

Abstract
N6-methyladenosine (m6A) modification is the most abundant RNA methylation modification and involves various biological processes, such as RNA splicing and degradation. Recent studies have demonstrated the feasibility of identifying m6A peaks using high-throughput sequencing techniques. However, such techniques cannot accurately identify specific methylated sites, which is important for a better understanding of m6A functions. In this study, we develop a novel machine learning-based predictor called M6APred-EL for the identification of m6A sites. To predict m6A sites accurately within genomic sequences, we trained an ensemble of three support vector machine classifiers that explore the position-specific information and physical chemical information from position-specific k-mer nucleotide propensity, physical-chemical properties, and ring-function-hydrogen-chemical properties. We examined and compared the performance of our predictor with other state-of-the-art methods of benchmarking datasets. Comparative results showed that the proposed M6APred-EL performed more accurately for m6A site identification. Moreover, a user-friendly web server that implements the proposed M6APred-EL is well established and is currently available at http://server.malab.cn/M6APred-EL/. It is expected to be a practical and effective tool for the investigation of m6A functional mechanisms.
Keywords: RNA methylation, support vector machine, ensemble learning, N6-methyladenosine, PS(k-mer)NP
Graphical Abstract
Introduction
Post-transcriptional modifications of RNA play a crucial role in understanding a variety of cellular processes, such as RNA splicing, RNA degradation, protein translation, stability, and immune tolerance.1, 2 N6-methyladenosine (m6A) is the most abundant RNA post-transcriptional modification.3 RNA m6A modification is catalyzed by a methyltransferase complex containing at least one subunit of METTL3 (methyltransferase-like 3). The event is reversible under catalysis of demethylases FTO and ALKBH5 and usually occurs at adenine (A) with the genetic motif GAC. Recent studies have demonstrated that m6A is also closely related to cancer and other human diseases.4 Therefore, it is of great importance to correctly identify m6A modification sites of RNA or genomic sequences containing GAC motifs. This would help us to understand and reveal in depth the functional mechanisms of m6A sites.
m6A modification has been detected in a variety of species, such as Saccharomyces cerevisiae,5 Arabidopsis thaliana,6 Homo sapiens,7 Mus musculus,7 and other species. In recent years, high-throughput sequencing techniques, such as MERIP-seq4 and m6A sequencing (m6A-seq),8 have identified m6A peaks. However, identifying m6A sites using next-generation sequencing techniques involves some intrinsic problems, such as low accuracy when detecting m6A sites and not being available for large-scale identification of genomic sequences.
In the past few years, machine learning-based methods have emerged as an attractive approach for m6A site identification. It is common to build predictive models with several machine learning algorithms. For instance, Schwartz et al.5 proposed the first machine learning-based method to predict m6A sites using features like local secondary structure stability, nucleotide composition, and relative position in sequences and training a logistic regression (LR) classifier to achieve promising predictive results. Later, Chen et al.9 established an m6A site predictor called “iRNA-Methyl” via pseudonucleotide composition and support vector machine (SVM). This predictor is reported to yield an accuracy of 65.59%, Matthew’s correlation coefficient (MCC) of 0.29 on a dataset containing 1,307 positives (m6A site-surrounding sequences), and 1,307 negatives (non-m6A site-surrounding sequences) in S. cerevisiae. To improve the predictive performance, Liu et al.10 proposed to incorporate auto-covariance and cross-covariance with physical-chemical properties for representations of RNA sequences and built a predictor named “pRNAm_PC” with SVM as the underlying prediction engine, successfully enhancing the accuracy to 69.74%. Moreover, Jia et al.11 incorporated bi-profile Bayes, dinucleotide composition, and k-nearest neighbor (KNN) scores for three feature extractions to establish a predictor named RNA-MethylPred, which yields better performance than iRNA-Methyl and pRNAm_PC. Likewise, a new predictor called “AthMethPre,” proposed by Zeng et al.,12 also employed SVM to train a classification model but used different features derived from positional flanking nucleotide sequences and a position-independent k-mer nucleotide spectrum. More recently, Zhou et al.13 developed a predictor called “SRAMP” by using an ensemble of three random forest (RF) classifiers respectively trained with three feature-encoding algorithms: sequence positional binary encoding, k-nearest neighbor encoding, and nucleotide pair spectrum encoding. It can be inferred that feature representation ability is the main focus of existing predictors to further improve predictive accuracy. Although much progress has been made, it is still a challenging task to extract sufficiently informative features to accurately distinguish m6A sites from non-m6A sites.
In this study, we proposed a novel sequence-based predictor called “M6APred-EL” for the identification of m6A sites within RNA sequences. As for feature representation, we proposed and used three types of feature descriptors to exploit physical-chemical information and position-specific information, including position-specific k-mer nucleotide propensity, physical-chemical properties, and ring-function-hydrogen-chemical properties, respectively. In the predictive model of M6APred-EL, we used an ensemble classifier as the underlying prediction engine. To construct the ensemble classifier, we trained three SVMs as base classifiers using the above three feature descriptors and then combined them as the ensemble classifier using a major voting strategy. Experimental results showed that our proposed predictive model outperformed existing methods in the literature under a benchmarking validation test, demonstrating the superiority of our predictor. Thus, it can be expected that our predictor can be an effective tool for identifying m6A sites.
Results and Discussion
Comparison of SVM and Other Classifiers
To measure the effectiveness of the underlying SVM, we compared its performance with other five commonly used machine learning algorithms, such as RF, LR, decision tree, nearest neighbors, and naive Bayes. The reason for using these algorithms as references is that they are all widely used in a lot fields of bioinformatics, including methylation site prediction14 and detection of tubule boundaries.15 For fair comparison, all classifiers were used under equal conditions; i.e., modeling with the same dataset (m6A dataset) and feature extraction method (i.e., ring-function-hydrogen-chemical properties without GAC [RFHC-GACs]). Algorithm performance is presented in Table 1. As shown in Table 1, the SVM algorithm achieved the best performance in terms of all four metrics compared with the other classifiers. The accuracy (Acc), sensitivity (Sn), specificity (Sp), and MCC of the SVM are 72.46%, 72.07%, 72.84%, and 0.4491, respectively. Compared with the runner-up LR classifier (Acc of 71.50%, Sn of 71.46%, Sp of 71.54%, and MCC of 0.43), the performance of SVM is 0.96%, 0.61%, 1.30%, and 1.91% higher in terms of Acc, Sn, Sp, and MCC, respectively. This demonstrates that the SVM has more classification power and effectiveness for distinguishing m6A sites from non-m6A sites than other classification algorithms.
Table 1.
Performance Comparison of SVM and Other Different Classifiers
| Classifiers | Acc (%) | Sn (%) | Sp (%) | MCC |
|---|---|---|---|---|
| Naive Bayes | 65.95 | 64.65 | 67.25 | 0.3192 |
| Decision tree | 65.19 | 66.41 | 63.96 | 0.3038 |
| RF | 67.44 | 69.63 | 65.26 | 0.3492 |
| LR | 71.50 | 71.46 | 71.54 | 0.4300 |
| Nearest neighbors | 65.72 | 69.47 | 61.97 | 0.3153 |
| SVM | 72.46 | 72.07 | 72.84 | 0.4491 |
Parameter Determination of the PS(k-mer)NP Descriptor
In this section, we compared five feature representation methods based on the optimized SVM model. These feature matrices were extracted by position-specific k-mer nucleotide propensity (PS(k-mer)NP), where , and the dimensions of them are 51, 50, 49, 48, and 47, respectively. To fairly compare the performance of the five classifiers, the benchmark dataset illustrated in Dataset was used, and their optimization value ranges of SVM parameters were controlled at the same level, which is elaborated in Ensemble of SVM. The predictive performance was evaluated with the benchmark dataset. As shown in Table 2, the PS(1-mer)NP achieved the highest Acc of 74.28%, Sn of 74.89%, and MCC of 0.4860. Compared with the PS(2-mer) NP, which has the best Sp of 74.9%, PS(1-mer)NP reached 0.46%, 2.14%, and 0.86% in terms of Acc, Sn, and MCC, respectively. To more intuitively compare the performance of these features, the receiver operating characteristic (ROC) and physical chemical (PC) curves are illustrated in Figure 1. As shown in Figure 1, similar results can be observed, showing that the PS(1-mer)NP is comparable with the PS(2-mer)NP, outperforming other PS(k-mer)NP features in terms of area under ROC (AUROC) and area under precision-recall curve (AUPRC). Therefore, PS(1-mer)NP is used in our predictive model.
Table 2.
Predictive Performance of the PS(k-mer)NP Descriptors with Varied k Values
| Feature Representation | Acc (%) | Sn (%) | Sp (%) | MCC |
|---|---|---|---|---|
| PS(1-mer)NP | 74.28 | 74.89 | 73.67 | 0.4860 |
| PS(2-mer)NP | 73.82 | 72.75 | 74.90 | 0.4774 |
| PS(3-mer)NP | 70.07 | 69.99 | 70.16 | 0.4020 |
| PS(4-mer)NP | 67.32 | 67.70 | 66.95 | 0.3469 |
| PS(5-mer)NP | 65.26 | 65.72 | 64.80 | 0.3054 |
Figure 1.

Performance of the PS(k-mer)NP Feature Descriptor with Varied k Values on Benchmarking Dataset
(A) ROC curves of the PS(k-mer)NP feature descriptor under different k values (k = 1, 2, 3, 4, 5). (B) PR curves of the PS(k-mer)NP feature descriptor under different k values (k = 1, 2, 3, 4, 5).
Comparison of the Ensemble Classifier and Its Three Base Classifiers
In our predictive model, we trained an ensemble classifier that combines three SVM classifiers, each of which we called “base classifier.” The three base classifiers were preliminarily trained using three feature descriptors, including RFHC-GACs, PC properties (PCPs), and PS(1-mer)NP. To validate the effectiveness of the ensemble classifier, we evaluated its performance with the 10-fold cross validation test. For comparison, its three base classifiers were also evaluated. The evaluation results are presented in Table 3.
Table 3.
Comparison of Voting Performance with Single Classifier
| Feature Descriptors | Acc (%) | Sn (%) | Sp (%) | MCC |
|---|---|---|---|---|
| PCP | 68.89 | 68.63 | 69.16 | 0.3781 |
| RFHC-GAC | 73.10 | 71.83 | 74.37 | 0.4627 |
| PS(1-mer)NP | 74.28 | 74.89 | 73.67 | 0.4860 |
| Ensemble classifier | 80.83 | 80.72 | 80.95 | 0.6167 |
As seen in Table 3, among the three base classifiers, the classifier trained using the PS(1-mer)NP features outperforms the other two base classifiers trained with PCPs and RFHC-GACs. To be specific, the Acc, Sn, Sp, and MCC of the PS(1-mer)NP-based classifier are 74.28%, 74.89%, 73.67%, and 0.486, respectively. Three of the metrics (Acc, Sn, and MCC) are higher than those of the other base classifiers. When the three base classifiers were combined to construct the ensemble classifier, we found that the performance improved significantly. The four metrics of the ensemble classifier increased to 80.83%, 80.72%, 80.95%, and 0.6167, which is 6.55%, 5.83%, 7.28% and 0.1307 higher, respectively, than the best-performing base classifier (based on PS(1-mer)NP). To compare the performance more intuitively, ROC and PC curves are plotted in Figure 2. As shown in Figure 2, we can observe that the ensemble classifier also exhibits a better performance in terms of AUROC and AUPRC. The two metrics of the ensemble classifiers are above 0.90, whereas that of the other three base classifiers is below 0.8. The significant improvement by the ensemble strategy is probably because that the outcomes of three basic classifiers exist the significant difference. Therefore, based on the difference ensemble theory, fusing these outcomes can effectively improve the performance.
Figure 2.

Performance of the Proposed Ensemble Classifier and Its Three Base Classifiers
(A) ROC curves of the proposed ensemble classifier and its three base classifiers trained with PCP, PS(1-mer)NP, and RFHC-GAC, respectively. (B) PR curves of the proposed ensemble classifier and its three base classifiers trained with PCP, PS(1-mer)NP, and RFHC-GAC, respectively.
Comparison of the Ensemble Strategy and Feature Fusion Strategy
The proposed predictive model in this study is integrated by three base classifiers that were trained with three feature descriptors, including RFH-GACs, PCPs, and PS(1-mer)NP. To further illustrate the superiority of the proposed ensemble classifier, we also constructed a classifier based on the feature fusion strategy that merges the above three feature descriptors into one. We compared our ensemble classifier with the newly constructed classifier on the benchmark dataset with 10-fold cross-validation. The evaluation results are illustrated in Figure 3. As seen from Figure 3, the performance of the newly constructed classifiers is 74.97%, 74.97%, 74.97%, and 0.50% in terms of Acc, Sn, Sp, and MCC, respectively. Compared with our ensemble classifier, our classifier performance is 5.86%, 5.75%, 5.98%, and 0.12% higher than the classifier based on feature fusion in terms of Acc, Sn, Sp, and MCC. This demonstrates that the ensemble strategy is more effective than the feature fusion strategy.
Figure 3.

Performance of the Ensemble Classifier and the Classifier Based on the Feature Fusion Strategy
Comparison with State-of-the-Art Predictors
To evaluate the performance of the proposed M6APred-EL, we compared our predictor with four state-of-the-art predictors, including iRNA-methyl,9 pRNAm_PC,10 RAM-ESVM,16 and RAM_NPPS.17 The reason to choose the above four predictors for comparison is that they have been reported to achieve outstanding predictive performance in m6A site identification. For fairness of comparison, all compared predictors were trained and validated on the same benchmarking dataset as presented in this study. The evaluation results are summarized in Table 4. It can be observed that, among the compared predictors, the proposed M6APred-EL obtained the best performances in terms of Acc, Sn, Sp, and MCC, with 80.83%, 80.72%, 80.95%, and 0.62%, respectively. Specifically, compared with the best of the existing predictors, RAM_NPPS, our Acc, Sn, Sp, and MCC are 1.18%, 2.3%, 0.08%, and 0.03% higher, respectively. The performance improvement by our predictor indicated that our predictor is more accurate than the state-of-the-art predictors to distinguish true m6A sites from non-m6A sites. Furthermore, the performance of Acc and MCC is higher than in previous research, and it illustrates that the predictor is more stable and reliable. This is extraordinary progress in biological research because a more reliable tool for the identification of biological macromolecules can enormously reduce experimental cost. In conclusion, the predictor can be expected to be a tool with high availability for the identification of m6A sites.
Table 4.
Performance of the Proposed M6APred-EL and Other State-of-the-Art Predictors on the Benchmarking Dataset
| Predictors | Acc (%) | Sn (%) | Sp (%) | MCC |
|---|---|---|---|---|
| iRNA-Methyl | 65.59 | 70.55 | 60.63 | 0.29 |
| pRNAm_PC | 69.74 | 69.72 | 69.75 | 0.4 |
| RAM-ESVM | 78.35 | 78.93 | 77.78 | 0.57 |
| RAM_NPPS | 79.65 | 78.42 | 80.87 | 0.59 |
| M6APred-EL (this study) | 80.83 | 80.72 | 80.95 | 0.62 |
Conclusion
RNA is considered to be related to several diseases,18, 19 including cancer.20, 21 In this study, we propose a novel predictor called M6APred-EL for the identification of m6A sites within RNA. To explore sufficient information to improve predictive performance, we used three feature representation approaches from position-specific nucleotide composition and physical-chemical properties. In particular, PS(k-mer)NP is a novel algorithm proposed in this study. In this study, PS(1-mer)NP is adopted as the basic feature extraction algorithm for its better performance in comparison with PS(k-mer)NPs within its five k values. For our predictor, we constructed an ensemble classifier by combining three SVM classifiers trained with the three types of features. Through a series of experimental analyses, we found that, after combining the three classifiers, performance improved significantly. This implies that the prediction outcomes differ to some extent, which is beneficial for forming an improved prediction model. To validate the effectiveness of the predictor M6APred-EL proposed in this study, we compare it with state-of-the-art predictors. The cross-validation results show that our predictor outperforms existing predictors by 1.18% and 0.03% in terms of Acc and MCC. It is anticipated that M6APred-EL will be a highly available and indispensable software tool for detecting m6A sites within RNA.
Materials and Methods
Dataset
A dataset originally proposed in Chen’s work9 is widely used as the benchmarking dataset for performance comparison of m6A site predictors. For fair comparison, we also employed this dataset to evaluate and compare the proposed predictor with existing predictors. This dataset contains a total of 2,614 sequences derived from Saccharomyces cerevisiae, of which 1,307 sequences are positive samples and an equal number of sequences are negative samples. Positive samples are sequences centered on true m6A sites, whereas negative samples are sequences centered on non-m6A sites. Notably, all positive and negative samples are 51 nt long. Moreover, the sequence identity of this dataset is less than 85%. As explained by Chen et al.,9 this can prevent biased performance evaluation of this dataset. More details regarding this dataset can be found in the study by Chen et al.9 The dataset can be downloaded from http://server.malab.cn/M6APred-EL/.
Prediction Framework of the Proposed Predictor
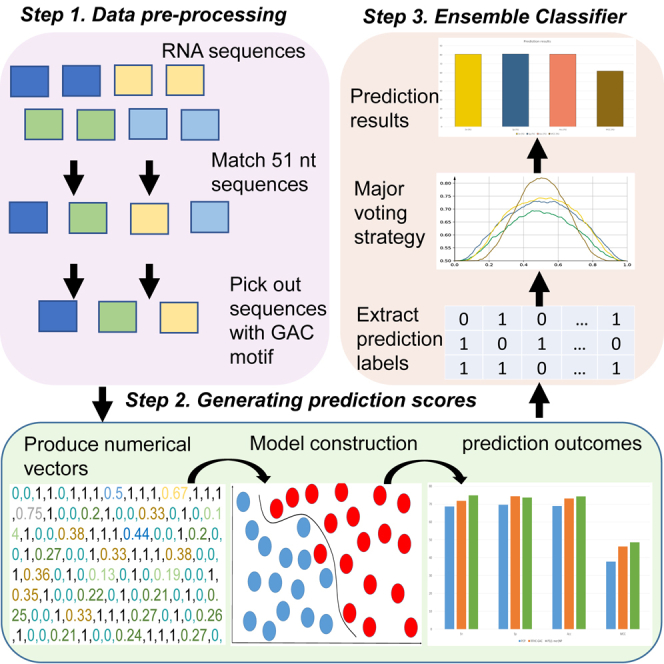
To precisely identify m6A sites within RNA sequences, in this study, we proposed a sequence-based predictor called M6APred-EL. The overall process is illustrated in Figure 4. The prediction procedure of M6APred-EL involves three steps. First, for given query RNA sequences, a 51-nt flanking window is used to scan the sequences; sub-sequences centered on GAC motifs are selected. Second, the resulting sequences are submitted to three types of feature representation approaches to extract numerical feature vectors, which are then submitted to three well-trained SVM models for prediction. Afterward, each model gives the prediction score for the corresponding feature vector. Notably, the prediction score is 0 or 1. If the score is 0, it indicates that the sequence is predicted to be a true m6A site; otherwise, it is a non-m6A site. Finally, we used a major voting strategy to combine the prediction scores of the three models and then obtained the final prediction score. With this strategy, if the scores of two of the three models are 0, then the final score of our ensemble classifier is 0, which indicates that the sequence is a true m6A site; otherwise, it is a non-m6A site. More details about feature representation methods and SVM can be found in Feature Representation and Ensemble of SVM, respectively.
Figure 4.

Framework of M6APred-EL
The procedure of m6A site identification is described in the following three steps. First, original input RNA sequences are scanned with a 51-nt window. Those sequences, including the GAC motif, were retained; the others were discarded. Second, the remaining sequences are submitted to three feature representation approaches and predicted by three well-trained SVM models to generate three prediction scores. Finally, the prediction result is generated by the major voting strategy.
Feature Representation
Feature representation, fusion,22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41 and selection42, 43, 44, 45, 46, 47, 48, 49 are the key steps in the machine learning process. In this paper, we propose and employ three feature representation algorithms, including PS(k-mer)NP, PCPs,10 and RFHC-GACs. The three algorithms are capable to extract features directly from RNA primary sequences. The dataset presented in this study contains equal-length samples (RNA sequences). Thus, each sequence sample is denoted as follows:
| (Equation 1) |
where L represents the sequence length, and represents one specific nucleotide in the i-th position of the sequence, which can be denoted as
| (Equation 2) |
The three sequence-based feature representation algorithms are described as follows.
PS(k-mer)NP
The position-specific theory has been successfully applied to many fields of bioinformatics. For instance, a previous study has revealed that the position-specific composition of tri-nucleotides is powerful for identifying promoters. Motivated by this work, we propose a new feature representation algorithm, PS(k-mer)NP, which is also based on the position-specific theory.
To compute the PS(k-mer)NP features, we first defined a k-mer nucleotide set as follows:
| (Equation 3) |
where represents k consecutive As, represents (k-1) consecutive As followed by a C, represents (k-1) consecutive As followed by two Cs, and so forth. Thus, there are a total of 4k nucleotide combinations. Then we computed position-specific k-mer nucleotide propensity on the whole dataset and generated a 4k(52-k) global frequency matrix represented as
| (Equation 4) |
with the element formulated as
| (Equation 5) |
where means the occurrence frequency of the i-th k-mer nucleotides at the j-th position in the positive samples and denotes the occurrence frequency of the i-th k-mer nucleotides at the j-th position in the negative samples . For the convenience of calculating, the algorithm can also be expressed as the vector as follows:
| (Equation 6) |
where T represents the transpose operator. is indicated by
| (Equation 7) |
where represents a sub-sequence with K bases long. This method measures the specificity of position-specific k-mer nucleotide on the entire dataset.
RFHC-GAC
There are four types of ribonucleotides: adenine (A), cytosine (C), guanine (G), and uracil (U). Previous studies have demonstrated that the four nucleotides have different chemical properties, such as rings, functional groups, and hydrogen bonds.50 As for the ring structures, A and G are purines that have two rings structures, whereas C and U are pyrimidines that have one ring only. In terms of secondary structures, A and U are assigned to one group because they both contain weak hydrogen bonds, whereas C and G are in the same group because they have strong hydrogen bonds. Regarding chemical functionality, A and C can be assigned to an amino group, whereas the others are classified into a keto group. To incorporate the chemical property information into feature representation, we constructed a four-dimensional vector .51, 52, 53 The algorithm can be formulated as follows:
| (Equation 8) |
where x denotes a nucleotide and A, C, G, and U can be represented as , , , and (0,1,0), respectively, considering that a certain degree of correlation between one nucleotide and the sequence to which it belongs. The density method is used to measure the relevance between frequency and position. The density of can be denoted by the following formula:
| (Equation 9) |
where L denotes sequence length, denotes the length from current nucleotide position to first nucleotide, and q is a symbol of . To intuitively illustrate the algorithm, for a given sequence example ‘CAAAGGUGAC’, it can be transferred into the following discrete vector like (1.0,0.5,0.67,0.75,0.2,0.33,0.14,0.38,0.44,0.2).
To further enhance the prediction ability, it is important to reduce similarity between positive and negative samples. Obviously, the GAC motif exists in the same position of both positive and negative samples. When we use these samples to train a prediction model, the similarity would affect the predictive performance. Thus, it is imperative to extract information through its flanking nucleotide without using the GAC motif in the center. Finally, the vector length is.
Features Based on Physical-Chemical Properties
The feature method based on physical-chemical properties10, 54 is proposed to incorporate the dinucleotide composition with physical-chemical properties and the transformation of auto-covariance and cross covariance. Here we used the following 10 physical-chemical properties: PC1, rise;55 PC2, roll;55 PC3, shift;55 PC4, slide;55 PC5, tilt;55 PC6, twist;55 PC7, enthalpy;55 PC8, entropy;56 PC9, stack energy;57 PC10, free energy.56 For an RNA sequence, there are kinds of dinucleotides. Each of the 16 dinucleotides has a set of 10 PC properties corresponding to specific values, as shown in Table 5.
Table 5.
The Original 10 Physical-Chemical Properties for 16 Dinucleotides
| Dinucleotides | PC1 | PC2 | PC3 | PC4 | PC5 | PC6 | PC7 | PC8 | PC9 | PC10 |
|---|---|---|---|---|---|---|---|---|---|---|
| AA | 3.18 | 7 | −0.08 | −1.27 | −0.8 | 31 | −6.82 | −18.4 | −13.7 | −0.9 |
| AC | 3.24 | 4.8 | 0.23 | −1.43 | 0.8 | 32 | −11.4 | −26.2 | −13.8 | −2.1 |
| AG | 3.3 | 8.5 | −0.04 | −1.5 | 0.5 | 30 | −10.48 | −19.2 | −14 | −1.7 |
| AU | 3.24 | 7.1 | −0.06 | −1.36 | 1.1 | 33 | −9.38 | −15.5 | −15.4 | −0.9 |
| CA | 3.09 | 9.9 | 0.11 | −1.46 | 1 | 31 | −10.44 | −27.8 | −14.4 | −1.8 |
| CC | 3.32 | 8.7 | −0.01 | −1.78 | 0.3 | 32 | −13.39 | −29.7 | −11.1 | −2.9 |
| CG | 3.3 | 12.1 | 0.3 | −1.89 | −0.1 | 27 | −10.64 | −19.4 | −15.6 | −2 |
| CU | 3.3 | 8.5 | −0.04 | −1.5 | 0.5 | 30 | −10.48 | −19.2 | −14 | −1.7 |
| GA | 3.38 | 9.4 | 0.07 | −1.7 | 1.3 | 32 | −12.44 | −35.5 | −14.2 | −2.3 |
| GC | 3.22 | 6.1 | 0.07 | −1.39 | 0 | 35 | −14.88 | −34.9 | −16.9 | −3.4 |
| GG | 3.32 | 12.1 | −0.01 | −1.78 | 0.3 | 32 | −13.39 | −29.7 | −11.1 | −2.9 |
| GU | 3.24 | 4.8 | 0.23 | −1.43 | 0.8 | 32 | −11.4 | −26.2 | −13.8 | −2.1 |
| UA | 3.26 | 10.7 | −0.02 | −1.45 | −0.2 | 32 | −7.69 | −22.6 | −16 | −1.1 |
| UC | 3.38 | 9.4 | 0.07 | −1.7 | 1.3 | 32 | −12.44 | −26.2 | −14.2 | −2.1 |
| UG | 3.09 | 9.9 | 0.11 | −1.46 | 1 | 31 | −10.44 | −19.2 | −14.4 | −1.7 |
| UU | 3.18 | 7 | −0.08 | −1.27 | −0.8 | 31 | −6.82 | −18.4 | −13.7 | −0.9 |
For given a RNA sequence, it can be formulated as the following dinucleotide vector:
| (Equation 10) |
where C represents the dinucleotide vector, each element is a dinucleotide pair, and L is the length of each sequence. Then the dinucleotide vector can be further encoded with the following matrix as
| (Equation 11) |
where denotes the j-th property value for dinucleotide in Equation 10. The property matrix is illustrated in Table 5, which needs to be normalized before being applied to extract features.
For the same line of PCs, the correlation between two nucleotides is separated by nucleotide intervals. To incorporate the auto-covariance transformation, the features are computed as
| (Equation 12) |
where ranges from 0 to 49 and represents the average of the m-th row in the matrix, which is illustrated in Equation 11.
Cross-covariance measures the correlation between two different nucleotides belonging to different properties. It can be formulated by the following:
| (Equation 13) |
To this end, we yielded features from auto-covariance and features from cross-covariance. In this study, we set as 4 because it contributes to the best predictive performance. Therefore, we have a total of features based on physical-chemical properties.
Ensemble of SVM
SVM is a powerful algorithm that has been widely used in bioinformatics fields.13, 58, 59, 60, 61, 62, 63, 64, 65, 66 The basic idea of SVM is to determine a separating hyperplane to maximize the margin between positive and negative samples. In particular, as for non-linear separable data, the SVM algorithm uses kernel functions to map a non-linear feature space to a high-dimensional one, where the mapped feature space is linearly separable. There are three kernel functions that are usually used: polynomial, radial basis function (RBF), and Gaussian. Here, RBF was used as the kernel function because its performance is better than the other two. More details regarding SVM and its kernel function can refer be found in Chou et al.,67 Cai et al.,68 and Cristianini and Shawe-Taylor.69 To achieve the best classification performance, we optimized two parameters of the SVM algorithm by using a grid search approach. The first parameter is the penalty coefficient (denoted as ), and the second one is , which is used to balance the kernel function in case of overfitting. The optimization ranges about C and are , respectively. The SVM is implemented and optimized in the Python package (version 3.5.2).
To construct an ensemble classifier, we first extracted the three types of features of the training dataset using the three feature descriptors, respectively. Afterward, for each training dataset encoded by one specific feature descriptor, we trained an SVM model. A total of three SVM models were obtained. For each trained model, we evaluated its predictive performance and produced the prediction scores. Finally, if the scores of two of the three models are 0, then the final score of our ensemble classifier is 0, which indicates that the sequence is a true m6A site; otherwise, it is a non-m6A site.
Performance Measurement
In this study, four commonly used metrics are employed to evaluate predictive performance, including Acc, Sp, Sn, and MCC. They are formulated as follows:
| (Equation 14) |
where TP, TN, FP, and FN are true positive, true negative, false positive, and false negative, respectively. In this study, TP represents the number of true m6A sites predicted correctly, TN represents the number of non-m6A sites predicted correctly, FP represents the number of non-m6A sites predicted incorrectly as true m6A sites, and FN represents the number of true m6A sites predicted incorrectly as non-m6A sites. MCC and Acc are two metrics used for evaluating the overall performance of a predictive model on the whole dataset, whereas SN and SP are used for measuring the performance on positive samples and negative samples, respectively.
Moreover, we used the 10-fold cross-validation method to measure the predictive performance of the predictor. The procedure of this validation method is briefly described as follows. First, a dataset is randomly partitioned into 10 subsets with equal size. Of the 10 subsets, nine subsets are chosen as the training data to train a predictive model, whereas the remaining single subset is retained as the validation data to test the model. This process is then repeated 10 times, with each of the 10 subsets used exactly once as the validation data. Last, the 10 results are averaged to obtain a final prediction estimation.
ROC Curve
The ROC curve is often used to measure the overall performance of a binary classifier system. The ROC curve is generated by plotting the true positive rate (TPR) against the false positive rate (FPR) under different classification thresholds. The TPR is also known as sensitivity above, whereas the FPR can be calculated as (1 − specificity). We also calculated the area under the ROC curve (AUC) to evaluate the performance of a predictor. The range of the AUC is from 0.5 to 1. When the AUC score of a predictor is near 1, the predictor is considered a perfect predictor; when the AUC score is 0.5, it corresponds to a random predictor. The larger the AUC, the better and more robust the model.
Precision-Recall Curve
Another measurement is the precision-recall curve, which measures the trade-off in precision and recall. Precision-recall curves plot precision (the fraction of TP in all predicted positives) against recall (sensitivity) at various threshold settings. The PR curve is more sensitive to FPs than the ROC curve.
Author Contributions
L.W. wrote the manuscript and designed the experiments. H. C. carried out the experimental analysis. R.S. improved the manuscript. All authors read and approved the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
The work was supported by the National Natural Science Foundation of China (61701340 and 61702361) and the State Key Laboratory of Medicinal Chemical Biology in China.
References
- 1.Karikó K., Buckstein M., Ni H., Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Wei W., Ji X., Guo X., Ji S. Regulatory Role of N6 -methyladenosine (m6 A) Methylation in RNA Processing and Human Diseases. J. Cell. Biochem. 2017;118:2534–2543. doi: 10.1002/jcb.25967. [DOI] [PubMed] [Google Scholar]
- 3.Nilsen T.W. Molecular biology. Internal mRNA methylation finally finds functions. Science. 2014;343:1207–1208. doi: 10.1126/science.1249340. [DOI] [PubMed] [Google Scholar]
- 4.Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz S., Agarwala S.D., Mumbach M.R., Jovanovic M., Mertins P., Shishkin A., Tabach Y., Mikkelsen T.S., Satija R., Ruvkun G. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155:1409–1421. doi: 10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo G.-Z., MacQueen A., Zheng G., Duan H., Dore L.C., Lu Z., Liu J., Chen K., Jia G., Bergelson J., He C. Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 2014;5:5630. doi: 10.1038/ncomms6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 8.Dominissini D., Moshitch-Moshkovitz S., Salmon-Divon M., Amariglio N., Rechavi G. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013;8:176–189. doi: 10.1038/nprot.2012.148. [DOI] [PubMed] [Google Scholar]
- 9.Chen W., Feng P., Ding H., Lin H., Chou K.C. iRNA-Methyl: Identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015;490:26–33. doi: 10.1016/j.ab.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z., Xiao X., Yu D.J., Jia J., Qiu W.R., Chou K.C. pRNAm-PC: Predicting N(6)-methyladenosine sites in RNA sequences via physical-chemical properties. Anal. Biochem. 2016;497:60–67. doi: 10.1016/j.ab.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Jia C.-Z., Zhang J.-J., Gu W.-Z. RNA-MethylPred: A high-accuracy predictor to identify N6-methyladenosine in RNA. Anal. Biochem. 2016;510:72–75. doi: 10.1016/j.ab.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Zeng X., Zhang X., Zou Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 2016;17:193–203. doi: 10.1093/bib/bbv033. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91. doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei L., Xing P., Shi G., Ji Z.L., Zou Q. Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017 doi: 10.1109/TCBB.2017.2670558. Published online February 16, 2017. [DOI] [PubMed] [Google Scholar]
- 15.Su R., Zhang C., Pham T.D., Davey R., Bischof L., Vallotton P., Lovell D., Hope S., Schmoelzl S., Sun C. Detection of tubule boundaries based on circular shortest path and polar-transformation of arbitrary shapes. J. Microsc. 2016;264:127–142. doi: 10.1111/jmi.12421. [DOI] [PubMed] [Google Scholar]
- 16.Chen W., Xing P., Zou Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017;7:40242. doi: 10.1038/srep40242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xing P., Su R., Guo F., Wei L. Identifying N6-methyladenosine sites using multi-interval nucleotide pair position specificity and support vector machine. Sci. Rep. 2017;7:46757. doi: 10.1038/srep46757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y., Zeng X., He Z., Zou Q. Inferring MicroRNA-Disease Associations by Random Walk on a Heterogeneous Network with Multiple Data Sources. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2017;14:905–915. doi: 10.1109/TCBB.2016.2550432. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J., Zhang Z., Chen Z., Deng L. Integrating Multiple Heterogeneous Networks for Novel LncRNA-disease Association Inference. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2017 doi: 10.1109/TCBB.2017.2701379. Published online May 4, 2017. [DOI] [PubMed] [Google Scholar]
- 20.Tang W., Wan S., Yang Z., Teschendorff A.E., Zou Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics. 2018;34:398–406. doi: 10.1093/bioinformatics/btx622. [DOI] [PubMed] [Google Scholar]
- 21.Liao Z., Li D., Wang X., Li L., Zou Q. Cancer diagnosis from isomiR expression with machine learning method. Curr. Bioinform. 2018;13:57–63. [Google Scholar]
- 22.He W., Jia C., Duan Y., Zou Q. 70ProPred: a predictor for discovering sigma70 promoters based on combining multiple features. BMC Syst. Biol. 2018;12(Suppl 4):44. doi: 10.1186/s12918-018-0570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu B., Liu F., Wang X., Chen J., Fang L., Chou K.C. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015;43(W1) doi: 10.1093/nar/gkv458. W65-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan C., Liu D., Huang R., Chen Z., Deng L. PredRSA: a gradient boosted regression trees approach for predicting protein solvent accessibility. BMC Bioinformatics. 2016;17:S8. doi: 10.1186/s12859-015-0851-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W., Liu J., Zhao M., Li Q. Predicting linear B-cell epitopes by using sequence-derived structural and physicochemical features. Int. J. Data Min. Bioinform. 2012;6:557–569. doi: 10.1504/ijdmb.2012.049298. [DOI] [PubMed] [Google Scholar]
- 26.Cheng X.-Y., Huang W.J., Hu S.C., Zhang H.L., Wang H., Zhang J.X., Lin H.H., Chen Y.Z., Zou Q., Ji Z.L. A global characterization and identification of multifunctional enzymes. PLoS ONE. 2012;7:e38979. doi: 10.1371/journal.pone.0038979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W., Niu Y., Zou H., Luo L., Liu Q., Wu W. Accurate prediction of immunogenic T-cell epitopes from epitope sequences using the genetic algorithm-based ensemble learning. PLoS ONE. 2015;10:e0128194. doi: 10.1371/journal.pone.0128194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D., Luo L., Zhang W., Liu F., Luo F. A genetic algorithm-based weighted ensemble method for predicting transposon-derived piRNAs. BMC Bioinformatics. 2016;17:329. doi: 10.1186/s12859-016-1206-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo L., Li D., Zhang W., Tu S., Zhu X., Tian G. Accurate Prediction of Transposon-Derived piRNAs by Integrating Various Sequential and Physicochemical Features. PLoS ONE. 2016;11:e0153268. doi: 10.1371/journal.pone.0153268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W., Chen Y., Tu S., Liu F., Qu Q. Drug side effect prediction through linear neighborhoods and multiple data source integration. IEEE Xplore. 2016;2016:427–434. [Google Scholar]
- 31.Zhang W., Zou H., Luo L., Liu Q., Wu W., Xiao W. Predicting potential side effects of drugs by recommender methods and ensemble learning. Neurocomputing. 2016;173:979–987. [Google Scholar]
- 32.Zhang W., Chen Y., Li D. Drug-Target Interaction Prediction through Label Propagation with Linear Neighborhood Information. Molecules. 2017;22:2056. doi: 10.3390/molecules22122056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W., Chen Y., Liu F., Luo F., Tian G., Li X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinformatics. 2017;18:18. doi: 10.1186/s12859-016-1415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W., Zhu X., Fu Y., Tsuji J., Weng Z. Predicting human splicing branchpoints by combining sequence-derived features and multi-label learning methods. BMC Bioinformatics. 2017;18(Suppl 13):464. doi: 10.1186/s12859-017-1875-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W., Qu Q., Zhang Y., Wang W. The linear neighborhood propagation method for predicting long non-coding RNA–protein interactions. Neurocomputing. 2018;273:526–534. [Google Scholar]
- 36.Zhang W., Yue X., Liu F., Chen Y., Tu S., Zhang X. A unified frame of predicting side effects of drugs by using linear neighborhood similarity. BMC Syst. Biol. 2017;11(Suppl 6):101. doi: 10.1186/s12918-017-0477-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W., Liu X., Chen Y., Wu W., Wang W., Li X. Feature-derived graph regularized matrix factorization for predicting drug side effects. Neurocomputing. 2018;287:154–162. [Google Scholar]
- 38.Li B., Tang J., Yang Q., Li S., Cui X., Li Y., Chen Y., Xue W., Li X., Zhu F. NOREVA: normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017;45(W1):W162–W170. doi: 10.1093/nar/gkx449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y.H., Yu C.Y., Li X.X., Zhang P., Tang J., Yang Q., Fu T., Zhang X., Cui X., Tu G. Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2018;46(D1):D1121–D1127. doi: 10.1093/nar/gkx1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mrozek D., Socha B., Kozielski S., Małysiak-Mrozek B. An efficient and flexible scanning of databases of protein secondary structures. J. Intell. Inf. Syst. 2016;46:213–233. [Google Scholar]
- 41.Mrozek D., Małysiak-Mrozek B., Siążnik A. search GenBank: interactive orchestration and ad-hoc choreography of Web services in the exploration of the biomedical resources of the National Center For Biotechnology Information. BMC Bioinformatics. 2013;14:73. doi: 10.1186/1471-2105-14-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zou Q., Wan S., Ju Y., Tang J., Zeng X. Pretata: predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016;10(Suppl 4):114. doi: 10.1186/s12918-016-0353-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou Q., Zeng J., Cao L., Ji R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing. 2016;173:346–354. [Google Scholar]
- 44.Liu B. BioSeq-Analysis: a platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2017 doi: 10.1093/bib/bbx165. Published online December 19, 2017. [DOI] [PubMed] [Google Scholar]
- 45.Pan Y., Wang Z., Zhan W., Deng L. Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics. 2018;34:1473–1480. doi: 10.1093/bioinformatics/btx822. [DOI] [PubMed] [Google Scholar]
- 46.Qiao Y., Xiong Y., Gao H., Zhu X., Chen P. Protein-protein interface hot spots prediction based on a hybrid feature selection strategy. BMC Bioinformatics. 2018;19:14. doi: 10.1186/s12859-018-2009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Q., Xiong Y., Dai H., Kumari K.M., Xu Q., Ou H.Y., Wei D.Q. PDC-SGB: Prediction of effective drug combinations using a stochastic gradient boosting algorithm. J. Theor. Biol. 2017;417:1–7. doi: 10.1016/j.jtbi.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 48.Mrozek D., Gosk P., Małysiak-Mrozek B. Scaling Ab initio predictions of 3D protein structures in Microsoft Azure cloud. J. Grid Comput. 2015;13:561–585. [Google Scholar]
- 49.Mrozek D., Daniłowicz P., Małysiak-Mrozek B. HDInsight4PSi: Boosting performance of 3D protein structure similarity searching with HDInsight clusters in Microsoft Azure cloud. Inf. Sci. 2016;349:77–101. [Google Scholar]
- 50.Bari A.T.M.G., Reaz M.R., Choi H.J., Jeong B.S. DNA encoding for splice site prediction in large DNA sequence. In: Hong B., Meng X., Chen L., Winiwarter W., Song W., editors. International Conference on Database Systems for Advanced Applications. Springer; 2013. pp. 46–58. [Google Scholar]
- 51.Chen W., Feng P., Tang H., Ding H., Lin H. Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics. 2016;107:255–258. doi: 10.1016/j.ygeno.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Chen W., Tang H., Lin H. MethyRNA: a web server for identification of N6-methyladenosine sites. J. Biomol. Struct. Dyn. 2017;35:683–687. doi: 10.1080/07391102.2016.1157761. [DOI] [PubMed] [Google Scholar]
- 53.Chen W., Feng P., Ding H., Lin H. Identifying N 6-methyladenosine sites in the Arabidopsis thaliana transcriptome. Mol. Genet. Genomics. 2016;291:2225–2229. doi: 10.1007/s00438-016-1243-7. [DOI] [PubMed] [Google Scholar]
- 54.Liu B., Weng F., Huang D.S., Chou K.C. iRO-3wPseKNC: Identify DNA replication origins by three-window-based PseKNC. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty312. Published online April 19, 2018. [DOI] [PubMed] [Google Scholar]
- 55.Pérez A., Noy A., Lankas F., Luque F.J., Orozco M. The relative flexibility of B-DNA and A-RNA duplexes: database analysis. Nucleic Acids Res. 2004;32:6144–6151. doi: 10.1093/nar/gkh954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freier S.M., Kierzek R., Jaeger J.A., Sugimoto N., Caruthers M.H., Neilson T., Turner D.H. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. USA. 1986;83:9373–9377. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goñi J.R., Pérez A., Torrents D., Orozco M. Determining promoter location based on DNA structure first-principles calculations. Genome Biol. 2007;8:R263. doi: 10.1186/gb-2007-8-12-r263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu B., Zhang D., Xu R., Xu J., Wang X., Chen Q., Dong Q., Chou K.C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics. 2014;30:472–479. doi: 10.1093/bioinformatics/btt709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen X., Yan C.C., Zhang X., You Z.H., Deng L., Liu Y., Zhang Y., Dai Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci. Rep. 2016;6:21106. doi: 10.1038/srep21106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang H., Chen W., Lin H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique. Mol. Biosyst. 2016;12:1269–1275. doi: 10.1039/c5mb00883b. [DOI] [PubMed] [Google Scholar]
- 61.Yang H., Tang H., Chen X.X., Zhang C.J., Zhu P.P., Ding H., Chen W., Lin H. Identification of Secretory Proteins in Mycobacterium tuberculosis Using Pseudo Amino Acid Composition. BioMed Res. Int. 2016;2016:5413903. doi: 10.1155/2016/5413903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin H., Liang Z.Y., Tang H., Chen W. Identifying sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017 doi: 10.1109/TCBB.2017.2666141. Published online February 8. 2017. [DOI] [PubMed] [Google Scholar]
- 63.Liu B., Wang S., Long R., Chou K.C. iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics. 2017;33:35–41. doi: 10.1093/bioinformatics/btw539. [DOI] [PubMed] [Google Scholar]
- 64.Liu B., Fang L., Liu F., Wang X., Chen J., Chou K.C. Identification of real microRNA precursors with a pseudo structure status composition approach. PLoS ONE. 2015;10:e0121501. doi: 10.1371/journal.pone.0121501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao Y., Zhang J., Deng L. Prediction of lncRNA-protein interactions using HeteSim scores based on heterogeneous networks. Sci. Rep. 2017;7:3664. doi: 10.1038/s41598-017-03986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lai H.Y., Chen X.X., Chen W., Tang H., Lin H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget. 2017;8:28169–28175. doi: 10.18632/oncotarget.15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chou K.-C., Cai Y.-D. Using functional domain composition and support vector machines for prediction of protein subcellular location. J. Biol. Chem. 2002;277:45765–45769. doi: 10.1074/jbc.M204161200. [DOI] [PubMed] [Google Scholar]
- 68.Cai Y.-D., Zhou G.-P., Chou K.-C. Support vector machines for predicting membrane protein types by using functional domain composition. Biophys. J. 2003;84:3257–3263. doi: 10.1016/S0006-3495(03)70050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cristianini N., Shawe-Taylor J. Cambridge University Press; 2000. An introduction to support vector machines and other kernel-based learning methods. [Google Scholar]