

Abstract
Circulating tumor cells (CTCs) have the potential to predict metastasis, and the capture of CTCs based on their surface markers is mostly applied for CTC detection. Considering that the CTCs with a metastatic phenotype preferably form a metastatic focus and that aptamers have the ability to bind targets with high specificity and affinity, we selected aptamers directed toward metastatic cells by subtractive Cell-SELEX technology using highly metastatic MDA-MB-231 cells as the target cell and low-metastatic MCF-7 cells as the negative cell for the capture of metastatic CTCs. Affinity and selectivity assays showed that aptamer M3 had the highest affinity, with a KD of 45.6 ± 1.2 nM, and had good specificity against several other types of metastatic cancer cells. Based on these findings, we developed an M3-based capture system for CTC enrichment, which has the capability to specifically capture the metastatic cells MDA-MB-231 mixed with non-metastatic MCF-7 cells and CTCs derived from the peripheral blood from metastatic breast cancer patients. A further comparative analysis with the anti-EpCAM probe showed that M3 probe captured epithelial feature-deletion metastatic cells. We developed an aptamer-based CTC capture system through the selection of aptamers by taking whole metastatic cells, not known molecules, as targets, which provided a new insight into CTC capture and Cell-SELEX application.
Keywords: circulating tumor cells, Cell-SELEX, aptamer, breast cancer, EpCAM
Graphical Abstract
Introduction
Breast cancer (BC) is the most frequently diagnosed malignancy among women worldwide, and metastasis is the main cause of cancer-related death in patients with BC.1 Despite significant advances in its clinical management, BC mortality remains high, due to the failure to discover early metastasis and predict recurrence.2 Therefore, there is an urgent need to develop effective strategies to monitor the metastasis and predict the recurrence of BC to increase survival.
Circulating tumor cells (CTCs) are rare cancer cells released from primary tumors into the bloodstream.3 The CTCs that exist in patients’ peripheral blood are closely related to the risk of metastasis and recurrence as well as the effect of anti-cancer therapy; therefore, CTCs have the potential to serve as predictive biomarkers.4, 5, 6 Hiraiwa et al.4 detected CTCs derived from gastric cancer patients and found that the counts were higher in metastatic patients than in non-metastatic ones and were significantly correlated with advanced tumor stages. In the case of BC, a CTC analysis is expected to have a more notable value in clinical settings, compared to other kinds of cancers, due to its dominant hematogenous metastasis feature.7 Yan et al.5 included 50 studies with 6,712 BC patients in a meta-analysis and found that CTC counts significantly reduced after treatment, compared to pretreatment, and that the decreased CTC counts predicted an increased overall survival.
Because CTCs are extremely rare, with numbers as low as one CTC among a few million white blood cells (WBCs) and a few billion red blood cells (RBCs) in 1 mL of whole blood,8, 9 how to effectively separate and enrich CTCs from patients’ blood becomes a prominent challenge for CTC analysis. Current CTC enrichment and isolation approaches are mainly based on CTCs’ physical and/or biochemical properties, which are distinguished from those of various blood cells.10 Using the physical methods, CTCs are isolated based on their size, density, and electric charge. However, these methods do not have sufficient specificity to acquire the required enrichment purity.11 The biochemical methods for capturing CTCs rely on the specific markers distributed on the tumor cell surface by immune reaction, such as an antigen-antibody reaction. The most widely used marker is the epithelial cell adhesion molecule (EpCAM), which is expressed by most tumor cells derived from the epithelium.12, 13 Although anti-EpCAM is utilized to capture the CTCs from different kinds of cancers, the strategy is still impractical in terms of specificity, due to the failure to capture EpCAM-deletion CTCs, such as the CTCs possessing stem cell features or those undergoing epithelial-to-mesenchymal transition (EMT), which might result in false negatives, because both tumor stem cells and EMT cells in circulating blood have a superior metastasis potential, compared to other epithelial-feature cells, and possibly act as functional seeds for initiating metastasis.14, 15, 16, 17 Perhaps, this is the main reason why some reports do not show a significant relationship between CTC counts and clinical metastasis.18 Accordingly, increasingly more studies focus on improving the enrichment efficacy by increasing the specificity using metastasis-related markers or a combination of multiple markers to enrich functional CTCs.19, 20, 21 For example, Satelli et al.19 used the EMT marker cell-surface vimentin (CSV) to detect CTCs from prostate cancer patients and demonstrated that the CSV-based method isolated and quantified CTCs with higher sensitivity (93.33%) and specificity (94.4%) compared to the EpCAM-based method, which had a sensitivity of 53.33% and a specificity of 83.33%. Liu et al.20 combined CA9 and CD147-capture antibodies for CTC enrichment in clear-cell renal cell carcinoma patients and achieved a 181% increase in the capture efficiency of CTCs, in comparison with the EpCAM antigen alone. Accordingly, the key issue in verifying CTCs’ clinical significance, perhaps, is to find and detect the CTCs with the ability to form a metastatic focus, and we call these functional CTCs.
Aptamers, also called chemical antibodies, are short, single-stranded oligonucleotides (single-stranded DNA [ssDNA] or single-stranded RNA [ssRNA]) with the ability to selectively recognize various target molecules, and they are usually generated through an in vitro selection procedure called the systematic evolution of ligands by exponential enrichment (SELEX) from a large library of random DNA or RNA molecules.22, 23 Compared to monoclonal antibodies, aptamers possess higher affinity and specificity, ease of chemical modification, and superior stability, making them the most advanced reagents for the detection of target molecules.24 Cell-SELEX is a cell-based SELEX technology that aims to select aptamers directed toward cell-surface molecules by using whole living cells as targets.25 Unlike other SELEX methods, Cell-SELEX enables the generation of cell-specific aptamers without any prior knowledge of the molecular features of the selected cells, making it possible to generate aptamers that recognize unknown cell-surface biomarkers.26 Specifically, the newly developed subtractive Cell-SELEX generates aptamers targeting specific phenotype cells by adding a subtractive step using the selected cells with given functional differences, such as differentiated cells and non-differentiated cells,27 virus-infected cells and uninfected cells,28 and metastatic cells and non-metastatic cells. In previous studies, we conducted this subtractive strategy using high-metastatic LoVo cells as the target and low-metastatic HCT-8 cells as the negative control, which resulted in the production of aptamers with the capability to bind specifically to metastatic colorectal cancer cells.29
Since it has been reported that not all CTCs that enter the blood circulation have the ability to join the final metastasis,30 developing capture probes against functional CTCs with a metastasis phenotype might be a better strategy for a clinical trial, compared to using universal probes such as anti-EpCAM. Fortunately, this can be achieved by generating aptamers via Cell-SELEX using selected cells with differentially metastatic phenotypes. Furthermore, a panel of aptamers against different targets on the same cells can be generated via a single Cell-SELEX process. Several studies have achieved an improved detection sensitivity for target cells using a group of aptamers.31, 32 As for a CTC analysis, the use of multi-aptamers directed toward a given phenotype of cells is expected to enhance the capture efficiency and accuracy. However, thus far, there are no reports on generating aptamers using Cell-SELEX for BC-derived CTC capture. Accordingly, we used metastatic BC MDA-MB-231 cells as the target cells and low metastatic MCF-7 cells as the negative cells to perform subtractive Cell-SELEX and generated five DNA aptamers that bind specifically to MDA-MB-231 cells. Furthermore, aptamer M3, with the highest affinity, was chosen as a specific probe to capture CTCs, and a highly specific enrichment of the target MDA-MB-231 cells and CTCs from BC-patient whole blood, especially the EpCAM-negative cells from the whole blood, was achieved.
Results and Discussion
Selection of the Aptamers Targeting MDA-MB-231 Cells by Cell-SELEX
Increasing reports show that only a small percentage of CTCs entering circulation are ultimately capable of forming metastases.30, 33, 34 Thus, developing a recognition system targeting these functional CTCs is expected to improve the capture efficiency of CTCs and, in turn, verify their clinical value. Here, we performed a subtractive Cell-SELEX using the human BC cell line MDA-MB-231, which has a high metastatic potential, as the target cells and the human BC cell line MCF-7 as the negative cells, aiming to select the metastasis-specific aptamers for the capture of CTCs from BC. Although the two cell lines’ metastatic potential has been reported in many literatures,35, 36, 37 we still detected the metastatic phenotype differences between the two types of cells using a Transwell assay before Cell-SELEX in order to confirm the functional difference of the two cell lines used in our lab. As shown in Figure S1, both the migration and invasion ability of MDA-MB-231 are much stronger than MCF-7 cells, which is consistent with the previous reports. For the first two selection rounds, we only applied the MDA-MB-231 cells for the positive selection to enrich the bound ssDNA sequences to ensure enough ssDNA sequences for the subsequent selection. From the third round, a negative selection with MCF-7 was added prior to the positive selection to remove the nonspecific sequences.
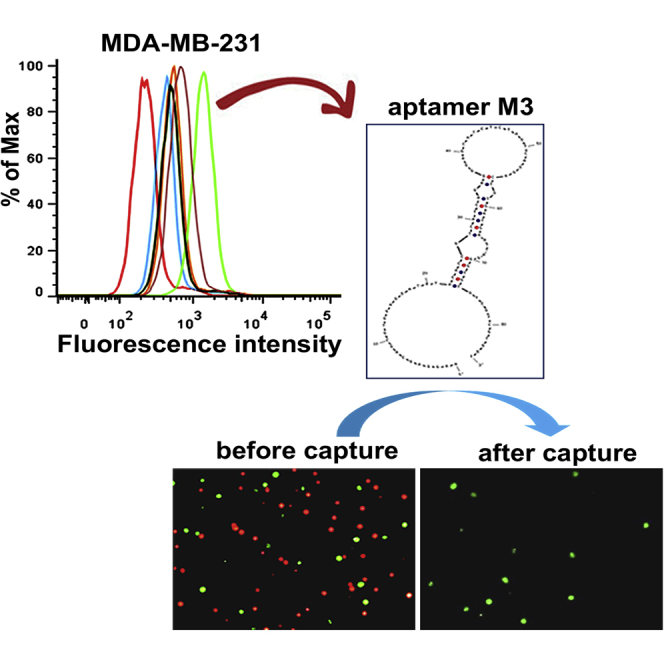
After ten rounds of selection, the enrichment progress of the third, sixth, eighth, and tenth selected pools was monitored by flow cytometry. As shown in Figure 1A, there was a clear right shift of the fluorescence peak of the MDA-MB-231 cells treated with the third round, compared to those treated with the ssDNA library, indicating that the ssDNA sequences that bound to the MDA-MB-231 cells were enriched. As the selection progressed, a steady right shift of the fluorescence peak was observed, while there was no further significant change from the sixth to tenth round, indicating that enrichment of the ssDNA sequences bound to MDA-MB-231 cells reached saturation. In contrast, for the MCF-7 cells, there was a left shift in the fluorescence peak after the third round, implying that counterselection removed the ssDNA sequences bound to the MCF-7 cells. A quantitative analysis of the fluorescence intensity showed a consistent result with that of flow cytometry, revealing that the enriched ssDNA sequences specifically bound to the target MDA-MB-231 cells but not the negative MCF-7 cells (Figure 1B). Based on the aforementioned findings, the tenth-round pool was considered the best candidate for further identification.
Figure 1.

Monitoring the Enrichment of Cell-SELEX Progression
(A) The FITC-labeled selected pools (3rd, 6th, 8th, and 10th) were incubated with MDA-MB-231 cells (target cells) and MCF-7 cells (negative cells) for 30 min at 4°C. Then the fluorescence intensity was determined by flow cytometry. The ssDNA library was used as negative control. At least three independent experiments were conducted. (B) Quantitative results based on flow cytometry. Fluorescence shift was calculated using the equation (Fround pool − Fcell)/(Flibrary − Fcell), where Fround pool, Flibrary, and Fcell refer to the fluorescence with the selected pool, library, and the cell itself, respectively. The values of fluorescence intensity represent the mean ± SD of three independent experiments.
The enriched pool of Cell-SELEX round 10 was first PCR amplified and cloned, and 50 clones were selected for sequencing. Based on the sequences’ repeatability and secondary structures, five candidate aptamers (named M1, M2, M3, M4, and M10) were chosen and synthesized with fluorescein isothiocyanate (FITC) labels for further characterization (Figure S2; Table S1). The binding abilities of these candidate aptamers to the target MDA-MB-231 cells and the control MCF-7 cells were determined by flow cytometry, which showed a greater fluorescent intensity with the MDA-MB-231 cells for all five candidate aptamers, compared to the ssDNA library (Figure 2A). In contrast, a weak fluorescent intensity was detected from the candidate aptamers with the MCF-7 cells, suggesting that these five aptamers specifically bound to the MDA-MB-231 cells but not to the MCF-7 cells. Among these, aptamers M3 and M10 showed a greater binding capability to the target MDA-MB-231 cells. Further fluorescence microscopy imaging (Figure 2B) revealed that, compared to the ssDNA library, the target MDA-MB-231 cells incubated with M3 and M10 showed obvious fluorescence signals on the cell surface, but this was not present for the MCF-7 cells, which further indicated the specific binding of M3 and M10 to the target MDA-MB-231 cells.
Figure 2.

Binding Assay of the Selected Aptamers
(A) Flow cytometry assays of the binding of FITC-labeled aptamers (M1, M2, M3, M4, and M10) with MDA-MB-231 cells (target cells) and MCF-7 cells (negative cells). (B) Fluorescence imaging of cells using QD605-labeled M3 and QD605-labeled M10. All pictures were taken under a fluorescence microscope with 200× magnification. The ssDNA library was used as negative control. At least three independent experiments were conducted.
Characterization of M3’s Properties for MDA-MB-231 Cell Capture
Because of the rarity of CTCs in the peripheral circulation, a high specificity and an excellent affinity are both mandatory requirements for effective CTC capture. High specificity ensures the capture of the functional CTCs, avoiding an inaccurate estimate in the clinic. An excellent affinity offers a strong enough binding to the target cells, which is especially necessary to resist the background interference from large amounts of blood cells. Moreover, recent studies show that CTCs take the form of clusters, which have more metastatic potential than single CTCs.38, 39, 40 CTC clusters are defined as a group of more than two or three tumor cells (up to >100), and their capture might require a probe with a stronger binding affinity. Compared to antibodies, aptamers generally display a strong binding affinity to their ligands, with dissociation constants on the order of picomoles to nanomoles.41, 42 For example, Jiang et al.41 revealed that the interaction between the protein immunoglobulin E and its aptamer matched, or even surpassed, that of the immunoglobulin E (IgE) and its monoclonal antibody. Based on the aforementioned data, we first chose aptamers M3 and M10 to determine the binding affinity with the target MDA-MB-231 cells. As shown in Figures 3A and S3, M3 and M10 both displayed high binding affinity to target MDA-MB-231 cells, with dissociation constant (KD) values in the nanomolar range, 45.6 ± 1.2 nM and 113 ± 2.4 nM, respectively. Considering that aptamer M3 had a better binding affinity, we selected aptamer M3 for further evaluation for capture use.
Figure 3.

Characterization of M3 Binding to MDA-MB-231 Cells
(A) Binding curve of aptamer M3 with MDA-MB-231 cells. The cells were incubated with increasing concentrations of FITC-labeled aptamer M3 and assessed by flow cytometry. The KD value of M3 was obtained by SigmaPlot software according to Y=Bmax × X/(KD + X). The ssDNA library was used as negative control. (B) Binding capacity of aptamer M3 with MDA-MB-231 cells at 4°C, 25°C, and 37°C. (C) Binding capacity of aptamer M3 with MDA-MB-231 cells treated with proteinase K or trypsin. The cells were treated with proteinase K or trypsin at 37°C for 3 min and 10 min, respectively, and the binding of the FITC-labeled aptamer M3 to the treated cells was then assessed by flow cytometry. At least three independent experiments were conducted.
To verify the cell selectivity of aptamer M3, aptamer M3 was further assessed with other BC cell lines, normal cell lines, and other cancer cell lines from different organs. First, we found that aptamer M3 did not bind to any normal cell lines, either from human or mouse, revealing that aptamer M3 possessed tumor cell-binding specificity (Figure S4; Table 1). Second, aptamer M3 showed obvious binding for some cancer cell lines, not only including ZR-75-30 and MDA-MB-436 from BC but also including LoVo from colorectal cancer, SGC-7901 and BGC823 from gastric cancer, and C6 from murine glioblastoma cancer. More notably, these cells are mostly derived from metastatic tumors,43, 44 suggesting that M3 had a highly specific binding ability to the cells with metastatic potential (Figures S5 and S6; Table 1). The aforementioned results confirmed that aptamer M3 has a high specificity and an excellent affinity, making it an effective molecular probe for capturing CTCs.
Table 1.
Binding Assay of Aptamer M3 to Different Cell Lines
| Cell Line/Aptamer | M3 |
|---|---|
| MDA-MB-231 | ++++ |
| MCF-7 | – |
| SK-BR-3 | – |
| BT474 | – |
| ZR-75-30 | + |
| MDA-MB-436 | + |
| LoVo | ++ |
| CL187 | – |
| SGC-7901 | ++ |
| BGC823 | + |
| A549 | – |
| C6 | + |
| HepG2 | + |
| HeLa | – |
| HEK293 | – |
| NIH 3T3 | – |
| CHO | – |
A threshold of the fluorescence intensity of flow cytometry analysis was set so that 95% of cells incubated with the FITC-labeled ssDNA library would have fluorescence intensity below it. The percentage of the cells with fluorescence above the set threshold was used to evaluate the binding capacity of the aptamer to the cells. Symbols: –, <14%; +, 15%–35%; ++, 36%–60%; ++++, >85%.
To effectively capture CTCs, the other characteristics of aptamer M3 were further analyzed. We performed the Cell-SELEX at 4°C to achieve more cell-surface-specific aptamers, as ssDNA is non-specifically taken up by cells through endocytosis at physiological temperatures.45 However, CTC capture is usually conducted at room temperature or under physiological circumstances; thus, we investigated the binding ability of M3 to target MDA-MB-231 cells at 25°C or 37°C. As shown in Figure 3B, there was little change in the fluorescence intensity of the target cells at 25°C or 37°C compared with that at 4°C, suggesting that M3 maintained its binding activity to the target cells at the detected temperatures, which provides very comfortable alternatives for aptamer-based CTC capture in practice.
As shown in Figure 2B, the M3-bound target was present on the MDA-MB-231 cell surface. To verify the biochemical characteristics of the target, we performed enzyme-treatment assays on the MDA-MB-231 cells using proteinase K or trypsin. As shown in Figure 3C, compared to the enzyme-free digestion, there was a detectable fluorescent intensity drop after the treatment with trypsin, in spite of no similar change with proteinase K, which suggested that aptamer M3’s target may be a protein. This molecular feature will be helpful to further identify the target of M3 and, in turn, improve the capture efficiency and verify the significance of the captured CTCs. In addition, considering that real CTC capture is manipulated in a whole-blood circumstance, in which the aptamers’ activity might be influenced because of degradation by nucleases,46 the stability of aptamer M3 in blood was investigated by incorporating M3 into human plasma. A DNA electrophoresis analysis showed that the aptamer M3 maintained its integrity (tight band) without obvious degradation during a 2-hr incubation with the plasma, and most of the aptamer still remained after 3 hr (Figure S7). Further, a flow cytometry assay revealed that M3 conserved its specific binding to the target cell MDA-MB-231 during at least a 2-hr incubation with the plasma. The good stabilities of M3 in terms of structure and function could meet the essential requirement for CTC capture. Given that more robust conditions may be encountered in the capture, the stability of the aptamer could be further enhanced by reducing its nuclease susceptibility via the modification of the nucleic acid backbone.47
Enrichment of Cancer Cells Using an Aptamer M3-Based Capture System
Taking advantage of M3’s high affinity and specificity to target cells, its ability to capture CTCs was evaluated by creating a capture system using M3-coated 96-well plates (Figure 4A). First, the system was applied to capture target MDA-MB-231 and non-target MCF-7 cells. As shown in Figure 4B, after undergoing the capture process, in the M3-coated wells, the green-stained target MDA-MB-231 cells were captured, but almost no red-stained non-target MCF-7 cells were captured; for the ssDNA library-coated wells, neither the target nor the non-target cells were observed. These results indicated that aptamer M3 specifically recognized the target cells and captured them under such a system. To further demonstrate the capture specificity of aptamer M3, we mixed the target MDA-MB-231 cells (pre-stained green) and the non-target MCF-7 cells (pre-stained red), at different percentages (target cells accounted for 50%, 25%, 15%, or 10%), to form mixed-cell suspensions. After capturing, the green-stained cells were present in the wells, but almost no red-stained cells were observed at the various mixing concentrations (Figure 4C), which demonstrated that the M3-based system had a strong ability for capturing target cells without the interference of non-target cells.
Figure 4.

Capture of CTCs Using the Aptamer M3-Based System
(A) Schematic workflow of CTC capture by the M3-based system. (B) Selective capture of MDA-MB-231 or MCF-7 cells by an aptamer M3-coated 96-well plate. After the aptamer M3 immobilized on the plate, the green-stained target MDA-MB-231 cells or the red-stained non-target MCF-7 cells were incubated for 30 min on the wells. Fluorescence microscopy was used to observe and take pictures before and after washing. The ssDNA library was used as negative control. (C) Fluorescent microscope image of MDA-MB-231 cells captured after mixing with MCF-7 cells at different spiked concentrations. All pictures were taken under a fluorescence microscope with 100× magnification. (D) Capture of CTCs in peripheral blood using an aptamer M3-coated 96-well plate. After capture, the cells were successively stained for DAPI, FITC-labeled anti-CD45, and PE-labeled anti-EpCAM and, finally, identified under the microscope. All pictures were taken under a fluorescence microscope with 400× magnification. (E) The analysis of positive rates of CTC capture in healthy volunteers, non-metastatic patients, and metastatic BC patients.
To further investigate the clinical utility of the M3-based system, we used aptamer M3 to capture CTCs in real blood samples, including six healthy volunteers and 25 BC patients. In Figure 4D, the cells without any staining were RBCs, and the cells positive for both DAPI and CD45 were WBCs, while the cells positive for DAPI and negative for CD45 were CTCs. Based on these data, no CTCs were found in all healthy volunteers’ blood samples, apart from WBCs and RBCs, while CTCs were detectable in blood from metastatic BC patients, with a higher detection percentage than that for non-metastatic BC patients (10/14, 71.4%, versus 1/11, 9.1%) (Figure 4E). Moreover, M3-probed CTCs were present in all eight blood samples from the diagnosed patients with distant metastasis, which was much higher than that from patients with lymph node metastasis alone (2/6, 33.3%) (Figure 4E; Table S2), which further indicated that M3 might have the potential to capture functional CTCs with metastatic activity.
Currently, most reports, including the commercial Food and Drug Administration (FDA)-approved capture system CellSearch, use epithelial antigens, such as EpCAM, for CTC enrichment, but the capture effectiveness for clinical utility is hard to satisfy.48 This is mostly because the method based on EpCAM fails to enrich the CTCs with low or no EpCAM expression, which usually have more metastatic potential due to the occurrence of EMT. To investigate the EpCAM expression patterns in various types of cells with different metastatic potential, we detected the EpCAM expression in several cell lines by flow cytometry. As shown in Figure S8, we did not find the obvious relationship between EpCAM expression levels and cells’ metastasis properties. For example, MDA-MB-436, LoVo, and SGC-7901 are high-metastatic-potential cell lines, but SGC-7901 cells displayed very strong EpCAM expression, while MDA-MB-436 and LoVo were nearly negative for EpCAM. The same case occurred in other several cell lines with low or no metastatic potential. Different from EpCAM probe, the binding of M3 probe to distinct types of cell lines presented a close agreement with cells’ metastatic activities, as described earlier (Tables 1 and S3). In this study, the selection of the aptamers was based on the difference between the metastatic tumor cells and the non-metastatic tumor cells. Thus, in theory, the obtained aptamers specifically recognized the metastatic tumor cells; indeed, aptamer M3 displayed its metastatic specificity in the aforementioned experiments. To further determine whether aptamer M3 targets different subpopulations from EpCAM-positive cells, we chose two metastatic BC cell lines, MDA-MB-231 and MDA-MB-436, to detect the targeting patterns of M3 and anti-EpCAM by flow cytometry. As shown in Figure 5A, for the MDA-MB-231 cells, approximately 39% of the cells were labeled by anti-EpCAM, whereas up to 83% of the cells were labeled by M3, and approximately 37% of the cells bound only to M3. These data mean that, if MDA-MB-231 cells are captured by an anti-EpCAM probe, then nearly 37% of the cells would be determined to be negative. For MDA-MB-436, the phenomenon was even more obvious: only about 8% of the cells were labeled by anti-EpCAM, whereas nearly 32% of the cells were labeled by M3, and up to 29% of the cells were only probed by M3, which means that if capture of MDA-MB-436 cells with EpCAM probe, a higher percentage of the detected cells would be defined as negative compared to that of MDA-MB-231 cells. Since both MDA-MB-231 and MDA-MB-436 have a high metastatic potential, and M3 labeled higher percentages of these cells compared to anti-EpCAM, it is reasonable to believe that M3 captured more active CTCs, although further validation experiments need to be done.
Figure 5.

Comparative Analysis of the Binding of Anti-EpCAM and the M3 Probe to MDA-MB-231 Cells
(A) Flow cytometry assay of anti-EpCAM and M3 co-labeled MDA-MB-231 and MDA-MB-436 cells. Two metastatic BC cell lines were first incubated with PE-labeled anti-EpCAM, followed by FITC-M3 and, finally, determined by flow cytometry. At least three independent experiments were conducted. (B) Analysis of the EpCAM expression on MDA-MB-231 cells mixed into whole blood captured by the M3-based system. The MDA-MB-231 cells were first mixed with whole blood and captured on an aptamer M3-coated 96-well plate. Then, the cells were successively stained for DAPI, FITC-labeled anti-CD45, and PE-labeled anti-EpCAM and, finally, identified under the microscope. All pictures were taken under a fluorescence microscope with 400× magnification.
To further confirm that the M3-based capture system also enriches M3-positive but EpCAM-negative MDA-MB-231 cells in the blood, we spiked MDA-MB-231 cells in healthy donor blood for cell-capture experiments. After M3 capture, followed by successively staining for FITC-labeled anti-CD45, phycoerythrin (PE)-labeled anti-EpCAM, and DAPI, the cells on the substrates of the M3-coated wells were identified under the microscope. As shown in Figure 5B, similar to the results from flow cytometry, among the cells captured by M3, there were also some EpCAM-negative cells apart from the EpCAM-positive ones, indicating that aptamer M3 captured metastatic cancer cells that did not express, or minimally expressed, EpCAM. The same results reappeared in the cells captured from clinical patient blood, obtaining some EpCAM-negative cells on M3-coated surface (Figure 4D). All these indicated that aptamer M3 might have a more favorable ability to capture CTCs compared to the widely used EpCAM-based approach.
Conclusions
We have successfully generated a group of five DNA aptamers that specifically recognized target MDA-MB-231 cells via subtractive Cell-SELEX technology. Among them, aptamer M3 demonstrated superior affinity, with a KD of 45.6 ± 1.2 nM, and had good specificity against metastatic cancer cells, including target cells and several other types of metastatic cancer cells. Furthermore, the cell enrichment results showed that M3 captured the target cells spiked into non-target cells or whole blood. In addition, aptamer M3 maintained its specific recognition capability for target cells at room or physiological temperature and maintained its stability in plasma. These characteristics of aptamer M3 enabled it to have great potential for the application of CTC enrichment. It is interesting to note that an M3-based probe not only detected CTCs from metastatic BC patients’ blood but also captured a high percentage of EpCAM-negative cells from metastatic target cells and clinical blood samples, implying that M3-bound CTCs might have a more significant prediction value for metastasis in the clinic. Based on this, we plan to sort the M3-bound cells to further confirm their metastatic potential. In this study, we obtained a group of aptamers, and all of them displayed the capability to bind to metastatic target cells. Thus, they might be used in combination to further increase the sensitivity of CTC capture. Although we implemented the target cell capture using the well plate system, the efficiency still needs to be enhanced, due to the limited contact frequency between the cells and the probe-loaded well surfaces. It is possible that improvements can be achieved using a microfluidic based system to increase the capture area by creating a nanomaterial-coated surface and by multiplying the contact between the cells and probes by allowing the cells to continuously flow on the chip surface.49, 50 Additionally, an RBC lysis step can be added prior to the capture for CTC pre-concentration.51 Compared to using probes derived from known markers for CTC capture, we applied Cell-SELEX technology to select the aptamers directed toward the cells with a metastatic phenotype as capture probes, regardless of the identification of the target molecules prior to use, which not only provides a new insight into CTC capture and CTC-related marker discovery but also extends the application of Cell-SELEX technology.
Materials and Methods
Cell Lines and Cell Culture
The human BC cell lines MDA-MB-231 (high metastatic potential, used as the target cell) and MCF-7 (low metastatic potential, used as the control cell) were used in this selection.35, 36, 37 The other 15 cell lines—i.e., human BC cell lines MDA-MB-436, BT474, SK-BR-3, and ZR-75-30; human colorectal cancer cell lines LoVo and CL187; human gastric cancer cell lines SGC-7901 and BGC823; human lung cancer cell line A549; human liver cancer cell line HepG2; human cervical cancer cell line HeLa; murine glioblastoma cancer cell line C6; the Chinese hamster ovary (CHO) cell line; murine fibroblast cell line NIH 3T3; and human cell line HEK293—were used in the cell-binding analysis. The cell lines BT474, LoVo, HepG2, HeLa, C6, and CHO were cultured in RPMI 1640 medium with 10% FBS and 100 U mL−1 penicillin-streptomycin, and the MDA-MB-436 cells were cultured in L15 medium with 10% FBS and 100 U mL−1 penicillin-streptomycin. The other ten cell lines were cultured in high-glucose DMEM with 10% FBS and 100 U mL−1 penicillin-streptomycin. The MDA-MB-436 cells were maintained in a 37°C and 100% air atmosphere, while the others were all incubated in a 37°C and 5% CO2 atmosphere.
Blood Collection
To prepare for the artificial blood sample environment, peripheral blood samples were collected in EDTA-K2 anticoagulant tubes from healthy volunteers, which were supplied by The First Affiliated Hospital of China Medical University (Shenyang, China). The blood samples from BC patients and healthy volunteers for CTC capture were from The First Affiliated Hospital of Fujian Medical University (Fuzhou, China). The study was approved by the ethics review committees at The First Affiliated Hospital of China Medical University and The First Affiliated Hospital of Fujian Medical University.
DNA Library and Primers
The ssDNA library and primers were previously used in our laboratory.29 The 85-nt library contained a 45-base nucleotide random sequence and two 20-nt primers flanked on each side (5′-AAGGAGCAGCGTGGAGGATA-45nt-TTAGGGTGTGTCGTCGTGGT-3′). The reverse primer was biotin labeled at the 5′ end in order to separate the ssDNA by streptavidin-coated Sepharose beads (GE Healthcare, Laurel, MD, USA). The forward primer was FITC-labeled at the 5′ end in order to monitor the progress of the selection by flow cytometry (Beckman Coulter, Sykesville, MD. USA). All sequences were chemically synthesized and purified by high-performance liquid chromatography (HPLC) (Sangon Biotechnology, Shanghai, China).
Cell-SELEX Procedure
In the first selection, the ssDNA library (12 nmol) was first dissolved in pre-cooled binding buffer (BB; containing PBS, 5 mM MgCl2, 4.5 g L−1 glucose, 1 mg mL−1 BSA, and 0.1 mg mL−1 salmon sperm DNA), was denatured at 95°C for 5 min, and was then cooled on ice for 10 min. The target MDA-MB-231 cells (1 × 107) were incubated with the ssDNA library at 4°C in an orbital shaker for 60 min and were then washed three times to remove the unbound sequences. The cells were scraped off, heated to 95°C for 10 min, and centrifuged. Afterward, the supernatant was collected as a template to be amplified by PCR with the labeled primers. The FITC-labeled ssDNA was isolated using streptavidin-coated Sepharose beads, was denatured by alkaline, and was then collected for the next round. The subtractive selection was performed from the third round, in which the ssDNA pool obtained from the previous round was first incubated with the control MCF-7 cells (1 × 106) at 4°C for 60 min, and then the supernatant containing the unbound sequences was collected for the positive selection. The selection stringency was enhanced gradually by increasing the content of BSA and salmon sperm DNA in the BB, increasing the number of control cells, extending the wash time, and reducing the incubation time for the target cells. A flow cytometry analysis was conducted to monitor the progress of the selection process. The ssDNA pool with the highest degree of enrichment was PCR amplified, and the product was cloned using the Mighty TA-Cloning Kit (TaKaRa, Tokyo, Japan) and then sequenced (Sangon Biotechnology). The Oligo Analyzer software was used to predict the secondary structures of the candidate aptamers and analyze the structure characteristics.
Flow Cytometry Binding Analysis
The enrichment of the selected pools during Cell-SELEX and the binding ability of the selected aptamers were both monitored by flow cytometry. The target MDA-MB-231 cells or the control MCF-7 (1 × 106) cells were incubated with 250 nM FITC-labeled selected pools or aptamers at 4°C for 30 min. Subsequently, the cells were washed and suspended in PBS, and then the fluorescence intensity was determined by flow cytometry. The ssDNA library was used as the negative control.
To calculate the equilibrium dissociation constants (KDs) of the selected aptamers, an increasing concentration of the FITC-labeled selected aptamers (0 to 1,024 nM) was incubated with the target MDA-MB-231 cells at 4°C for 30 min and was analyzed by flow cytometry. The KD value of each aptamer was obtained by SigmaPlot software according to Y = Bmax × X/(KD + X) (Jandel, San Rafael, CA, USA).
To determine the cell selectivity of the selected aptamers, all the cell lines described earlier were subjected to the aptamer binding assay using flow cytometry.
Fluorescence Microscopy for Binding Analysis
To determine the binding activity of the aptamers, MDA-MB-231 and MCF-7 cells were seeded in chamber slides and cultured overnight. After being washed twice with PBS, the cells were incubated with 250 nM of the biotin-labeled aptamers or the ssDNA library at 4°C for 1 hr. The cells were washed twice and incubated with QD605-streptavidin at a final concentration of 5 nM (Wuhan Jiayuan Quantum Dots, Wuhan, China) for 20 min at room temperature and then were fixed with 4% formaldehyde for 20 min and stained with DAPI for 5 min to counterstain the nucleus. Finally, the fluorescence images of the cells were obtained by fluorescence microscopy (Olympus IX51, Tokyo, Japan).
Protease Treatment Assay
To verify the feature of the M3’s target molecules, the target MDA-MB-231 cells were treated with 0.25% trypsin or 0.1 mg/mL proteinase K at 37°C for 5 or 10 min, respectively. Then, the culture medium containing FBS was added to stop the enzyme’s activity, and the cells were centrifuged and resuspended in PBS for the M3 binding assay by flow cytometry.
Plasma Stability of M3
To detect the stability of M3 in plasma, we first centrifuged human whole blood to obtain the supernatant and then added M3 (0.025 nmol) to the supernatant at 37°C for 0.5, 1, 2, or 3 hr. Finally, the M3 in plasma was separated by 3% agarose gel electrophoresis, and the DNA integrity was observed under UV spectrophotometry. The binding capacity of M3 to MDA-MB-231 cells in plasma was detected by flow cytometry after different times of incubation.
Cell Capture Assay
For the cancer cell capture experiments, 96-well streptavidin plates were used. The wells were incubated with biotin-labeled M3 or the ssDNA library for 1 hr, followed by washing three times with PBS. To facilitate the visualization and resolution, the target MDA-MB-231 cells and the control MCF-7 cells were first stained with Cell-Tracker Dye (green for target cells and red for control cells) and were resuspended in BB. Next, the cell suspension was added to the wells, and they were coated with M3 or the ssDNA library for 30 min. After the supernatant was removed, the wells were washed three times with ice-cold PBS prior to observation under fluorescence microscopy.
To further verify the capture specificity of M3 to the target cells, we mixed the target MDA-MB-231 cells (stained green) and the control MCF-7 cells (stained red) in a decreased proportion (target cells accounted for 50%, 25%, 25%, or 10%), and then the target cell capture experiment was performed using M3 as described earlier.
Capture of BC Cells in Real Peripheral Blood Samples
To evaluate the target capture capacity of aptamer M3 in a complex biological environment, MDA-MB-231 cells were prepared as described earlier and were mixed with whole blood. The mixed cells were added to wells coated with the M3 aptamer for 1 hr. After the supernatant was removed, the wells were washed three times with ice-cold PBS and were immediately fixed for 15 min in 4% paraformaldehyde. The cells were first incubated with DAPI for 5 min at room temperature and were then washed and incubated with FITC-labeled anti-CD45 for 1 hr at 4°C. Finally, the cells were incubated with PE-labeled anti-EpCAM for 1 hr at 4°C, and the fluorescence images of the cells were obtained using fluorescence microscopy.
Blood samples (5 mL each) for the study were drawn from the vein of patients or healthy volunteers. CTC capture experiments were performed as described earlier.
Binding Assay of Anti-EpCAM and/or M3 to Different Cell Lines by Flow Cytometry
The cells (MDA-MB-231, MCF-7, MDA-MB-436, LoVo, CL187, SGC-7901, A549, and HEK293) were suspended in BB and were incubated with PE-labeled anti-EpCAM for 1 hr at 4°C. The cells were washed after incubation, and MDA-MB-231 and MDA-MB-436 cells were incubated with FITC-labeled M3 for 30 min at 4°C. After then, the fluorescence intensity of all the cell lines was determined by flow cytometry.
Statistical Analysis
The data are expressed as the means ± SD of three independent experiments, and the statistical evaluations were conducted using unpaired two-tailed Student’s t tests. The statistical analyses were performed using GraphPad Prism software.
Author Contributions
W.-M.L. conceived the work, designed and performed the experiments, analyzed the data, and wrote the manuscript. L.-L.Z. designed and performed the experiments and analyzed the data. M.Z. collected the blood samples and analyzed the data. J.F. conceived the work, wrote the manuscript, and critically viewed and supervised the study.
Conflicts of Interest
The authors report no conflicts of interest.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (nos. 81502703, 81672920, and 21375149) and the Program for Excellent Talents of China Medical University (no. YQ20160007).
Footnotes
Supplemental Information includes Supplemental Materials and Methods, eight figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.omtn.2018.07.008.
Supplemental Information
References
- 1.Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Bettaieb A., Paul C., Plenchette S., Shan J., Chouchane L., Ghiringhelli F. Precision medicine in breast cancer: reality or utopia? J. Transl. Med. 2017;15:139–152. doi: 10.1186/s12967-017-1239-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fidler I.J. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 4.Hiraiwa K., Takeuchi H., Hasegawa H., Saikawa Y., Suda K., Ando T., Kumagai K., Irino T., Yoshikawa T., Matsuda S. Clinical significance of circulating tumor cells in blood from patients with gastrointestinal cancers. Ann. Surg. Oncol. 2008;15:3092–3100. doi: 10.1245/s10434-008-0122-9. [DOI] [PubMed] [Google Scholar]
- 5.Yan W.T., Cui X., Chen Q., Li Y.F., Cui Y.H., Wang Y., Jiang J. Circulating tumor cell status monitors the treatment responses in breast cancer patients: a meta-analysis. Sci. Rep. 2017;7:43464. doi: 10.1038/srep43464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rack B., Schindlbeck C., Jückstock J., Andergassen U., Hepp P., Zwingers T., Friedl T.W., Lorenz R., Tesch H., Fasching P.A., SUCCESS Study Group Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst. 2014;106:dju066. doi: 10.1093/jnci/dju066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Disibio G., French S.W. Metastatic patterns of cancers: results from a large autopsy study. Arch. Pathol. Lab. Med. 2008;132:931–939. doi: 10.5858/2008-132-931-MPOCRF. [DOI] [PubMed] [Google Scholar]
- 8.Kim M.Y., Oskarsson T., Acharyya S., Nguyen D.X., Zhang X.H.F., Norton L., Massagué J. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagrath S., Sequist L.V., Maheswaran S., Bell D.W., Irimia D., Ulkus L., Smith M.R., Kwak E.L., Digumarthy S., Muzikansky A. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song Y., Tian T., Shi Y., Liu W., Zou Y., Khajvand T., Wang S., Zhu Z., Yang C. Enrichment and single-cell analysis of circulating tumor cells. Chem. Sci. (Camb.) 2017;8:1736–1751. doi: 10.1039/c6sc04671a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bystricky B., Mego M. Circulating tumor cells in breast cancer patients. Neoplasma. 2016;63:18–29. doi: 10.4149/neo_2016_003. [DOI] [PubMed] [Google Scholar]
- 12.Sieuwerts A.M., Kraan J., Bolt J., van der Spoel P., Elstrodt F., Schutte M., Martens J.W., Gratama J.W., Sleijfer S., Foekens J.A. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J. Natl. Cancer Inst. 2009;101:61–66. doi: 10.1093/jnci/djn419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bednarz-Knoll N., Alix-Panabières C., Pantel K. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 2012;31:673–687. doi: 10.1007/s10555-012-9370-z. [DOI] [PubMed] [Google Scholar]
- 14.Liao T.T., Yang M.H. Revisiting epithelial-mesenchymal transition in cancer metastasis: the connection between epithelial plasticity and stemness. Mol. Oncol. 2017;11:792–804. doi: 10.1002/1878-0261.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorges T.M., Tinhofer I., Drosch M., Röse L., Zollner T.M., Krahn T., von Ahsen O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer. 2012;12:178. doi: 10.1186/1471-2407-12-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruscetti M., Quach B., Dadashian E.L., Mulholland D.J., Wu H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 2015;75:2749–2759. doi: 10.1158/0008-5472.CAN-14-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polyak K., Weinberg R.A. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 18.Krishnamurthy S., Cristofanilli M., Singh B., Reuben J., Gao H., Cohen E.N., Andreopoulou E., Hall C.S., Lodhi A., Jackson S., Lucci A. Detection of minimal residual disease in blood and bone marrow in early stage breast cancer. Cancer. 2010;116:3330–3337. doi: 10.1002/cncr.25145. [DOI] [PubMed] [Google Scholar]
- 19.Satelli A., Batth I., Brownlee Z., Mitra A., Zhou S., Noh H., Rojas C.R., Li H., Meng Q.H., Li S. EMT circulating tumor cells detected by cell-surface vimentin are associated with prostate cancer progression. Oncotarget. 2017;8:49329–49337. doi: 10.18632/oncotarget.17632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S., Tian Z., Zhang L., Hou S., Hu S., Wu J., Jing Y., Sun H., Yu F., Zhao L. Combined cell surface carbonic anhydrase 9 and CD147 antigens enable high-efficiency capture of circulating tumor cells in clear cell renal cell carcinoma patients. Oncotarget. 2016;7:59877–59891. doi: 10.18632/oncotarget.10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie J., Zhao R., Gu S., Dong H., Wang J., Lu Y., Sinko P.J., Yu T., Xie F., Wang L. The architecture and biological function of dual antibody-coated dendrimers: enhanced control of circulating tumor cells and their hetero-adhesion to endothelial cells for metastasis prevention. Theranostics. 2014;4:1250–1263. doi: 10.7150/thno.8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 23.Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 24.Jo H., Ban C. Aptamer-nanoparticle complexes as powerful diagnostic and therapeutic tools. Exp. Mol. Med. 2016;48:e230. doi: 10.1038/emm.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cerchia L., de Franciscis V. Targeting cancer cells with nucleic acid aptamers. Trends Biotechnol. 2010;28:517–525. doi: 10.1016/j.tibtech.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Fang X., Tan W. Aptamers generated from cell-SELEX for molecular medicine: a chemical biology approach. Acc. Chem. Res. 2010;43:48–57. doi: 10.1021/ar900101s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C., Zhang M., Yang G., Zhang D., Ding H., Wang H., Fan M., Shen B., Shao N. Single-stranded DNA aptamers that bind differentiated but not parental cells: subtractive systematic evolution of ligands by exponential enrichment. J. Biotechnol. 2003;102:15–22. doi: 10.1016/s0168-1656(02)00360-7. [DOI] [PubMed] [Google Scholar]
- 28.Tang Z., Parekh P., Turner P., Moyer R.W., Tan W. Generating aptamers for recognition of virus-infected cells. Clin. Chem. 2009;55:813–822. doi: 10.1373/clinchem.2008.113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W.M., Bing T., Wei J.Y., Chen Z.Z., Shangguan D.H., Fang J. Cell-SELEX-based selection of aptamers that recognize distinct targets on metastatic colorectal cancer cells. Biomaterials. 2014;35:6998–7007. doi: 10.1016/j.biomaterials.2014.04.112. [DOI] [PubMed] [Google Scholar]
- 30.Luzzi K.J., MacDonald I.C., Schmidt E.E., Kerkvliet N., Morris V.L., Chambers A.F., Groom A.C. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998;153:865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao X., Li S., Chen L., Ding H., Xu H., Huang Y., Li J., Liu N., Cao W., Zhu Y. Combining use of a panel of ssDNA aptamers in the detection of Staphylococcus aureus. Nucleic Acids Res. 2009;37:4621–4628. doi: 10.1093/nar/gkp489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W.M., Fang J. Quantitative multi-targeted imaging of metastatic colorectal cancer cells using aptamer probes in combination. Chem. J. Chin. Univ. 2016;7:1262–1268. [Google Scholar]
- 33.Chambers A.F., Groom A.C., MacDonald I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 34.O’Flaherty J.D., Gray S., Richard D., Fennell D., O’Leary J.J., Blackhall F.H., O’Byrne K.J. Circulating tumour cells, their role in metastasis and their clinical utility in lung cancer. Lung Cancer. 2012;76:19–25. doi: 10.1016/j.lungcan.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Brinkley B.R., Beall P.T., Wible L.J., Mace M.L., Turner D.S., Cailleau R.M. Variations in cell form and cytoskeleton in human breast carcinoma cells in vitro. Cancer Res. 1980;40:3118–3129. [PubMed] [Google Scholar]
- 36.Fusella F., Seclì L., Busso E., Krepelova A., Moiso E., Rocca S., Conti L., Annaratone L., Rubinetto C., Mello-Grand M. The IKK/NF-κB signaling pathway requires Morgana to drive breast cancer metastasis. Nat. Commun. 2017;8:1636. doi: 10.1038/s41467-017-01829-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwak B., Lee J., Lee J., Kim H.S., Kang S., Lee Y. Spiral shape microfluidic channel for selective isolating of heterogenic circulating tumor cells. Biosens. Bioelectron. 2018;101:311–316. doi: 10.1016/j.bios.2017.10.036. [DOI] [PubMed] [Google Scholar]
- 38.Aceto N., Bardia A., Miyamoto D.T., Donaldson M.C., Wittner B.S., Spencer J.A., Yu M., Pely A., Engstrom A., Zhu H. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–1122. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fabisiewicz A., Grzybowska E. CTC clusters in cancer progression and metastasis. Med. Oncol. 2017;34:12. doi: 10.1007/s12032-016-0875-0. [DOI] [PubMed] [Google Scholar]
- 40.Au S.H., Storey B.D., Moore J.C., Tang Q., Chen Y.L., Javaid S., Sarioglu A.F., Sullivan R., Madden M.W., O’Keefe R. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. USA. 2016;113:4947–4952. doi: 10.1073/pnas.1524448113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y., Zhu C., Ling L., Wan L., Fang X., Bai C. Specific aptamer-protein interaction studied by atomic force microscopy. Anal. Chem. 2003;75:2112–2116. doi: 10.1021/ac026182s. [DOI] [PubMed] [Google Scholar]
- 42.Ye M., Hu J., Peng M., Liu J., Liu J., Liu H., Zhao X., Tan W. Generating aptamers by cell-SELEX for applications in molecular medicine. Int. J. Mol. Sci. 2012;13:3341–3353. doi: 10.3390/ijms13033341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li T., Liu Y., Xiao H., Xu G. Long non-coding RNA TUG1 promotes cell proliferation and metastasis in human breast cancer. Breast Cancer. 2017;24:535–543. doi: 10.1007/s12282-016-0736-x. [DOI] [PubMed] [Google Scholar]
- 44.Zhan Y., Wang N., Liu C., Chen Y., Zheng L., He L. A novel taspine derivative, HMQ1611, suppresses adhesion, migration and invasion of ZR-75-30 human breast cancer cells. Breast Cancer. 2014;21:334–340. doi: 10.1007/s12282-012-0392-8. [DOI] [PubMed] [Google Scholar]
- 45.Li W., Chen H., Yu M., Fang J. Targeted delivery of doxorubicin using a colorectal cancer-specific ssDNA aptamer. Anat. Rec. (Hoboken) 2014;297:2280–2288. doi: 10.1002/ar.22990. [DOI] [PubMed] [Google Scholar]
- 46.Yu Y., Liang C., Lv Q., Li D., Xu X., Liu B., Lu A., Zhang G. Molecular selection, modification and development of therapeutic oligonucleotide aptamers. Int. J. Mol. Sci. 2016;17:358. doi: 10.3390/ijms17030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi H., He X., Cui W., Wang K., Deng K., Li D., Xu F. Locked nucleic acid/DNA chimeric aptamer probe for tumor diagnosis with improved serum stability and extended imaging window in vivo. Anal. Chim. Acta. 2014;812:138–144. doi: 10.1016/j.aca.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 48.Zamay A.S., Zamay G.S., Kolovskaya O.S., Zamay T.N., Berezovski M.V. Aptamer-based methods for detection of circulating tumor cells and their potential for personalized diagnostics. Adv. Exp. Med. Biol. 2017;994:67–81. doi: 10.1007/978-3-319-55947-6_3. [DOI] [PubMed] [Google Scholar]
- 49.Qian W., Zhang Y., Chen W. Capturing cancer: emerging microfluidic technologies for the capture and characterization of circulating tumor cells. Small. 2015;11:3850–3872. doi: 10.1002/smll.201403658. [DOI] [PubMed] [Google Scholar]
- 50.Xu Y., Yang X., Wang E. Review: aptamers in microfluidic chips. Anal. Chim. Acta. 2010;683:12–20. doi: 10.1016/j.aca.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Wu P., Sokoll L.J., Kudrolli T.A., Chowdhury W.H., Ma R., Liu M.M., Rodriguez R., Lupold S.E. A novel approach for detecting viable and tissue-specific circulating tumor cells through an adenovirus-based reporter vector. Prostate. 2014;74:1286–1296. doi: 10.1002/pros.22845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.