant p values are shown above the bars: *p<0.05, **p<0.01 with respect to WT vehicle-treated animals.
Tregs are positive for CD4 and CXCR3; therefore, we assessed the level of expression of their specific marker Foxp3 in the liver. Interestingly, the gene expression of Foxp3 was decreased in both uninjured and injured miR-155 deficient animals compared to WT mice, suggesting that a reduction in Treg cells (CD4+CXCR3+) may be involved in exacerbated acute liver injury in miR-155−/− mice (Figure 2C).
Effects of miR-155 deficiency in CD4+ T helper cells and CD4+CD25+ Treg cells
To better understand the role of miR-155 in T helper cells we isolated and cultured CD4+ splenocytes of WT and miR-155−/− mice treated with ConA and assessed their cytokine expression profile. Interestingly, as shown in figure 3A, miR-155 deficient CD4+ cells produced less IFN-γ, TNFα and IL-5 than WT CD4+ cells, at 24 or 48 hours. No differences were observed in the levels of IL-4, IL-2 and IL-10. Interestingly, CD4+ splenocytes deficient in miR-155 showed a lower expression of Foxp3 (Figure 3B), reinforcing the idea that miR-155 expression is important in Treg differentiation (31,32).
Figure 3. Deficiency of miR-155 alters CD4+ Thelper and CD4+ CD25+ Treg cells.
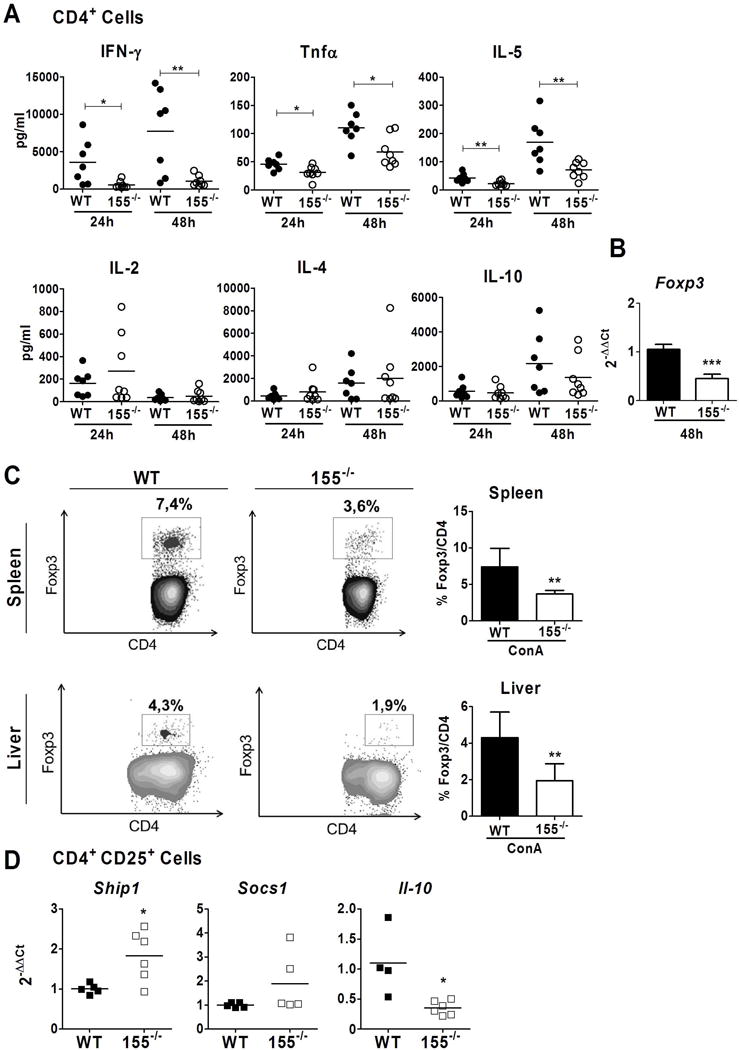
(A) Splenic CD4+ cells were isolated from WT (n=7) and miR-155−/− (n=8) mice. The concentrations of several cytokines were measured in CD4+ culture supernatant at 24 and 48 hours after stimulation. (B) Gene expression of Foxp3 in CD4+ cells at 48 hours after anti-CD3 stimulation (C) Foxp3 expression was analysed by flow cytometry in spleens and livers of WT (n=5) and miR-155−/− (n=6) ConA-treated mice after 8 hours. (D) Splenic CD4+ CD25+ Treg cells were isolated from WT (n=6) and miR-155−/− (n=6) mice and stimulated for 6 days. Gene expression of Socs1, Ship1 and Il-10 were analysed by qPCR. Significant p values are shown above the bars: *p<0.05, **p<0.01, ***p<0.0001
We further investigated the effect of miR-155 deficiency in Treg cells. First, we evaluated the number of Tregs in the spleen and those recruited to the liver. As shown in figure 3C miR-155−/− animals had a significant decrease in CD4+ Foxp3+ cells in both the spleen and the liver. Next, we evaluated miR-155 gene targets involved in Treg cell development such as Socs1 and Ship1 (14,16,17,33). While Ship1 expression was found to be increased in CD4+ CD25+ Treg cells deficient in miR-155 compared to WT cells, no differences in Socs1 were observed (Figure 3D). In addition the expression of the anti-inflammatory cytokine Il-10 was lower compared to WT Treg cells.
Altogether, these results show that miR-155 deficiency induces an increase in Ship1 and a defective recruitment of Tregs to the liver; supporting former studies showing that miR-155 deficiency causes a defective Treg development and function.
Inhibition of Ship1 activity in miR-155 deficient mice
Ship1 is a well-known target gene of miR-155 (17). To evaluate the role of Ship1 in liver damage we used the 3AC (herein referred as iSHIP), a selective inhibitor of SHIP1 polyphosphatase activity in WT and miR-155 deficient animals. As expected, ConA induced a higher level of liver transaminases in vehicle-treated miR-155−/− animals compared to WT. The increase in transaminase levels in miR-155−/− animals was blunted by iSHIP treatment, however, there were no significant changes in transaminase levels when compared to vehicle-treated miR-155−/− animals (Figure 4A). Moreover, Ship1 inhibition did not show changes in liver necrosis as assessed by TUNEL assay in miR-155 deficient or WT animals compared to vehicle-treated mice (Figure 4B). Altogether, Ship1 inhibition resulted in a trend towards decreased ALT with no changes in necrosis. Next, we evaluated gene expression of several inflammatory mediators in the liver. Remarkably, we observed that Ship1 blockade in ConA-treated miR-155−/− animals decreased the expression of Icam1, CCl2, Cxcl1, Cxcl5 and Cxcl10 with respect to vehicle-treated miR-155−/− animals (Figure 4C). As expected iSHIP treatment restored the deficient recruitment of Foxp3+ cells to the liver in animals deficient for miR-155. As shown in figure 4D iSHIP treated-miR155−/− mice had a significant increase in Foxp3 recruitment when compared to vehicle-treated miR-155−/− animals.
Figure 4. Inhibition of SHIP1 in miR-155 deficient mice.
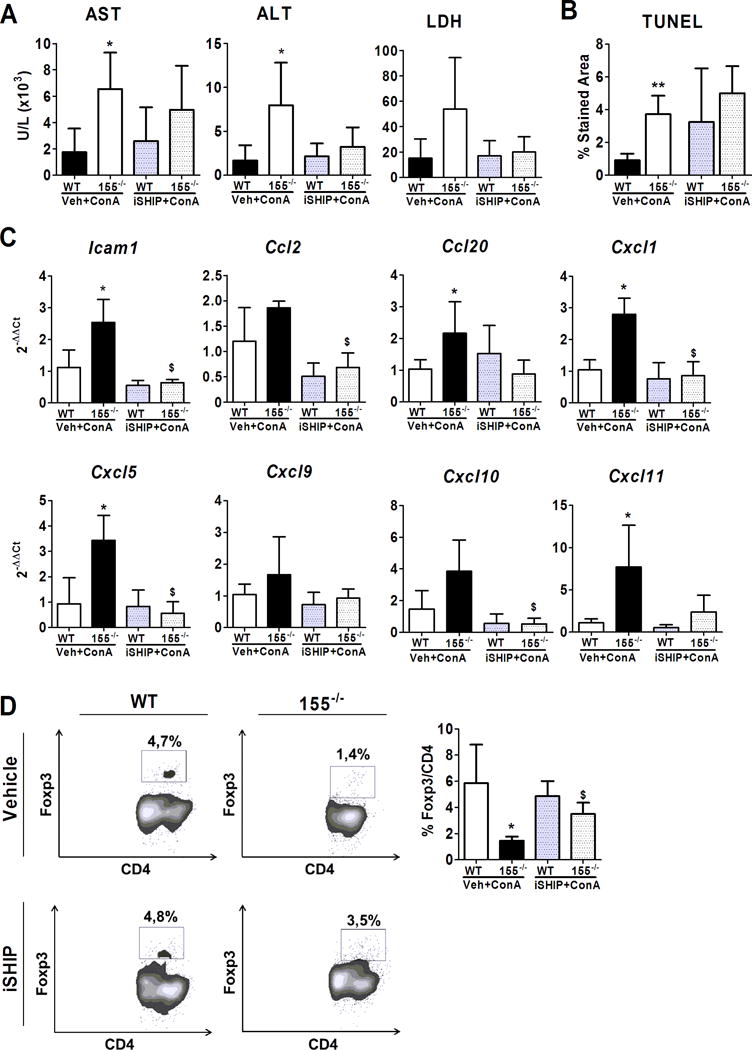
WT and miR-155−/− mice were treated with SHIP1 inhibitor (iSHIP) (WT, n=5; miR-155−/−, n=4) or with vehicle (WT, n=5; miR-155−/− n=4) during 2 days followed by 10 mg/Kg ConA treatment. Mice were analyzed after 18 hours. (A) Level of liver transaminases and LDH in serum. (B) Quantification of TUNEL staining. (C) Gene expression of several inflammatory mediators and cytokines in liver tissue assessed by qPCR and expressed as 2−ΔΔCt fold change respect to WT-vehicle plus ConA treated animals. (D) Liver recruitment of CD4+ Foxp3+ cells was analyzed by flow cytometry. Flow cytometry plots show representative CD4+ Foxp3+ staining. Graph shows mean values for CD4+ Foxp3+ quantification. Significant p values are shown above the bars: *p<0.05 respect to WT-vehicle plus ConA treated animals; $ p<0.05 respect to miR-155−/− vehicle plus ConA treated animals.
These results suggest that elevated Ship1 in miR-155 deficient animals may play a role in the deficient Treg function and recruitment to the liver.
Restoring miR-155 expression in inflammatory cells affects liver injury in miR-155−/− mice
To understand the role of miR-155 in liver parenchyma and in inflammatory cells in the context of acute liver damage, we performed BM transplantation in WT animals (CD45.1+ BM cells) and in miR-155−/− animals (CD45.2+ BM cells). Three weeks after BM transplantation a chimerism analysis was performed. WT animals transplanted with miR-155−/− BM showed only 50% of miR-155−/− CD45.2+ cells. This poor engraftment precluded evaluating the contribution of miR-155 in the liver parenchyma to acute liver damage.
On the other hand, miR-155−/− animals transplanted with WT-BM had 71.8 ± 8.2% of CD45.1+ WT-BM cells. We selected miR-155 deficient animals transplanted with WT-BM to better understand the role of miR-155 in inflammatory cells in liver injury. As a control group we used miR-155−/− animals transplanted with miR-155−/− BM. Eight weeks after hematopoietic reconstitution the mice were treated with 10 mg/kg of ConA during 18 hours.
First, the liver expression of miR-155 on transplanted animals was assessed. The baseline liver expression of miR-155 in miR-155−/− WT-BM mice was lower than in WT animals. Surprisingly, after ConA treatment, the miR-155−/− animals with WT-BM showed a marked increase in miR-155 expression, which was higher than in WT animals (Supplementary Figure 5). This suggests that increased liver expression of miR-155 after ConA damage may be due to the expression of miR-155 in inflammatory cells.
As shown in figure 5, WT-BM transplantation into miR-155−/− mice reduce the serum levels of AST, ALT and LDH when compared with miR-155−/−-miR-155−/−BM albeit not significant (Figure 5A). Regarding liver histology, miR-155−/− mice receiving WT-BM showed smaller necrotic areas compared with miR-155−/− mice transplanted with miR-155−/−BM (Figure 5B). However, recruitment of CD45+ inflammatory cells and neutrophils (MPO staining) did not change between the two groups (Figure 5B). The reduction in liver necrosis observed in the WT-BM transplanted animals, was confirmed by Western blot with the evaluation of the expression of several necrosis signaling proteins. We observed a marked reduction in the level of RIPK3, cleaved caspase-3 and pJNK in the livers of miR-155−/−- WT-BM; while RIPK1 did not change (Figure 5C). On the other hand, the assessment of liver apoptosis by TUNEL assay showed a decrease positive staining in miR-155−/− mice transplanted with WT-BM compared to miR-155−/−-miR-155−/− BM animals but this difference was not statistically significant (Figure 5D). Nevertheless, a significant reduction was observed in the activity of caspases 3 and 7 in miR-155−/− mice transplanted with WT-BM, suggesting that correct expression of miR-155 in inflammatory cells reduces apoptosis (Figure 5E).
Figure 5. Restoration of miR-155 expression in bone marrow decreases liver injury in a ConA model.
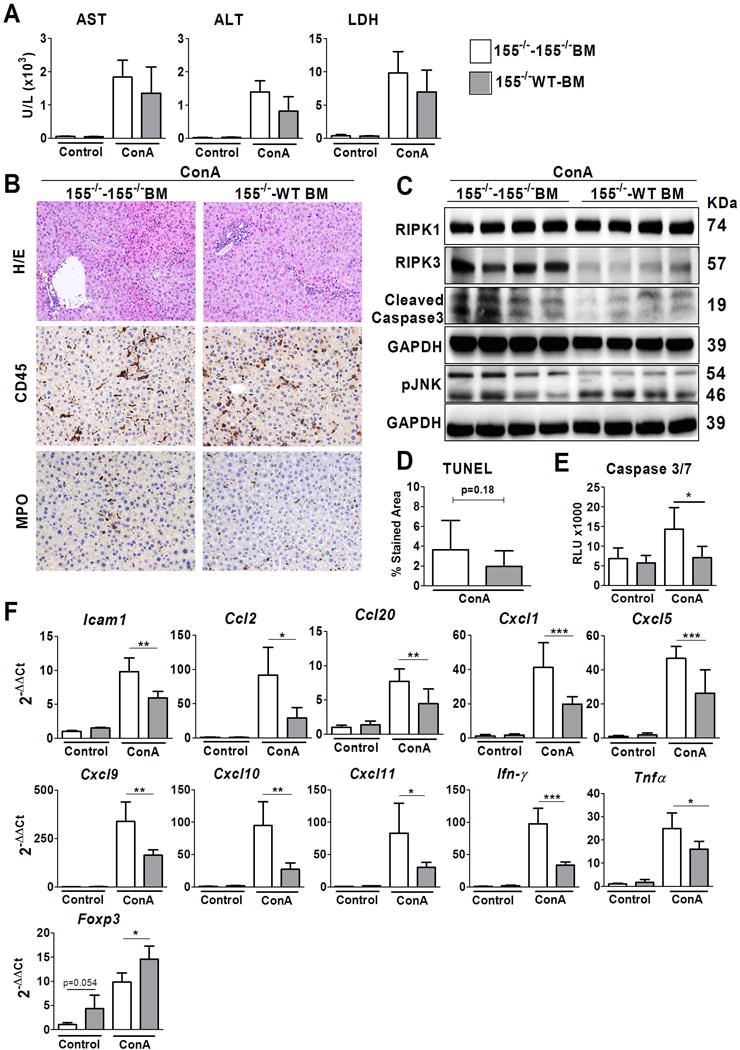
Experiments were performed in miR-155−/− animals transplanted with BM expressing miR-155 (WT-BM) or deficient in miR-155 (miR-155−/− BM) treated either with vehicle (NaCl) or with 10mg/Kg of ConA during 18 hours. Four groups were established: miR155−/− -miR155−/−BM NaCl-treated mice (n=4); miR-155−/− WT-BM NaCl-treated mice (n=5); miR-155−/−-miR-155−/−BM ConA-treated mice (n=5); and miR-155−/− WT-BM ConA-treated mice (n=5) (A) AST, ALD and LDH levels were measured in serum (B) Representatives images of haematoxylin/eosin (X100magnification), CD45 staining and MPO staining of livers of ConA-treated mice (X200 magnification) (C) Protein levels of RIPK1, RIPK3, cleaved Caspase 3 and pJNK were determined by Western blot. GAPDH was used as a loading control. (D) TUNEL assay staining quantification of ConA treated animals (E) Quantification of Caspase 3/7 activity on whole liver. (F) Hepatic gene expression of several genes of inflammatory mediators measured by qPCR. Significant p values are shown above the bars: *p<0.05, **p<0.01.
Notably, the gene expression level of several inflammatory mediators (i.e. Icam1, Ccl2, Ccl20, Cxcl1, 5, 9, 10, 11, Ifn-γ and Tnfα) was reduced in miR-155−/− mice transplanted with WT-BM compared to miR-155−/−-miR-155−/− BM (Figure 5F). Importantly, Foxp3 expression was increased in miR-155−/− WT-BM mice, supporting the notion that Treg recruitment may be involved in the degree of liver injury. Overall, these results show that transplant of WT BM into miR-155−/− mice partially rescue their phenotype in acute liver injury, and strongly suggest that miR-155 expression in inflammatory cells modulates liver inflammation and liver damage.
Expression of miR-155 in human samples
ConA treatment produces a liver injury similar to AIH, and is frequently used as a mouse model of AIH. Therefore, we evaluated the expression of miR-155 in liver tissue and circulating PBMC in patients with AIH as well as in cirrhotic patients. As shown in figure 6, expression of miR-155 has been found to be increase in liver tissue of patients with cirrhosis as compared to control livers. The increase was similar in compensated (3.37 fold ± 3.48) and decompensated patients (3.52 fold ± 1.70). Notably, patients with AIH showed and important increase in miR-155 expression in liver tissue with 7.6 fold ± 5.6 compared to control livers, and also with a significant increase compared to cirrhotic patients (Figure 6A). Experimental data from the ConA mouse model has suggested that a correct level of miR-155 expression in inflammatory cells may be important in modulating liver damage and inflammatory response; therefore we evaluated the expression of miR-155 in PBMC. As shown in Figure 6B, patients with AIH or with liver cirrhosis showed a decreased expression of miR-155 in PBMC compared to healthy controls. No significant differences were observed between compensated and decompensated cirrhotic patients (Figure 6B). On the other hand, we evaluated if immunosuppressive treatment affected miR-155 expression in PBMC of AIH patients. Our data show that AIH patients under immunosuppressive treatment and before treatment showed similar miR-155 expression levels in PBMCs (Figure 6C). Altogether these results suggest that changes in miR-155 expression in both liver and inflammatory cells occur in liver injury and diseases.
Figure 6. miR-155 expression in human samples.
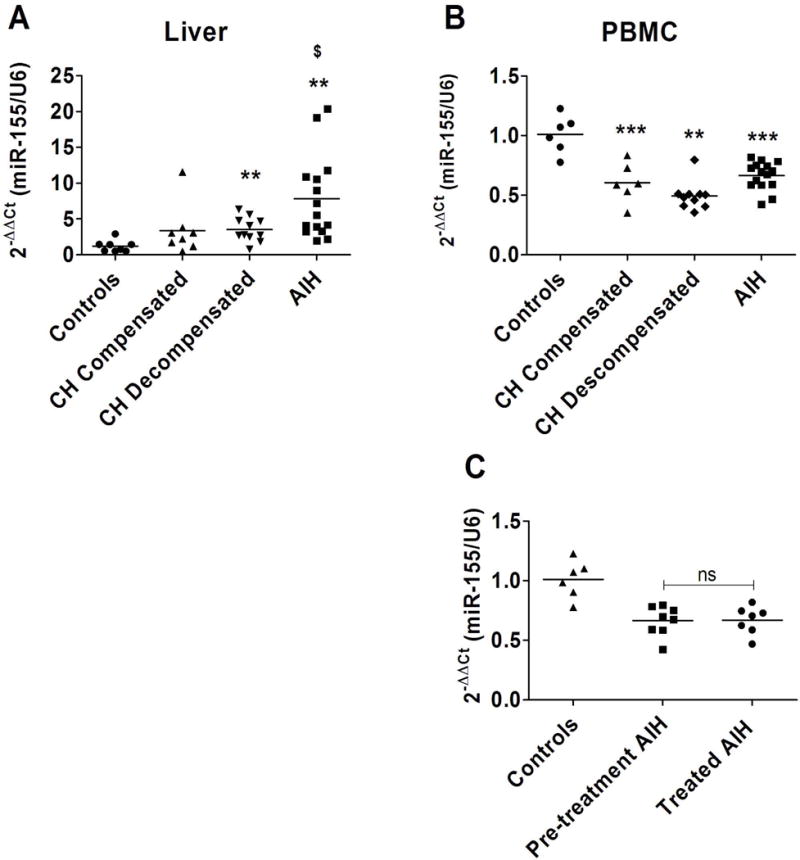
(A) Hepatic expression of miR-155 evaluated by qPCR in liver samples of compensated cirrhosis (n=8), decompensated cirrhosis (n=11) and AIH patients (n=15) and control livers (n=8) * p<0.05; ** p<0.01 vs. control livers; $ p<0.05 vs. decompensated and compensated cirrhosis (B) Expression of miR-155 in peripheral blood mononuclear cells (PBMC) of AIH patients (n=15), decompensated cirrhotic patients (n=10), compensated cirrhotic patients (n=6) and control individuals (n=6) ** p<0.01, ***p<0.001 vs. control individuals. (C) Expression of miR-155 in PBMC of debutant AIH patients before treatment (Pre-treatment AIH, n=8) and AIH patients undergoing immunosuppressive treatment (Treated AIH, n=7) and control individuals (n=6).
DISCUSSION
This study shows that the expression of miR-155 is altered in both liver tissue and inflammatory cells in liver injury and disease. With the use of miR-155 deficient mice we found that miR-155 plays an important role in T lymphocyte-mediated liver injury, modulating the extent of liver injury, Treg cells recruitment and cytokine production by T helper cells. Interestingly, on restoration of miR-155 expression in inflammatory cells by WT-BM transplantation in miR-155−/− animals, the extent of liver damage and cytokine expression was reduced. These results suggest that the expression of miR-155 in inflammatory cells may be important for the correct inflammatory response in liver disease.
miR-155 is expressed in most cell types and tissues, being implicated in a wide range of diseases. Therefore, understanding the role of miR-155 is challenging, particularly in multifactorial and/or complex conditions such as liver diseases. Recent studies have shown that miR-155 expression in liver tissue is increased in chronic liver disease and it can also be detected in the circulation (4,19,22,34). These studies not only suggest that miR-155 may play a role in the progression of liver diseases, but also that it may be a potential biomarker. In a mouse model of chronic liver injury miR-155 deficiency was shown to mitigate liver inflammation and steatosis (35). Although in our study the expression of miR-155 is protective in acute liver injury, these results are not necessarily contradictory, since the role of miR-155 in acute or in chronic conditions may be different due to a context-dependent role of miR-155. In addition, participation of cell types expressing miR-155 may change along the different phases of disease progression, and second, the expression of target genes may differ in the context of chronic or acute liver injury. Thus, therapeutic strategies aimed at modifying miR-155 expression should take into account miR-155 expression as well as it role along the different stages of liver disease.
To evaluate the role of miR-155 in liver injury we evaluated the impact of miR-155 deficiency in two animal models of acute liver injury with different mechanism of action. APAP-induced liver injury triggers death receptor-independent hepatocyte necrosis, which activates liver monocytes thus attracting other inflammatory cells (36). On the contrary, ConA treatment mainly induces an immune-mediated liver damage with a rapid recruitment of T lymphocytes to the liver triggered by activation of Kupffer cells, NK cells and NKT cells, followed by hepatocyte death mediated by the activation of death receptors (27,37). Interestingly, while miR-155−/− mice treated with APAP did not show enhanced liver damage, miR-155−/− mice where more susceptible to ConA-induced liver injury. These results are in agreement with previous reports suggesting that miR-155 may have an important role in inflammatory cell function and suggest the possible role of miR-155 expression in T lymphocytes for achieving correct inflammatory response (6,8,38,39).
Independently of the direct or indirect effects of miR-155 on the regulation of ConA-induced inflammatory response, there is increasing evidence that miR-155 might play an important role in cell death (40). Thus, we next investigated the mode of cell death during ConA administration. Interestingly, we observed increased expression of not only necrotic markers such as RIPK1 and RIPK3 but also caspase-3 cleavage. Thus, in keeping with findings of previous studies (40,41), miR-155 might repress RIPK and cleaved caspase-3-dependent cell death. Furthermore, increased JNK activation was observed in miR-155-deficient livers, supporting a previous report suggesting that miR-155-inducing signals use the JNK pathway (42).
One of the main findings of the study is the reduced recruitment of CD4+CXCR3+ cells in ConA-treated miR-155 deficient mice. The CXCR3 receptor is mainly expressed in Th1, but it is also expressed in tissue-homing Tregs (43). In addition evaluation of Foxp3+ cells in spleen and liver showed a reduced number in both tissues in miR-155−/− mice, suggesting that miR-155 deficiency decreases the number of total Tregs, and the number of Tregs recruited to the liver. These results are in agreement with reports showing that Tregs are reduced in spleen and thymus of miR-155-deficient mice and suggest that miR-155 expression may be involved in the development and function of regulatory T cells (31,32). Moreover, we also showed reduced liver expression of Foxp3 in miR-155−/− ConA-treated mice, which is specifically expressed in Treg cells. Interestingly, WT-BM transplant in miR-155−/− animals restored Foxp3 expression further suggesting the role of miR-155 in maintaining Treg population.
In order to evaluate if T helper lymphocytes response may be altered by miR-155 expression, we isolated CD4+ cells after ConA challenge and evaluated their cytokine profile. We observed a reduction in pro and anti-inflammatory cytokines and also a reduction of Foxp3 gene expression. As previously described, IFNγ production was reduced in CD4+ miR-155 deficient cells, although no differences were observed in IFNγ expression in whole liver tissue of miR-155−/− mice. These results suggest that deficiency of miR-155 may not only alter T helper recruitment but also the response to injury by regulating cytokine production in the context of liver injury. Nonetheless, these results do not imply that miR-155 post-transcriptionally regulates cytokine expression, because it has been widely reported the role of miR-155 in the regulation, the maturation and function of inflammatory cells, thus altering CD4+ cell inflammatory response (9,32,33,44,45).
To further understand the phenotype of miR-155 deficient-Treg cells we isolated and cultured CD4+CD25+ from WT and miR-155−/− mice. As expected the target gene Ship1 was found overexpressed in miR-155 deficient cells while Il-10 was reduced. The inositol phosphatase Ship1 is largely know to be a regulator of immune system, particularly in Treg cells (16,46,47). The Ship1 inhibitor 3AC (iSHIP) has been previously described to promote a boost of the immune system and to improve the immunoregulatory capacity of cells (48,49). In order to understand the role of miR-155 and Ship1 in acute liver damage, we blocked Ship1 activity in miR-155−/− and WT mice.
Our results show a reduction, in the increase of liver transaminases of miR-155−/− treated with iSHIP when compared to WT mice. Furthermore, iSHIP decreased the liver gene expression level of proinflammatory cytokines in miR-155−/− mice to a similar level as WT animals. As expected, the recruitment of Foxp3+ cells was increased in iSHIP-treated miR-155−/− animals. Thus, we observed that the inhibition of Ship1 is able to inhibit the exacerbated inflammatory response observed in miR-155−/− ConA-treated mice.
To further elucidate if the expression of miR-155 in inflammatory cells may determine liver injury we performed BM transplantation, which allowed evaluation of the role of miR-155 in the expression of inflammatory cells in liver injury. Although chimerism of engrafted WT-BM was 72%, the effects were remarkable. Interestingly, there were no changes in global infiltration assessed by CD45, but cytokine expression was reduced in miR-155−/− WT-BM mice while the expression of Foxp3 was increased. Moreover, WT-BM transplant into miR-155-deficient animals attenuated liver injury. These results suggest that miR-155 expression in inflammatory cells may play an important role in the modulation of liver injury and inflammatory response.
Our results do not exclude the possible role of miR-155 in liver parenchyma. On the contrary, we show that miR-155 is expressed in human and mouse liver tissue and its expression increases in ConA-derived acute liver injury and in acute and in chronic liver diseases. This observation together with previous results suggest that miR-155 may have a pleiotropic role in the context of liver injury and disease, and that a correct expression in both liver tissue and inflammatory cells may be important. In an attempt to unveil the contribution of miR-155 expression in liver parenchyma to acute liver damage, we transplanted WT animals with a miR-155-deficient BM. Unfortunately, miR-155-deficient BM cells showed poor engraftment with 50% of chimerism, thereby precluding its evaluation. Therefore, future studies are needed to elucidate the contribution of miR-155 expression in hepatocytes to acute liver injury.
Results in human samples indicate a dysregulation of miR-155 expression in both liver tissue and inflammatory cells in liver diseases. Interestingly, while the expression of miR-155 in human liver tissue is increased, a marked reduction in miR-155 expression was found in circulating PBMC. Unfortunately, paired samples of liver tissue and PBMC were not obtained, and therefore it cannot be excluded that changes of miR-155 expression in the liver tissue and in PBMC may not be concomitant. Nevertheless, these results together with the experimental data suggest that in human diseases the expression of miR-155 in inflammatory cells is affected and may influence liver injury. Although the results obtained in humans clearly show significant changes in miR-155 expression, larger patient cohorts should be analysed in order to assess the potential association of miR-155 expression with clinical parameters and disease outcome.
Overall, this study provides evidences that miR-155 is dysregulated in both liver tissue and inflammatory cells in a liver disease context. Moreover, our results suggest that correct expression of miR-155 in inflammatory cells may be critical for effective inflammatory response to injury, particularly in immune-mediated liver injury. These results highlight the importance of the regulation of miRNAs in different cell types and tissues in the context of liver diseases, and suggest their importance in disease progression.
Supplementary Material
Figure 7. Expression of miR-155 in inflammatory cells modulates ConA-liver injury.
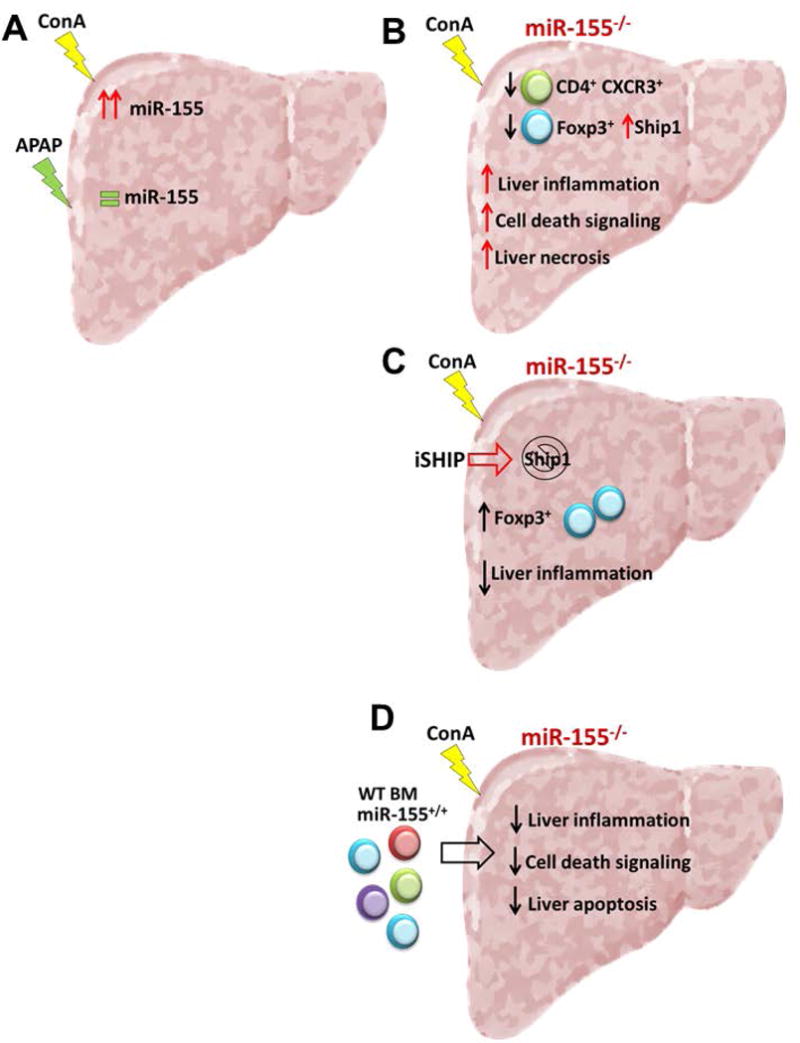
(A) Liver expression of miR-155 is highly increased in response to ConA treatment, while APAP treatment does not change miR-155 liver expression. (B) miR-155 deficiency increases ConA liver injury, liver inflammation and liver necrosis, showing a reduced CD4+ CXCR3+ and Treg cells recruitment in the liver. Treg cells from miR-155 deficient mice have an increased expression of miR-155 target gene Ship1. (C) Ship1 inhibition in miR-155 deficient animals reduces liver inflammation and increases Treg recruitment to the liver. (D) Restoration of mir-155 expression in inflammatory cells in a miR-155 deficient animal reduces liver inflammation, cell death signaling and liver apoptosis.
Acknowledgments
Financial support statement:
This work was supported by grants from the Fondo de Investigación Sanitaria Carlos III (FIS), co-financed by the Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea, “Una manera de hacer Europa” (FIS PI14/00320, PI 16/0043, FIS PI12/01265 to PS-B, PG, JC) and from the NIH National Institute on Alcohol Abuse and Alcoholism grant 1U01AA021908-01-33490, 1U01AA020821 and The European Foundation for Alcohol Research (ERAB) Grant EA1653 to PS-B. PS-B and BA-B are funded by the Instituto de Salud Carlos III, Miguel Servet (CP11/00071) and PFIS respectively and co-financed by the Fondo Europeo de Desarrollo Europeo (FEDER), Unión Europea, “Una manera de hacer Europa”. PG is funded by the Agencia de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) 2014 SGR 708, Centro de Investigación en Red Enfermedades Hepáticas y Digestivas (CIBEReHD) and the Institució Catalana de Recerca i Estudis Avançats (ICREA). FJC is funded by the MINECO Retos SAF2016-78711 and the Ramón y Cajal Fellowship (RYC-2014-15242).
Abbreviations
- miRNA
microRNA
- NK
natural killer cell
- AIH
autoimmune hepatitis
- PBMC
peripheral blood mononuclear cells
- miR-155−/−
miR-155 knockout mice
- WT
wild type
- ConA
concanavalin A
- APAP
acetaminophen
- BM
bone marrow
- qPCR
quantitative PCR
- ALT
alanine transaminase
- AST
aspartate transaminase
- LDH
lactate dehydrogenase
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- RIPK
receptor-interacting serine/threonine-protein kinase
- JNK
c-Jun N-terminal kinases
- MPO
myeloperoxidase
- iSHIP
SHIP1 inhibitor
Footnotes
Author’s contributions: DB contributed to the design of the study, and performed and analysed the experiments and interpreted the data. BA-B, SC-S, MC, LP and JV helped to perform the experiments and to interpret the data. IG, LL and EP included the patients, collected all the human samples and helped in the analysis and interpretation of human data. FH and FJC performed experiments and interpreted the results. JB, RB, JC and PG helped in the interpretation of the data and critically reviewing the final version. PS-B conceived the design of this study and was involved in the analysis, interpretation and drafting of the final version of the manuscript.
Conflict of interest statement: Nothing to declare.
References
- 1.Akbari Moqadam F, Pieters R, den Boer ML. The hunting of targets: challenge in miRNA research. Leukemia. 2013;27:16–23. doi: 10.1038/leu.2012.179. [DOI] [PubMed] [Google Scholar]
- 2.Erhard F, Haas J, Lieber D, Malterer G, Jaskiewicz L, Zavolan M, et al. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014;24:906–19. doi: 10.1101/gr.166702.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bala S, Szabo G. MicroRNA Signature in Alcoholic Liver Disease. Int. J. Hepatol. 2012;2012:498232. doi: 10.1155/2012/498232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaya D, Coll M, Rodrigo-Torres D, Vila-Casadesús M, Altamirano J, Llopis M, et al. Integrative microRNA profiling in alcoholic hepatitis reveals a role for microRNA-182 in liver injury and inflammation. Gut. 2016;65:1535–45. doi: 10.1136/gutjnl-2015-311314. [DOI] [PubMed] [Google Scholar]
- 5.Borel F, Konstantinova P, Jansen PLM, Parkin DM, Bosch FX, Ribes J, et al. Diagnostic and therapeutic potential of miRNA signatures in patients with hepatocellular carcinoma. J. Hepatol. 2012;56:1371–83. doi: 10.1016/j.jhep.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mashima R. Physiological roles of miR-155. Immunology. 2015;145:323–333. doi: 10.1111/imm.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vigorito E, Kohlhaas S, Lu D, Leyland R. miR-155: an ancient regulator of the immune system. Immunol. Rev. 2013;253:146–157. doi: 10.1111/imr.12057. [DOI] [PubMed] [Google Scholar]
- 9.Seddiki N, Brezar V, Ruffin N, Lévy Y, Swaminathan S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology. 2014;142:32–38. doi: 10.1111/imm.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumjohann D, Mark Ansel K. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Publ Gr. 2013;13 doi: 10.1038/nri3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner ML, Schnorfeil FM, Brocker T. MicroRNAs Regulate Dendritic Cell Differentiation and Function. J. Immunol. 2011;187:3911–3917. doi: 10.4049/jimmunol.1101137. [DOI] [PubMed] [Google Scholar]
- 12.Czyzyk-Krzeska MF, Zhang X. MiR-155 at the heart of oncogenic pathways. Oncogene. 2014;33:677–8. doi: 10.1038/onc.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranganath P. MicroRNA-155 and Its Role in Malignant Hematopoiesis. Biomark Insights. 2015;10:95. doi: 10.4137/BMI.S27676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu L-F, Gasteiger G, Yu I-S, Chaudhry A, Hsin J-P, Lu Y, et al. A Single miRNA-mRNA Interaction Affects the Immune Response in a Context- and Cell-Type-Specific Manner. Immunity. 2015;43:52–64. doi: 10.1016/j.immuni.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huffaker TB, O’connell RM. miR-155-SOCS1 as a Functional Axis: Satisfying the Burden of Proof. Immunity. 2015;43:3–4. doi: 10.1016/j.immuni.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 16.Locke NR, Patterson SJ, Hamilton MJ, Sly LM, Krystal G, Levings MK. SHIP regulates the reciprocal development of T regulatory and Th17 cells. J. Immunol. 2009;183:975–83. doi: 10.4049/jimmunol.0803749. [DOI] [PubMed] [Google Scholar]
- 17.O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang B, Majumder S, Nuovo G, Kutay H, Volinia S, Patel T, et al. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50:1152–1161. doi: 10.1002/hep.23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, et al. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286:1436–1444. doi: 10.1074/jbc.M110.145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeligar S, Tsukamoto H, Kalra VK. Ethanol-Induced Expression of ET-1 and ET-BR in Liver Sinusoidal Endothelial Cells and Human Endothelial Cells Involves Hypoxia-Inducible Factor-1 and MicroRNA-199. J. Immunol. 2009;183:5232–5243. doi: 10.4049/jimmunol.0901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng YQ, Ren JP, Zhao J, Wang JM, Zhou Y, Li GY, et al. MicroRNA-155 regulates interferon- γ production in natural killer cells via Tim-3 signalling in chronic hepatitis C virus infection. Immunology. 2015;145:485–497. doi: 10.1111/imm.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bala S, Csak T, Saha B, Zatsiorsky J, Kodys K, Catalano D, et al. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J Hepatol. 2016 doi: 10.1016/j.jhep.2016.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lippai D, Bala S, Csak T, Kurt-Jones EA, Szabo G. Chronic Alcohol-Induced microRNA-155 Contributes to Neuroinflammation in a TLR4-Dependent Manner in Mice. PLoS One. 2013;8:1–10. doi: 10.1371/journal.pone.0070945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Han C, Zhang J, Song K, Wang Y, Wu T. Deletion of Mir155 prevents fas-induced liver injury through up-regulation of Mcl-1. Am J Pathol. 2015;185:1033–1044. doi: 10.1016/j.ajpath.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller AM, Gilchrist DS, Nijjar J, Araldi E, Ramirez CM, Lavery CA, et al. MiR-155 Has a Protective Role in the Development of Non-Alcoholic Hepatosteatosis in Mice. PLoS One. 2013;8 doi: 10.1371/journal.pone.0072324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Affo S, Dominguez M, Lozano JJ, Sancho-Bru P, Rodrigo-Torres D, Morales-Ibanez O, et al. Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut. 2013;62:452–460. doi: 10.1136/gutjnl-2011-301146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H-X, Liu M, Weng S-Y, Li J-J, Xie C, He H-L, et al. Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J. Gastroenterol. 2012;18:119–25. doi: 10.3748/wjg.v18.i2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heymann F, Hamesch K, Weiskirchen R, Tacke F. The concanavalin A model of acute hepatitis in mice. Lab. Anim. 2015;49:12–20. doi: 10.1177/0023677215572841. [DOI] [PubMed] [Google Scholar]
- 29.Tujios S, Fontana RJ. Mechanisms of drug-induced liver injury: from bedside to bench. Nat. Rev. Gastroenterol. Hepatol. 2011;8:202–211. doi: 10.1038/nrgastro.2011.22. [DOI] [PubMed] [Google Scholar]
- 30.Martin-Murphy BV, Kominsky DJ, Orlicky DJ, Donohue TM, Ju C. Increased susceptibility of natural killer T-cell-deficient mice to acetaminophen-induced liver injury. Hepatology. 2013;57:1575–1584. doi: 10.1002/hep.26134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu L-F, Thai T-H, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J. Immunol. 2009;182:2578–2582. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- 33.Yao R, Ma Y-L, Liang W, Li H-H, Ma Z-J, Yu X, et al. MicroRNA-155 Modulates Treg and Th17 Cells Differentiation and Th17 Cell Function by Targeting SOCS1. PLoS One. 2012;7:e46082. doi: 10.1371/journal.pone.0046082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology. 2012;56:1946–1957. doi: 10.1002/hep.25873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bala S, Csak T, Saha B, Zatsiorsky J, Kodys K, Catalano D, et al. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016;64:1378–87. doi: 10.1016/j.jhep.2016.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krenkel O, Mossanen JC, Tacke F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg. Nutr. 2014;3:331–43. doi: 10.3978/j.issn.2304-3881.2014.11.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tiegs G, Hentschel J, Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Invest. 1992;90:196–203. doi: 10.1172/JCI115836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jablonski KA, Gaudet AD, Amici SA, Popovich PG, Guerau-De-Arellano M. Control of the Inflammatory Macrophage Transcriptional Signature by miR-155. PLoS One. 2016;11 doi: 10.1371/journal.pone.0159724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Cheng Y, Cui W, Li M, Li B, Guo L. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol. 2014;266:56–63. doi: 10.1016/j.jneuroim.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, van Mil A, Vrijsen K, Zhao J, Gao L, Metz CHG, et al. MicroRNA-155 prevents necrotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J Cell Mol Med. 2011;15:1474–82. doi: 10.1111/j.1582-4934.2010.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gironella M, Seux M, Xie M-J, Cano C, Tomasini R, Gommeaux J, et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci. 2007;104:16170–16175. doi: 10.1073/pnas.0703942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–9. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen D, Bromberg JS. T Regulatory Cells and Migration. Am J Transplant. 2006;6:1518–1523. doi: 10.1111/j.1600-6143.2006.01372.x. [DOI] [PubMed] [Google Scholar]
- 44.O’Connell RM, Kahn D, Gibson WSJ, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–19. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thai T-H, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the Germinal Center Response by MicroRNA-155. Science (80-) 2007:316. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 46.Collazo MM, Wood D, Paraiso KHT, Lund E, Engelman RW, Le C-T, et al. SHIP limits immunoregulatory capacity in the T-cell compartment. Blood. 2009;113:2934–2944. doi: 10.1182/blood-2008-09-181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collazo MM, Paraiso KHT, Park M-Y, Hazen AL, Kerr WG. Lineage extrinsic and intrinsic control of immunoregulatory cell numbers by SHIP. Eur J Immunol. 2012;42:1785–1795. doi: 10.1002/eji.201142092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brooks R, Iyer S, Akada H, Neelam S, Russo CM, Chisholm JD, et al. Coordinate Expansion of Murine Hematopoietic and Mesenchymal Stem Cell Compartments by SHIPi. Stem Cells. 2015;33:848–858. doi: 10.1002/stem.1902. [DOI] [PubMed] [Google Scholar]
- 49.Gumbleton M, Sudan R, Fernandes S, Engelman RW, Russo CM, Chisholm JD, et al. Dual enhancement of T and NK cell function by pulsatile inhibition of SHIP1 improves antitumor immunity and survival. Sci Signal. 2017;10:eaam5353. doi: 10.1126/scisignal.aam5353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.