

Summary
Regulation of AMPA receptor (AMPAR) trafficking is a key modulator of excitatory synaptic transmission; however, intracellular vesicular transport of newly synthesized AMPARs has been little studied due to technical limitations. By combining molecular tools with imaging strategies in cultured rat hippocampal neurons, we found that vesicles containing newly synthesized, GluA1-subunit-containing AMPARs are transported antero- and retrogradely at a mean speed of 1.5 μm.s−1. Synaptic activity and variations in intracellular calcium levels bidirectionally modulate GluA1 transport. Chemical long-term potentiation (cLTP) initially induces a halt in GluA1 transport, followed by a sustained increase, while acute glutamate uncaging on synaptic spines arrests vesicular movements. GluA1 phosphomimetic mutants preferentially travel to the dendritic tip, probably to replenish extrasynaptic pools, distal to the soma. Our findings indicate that AMPAR intracellular transport is highly regulated during synaptic plasticity and likely controls AMPAR numbers at the plasma membrane.
Keywords: intracellular transport, GluA1-containing AMPARs, synaptic activity, plasticity, long-term potentiation, calcium, GCaMP6f, phosphorylation, videomicroscopy
Graphical Abstract
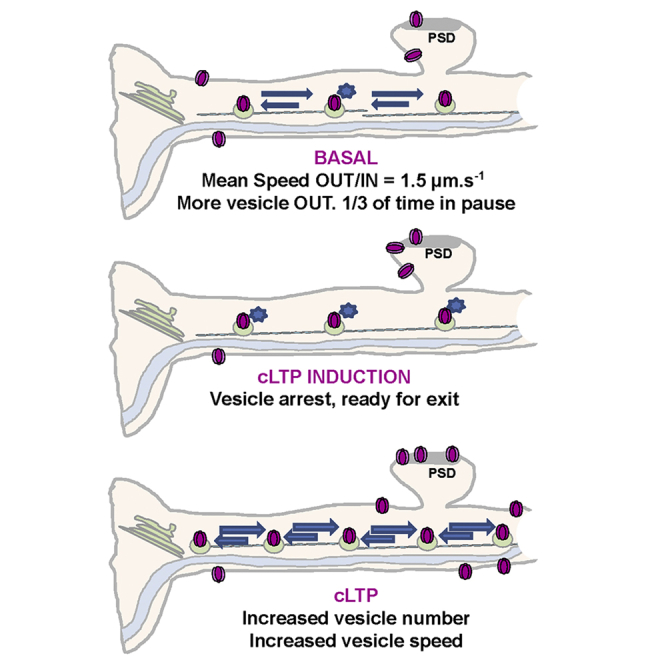
Highlights
-
•
AMPARs are transported bidirectionally and intracellularly in dendrites ∼1.5 μm.s−1
-
•
Acute increase in intracellular Ca2+ during early LTP arrests GluA1-AMPAR vesicles
-
•
20 min after LTP induction, number and speed of GluA1-AMPAR vesicles are increased
-
•
GluA1 phosphorylation at S831/S845 increases vesicle number and outward transport
Hangen et al. show that, under basal conditions, GluA1-AMPAR-containing vesicles are transported similarly outward and inward ∼1.5 μm.s−1, spending a third of their time in a paused state. They find that transport is differentially regulated during the different phases of LTP by calcium and GluA1 phosphorylation-dependent processes.
Introduction
AMPA (alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid)-type glutamate receptors (AMPARs) mediate most fast excitatory transmission in the vertebrate CNS. Control of postsynaptic AMPAR number regulates synaptic efficacy (Henley and Wilkinson, 2016, Huganir and Nicoll, 2013) and results from a dynamic equilibrium between AMPARs stored in intracellular pools and at the synaptic or extrasynaptic plasma membrane (PM). Regulation of synaptic AMPAR abundance underlies many forms of synaptic plasticity, the cellular mechanism thought to mediate memory and learning, and is set by three trafficking steps: (1) lateral diffusion in the PM (Choquet and Triller, 2013), (2) endo- and exocytosis (Wang et al., 2008), and (3) motor-dependent intracellular transport (Kim and Lisman, 2001, Setou et al., 2002). The transport of newly synthesized AMPARs from the Golgi apparatus (GA)—either in the cell soma or from Golgi outposts—into dendrites is likely a critical determinant of the number of AMPARs at the PM and synapses. However, the mechanisms of intracellular AMPAR transport and their effect on synaptic strength are largely unknown.
The complex morphology of neurons requires AMPARs exiting the GA to travel up to hundreds of microns along the dendritic shaft to reach the PM and the synapse. Stringent quality-control mechanisms at the level of the endoplasmic reticulum (ER) allow only correctly folded receptors to be exported to the PM (Greger et al., 2007). Although synthesis of AMPARs at Golgi outposts in dendrites has been reported (Hanus and Schuman, 2013, Ju et al., 2004), most receptors are synthesized in the somatic GA. AMPAR subunits can have a glycosylation profile corresponding to mature proteins processed by the somatic Golgi (Hanus et al., 2016).
After budding from the Golgi, secretory vesicles containing AMPARs traffic to the PM, presumably through direct or indirect interactions with microtubules and microtubule-based motor proteins such as kinesin and/or dynein (Hirokawa and Takemura, 2005, Kapitein et al., 2010). Electrophysiological studies indicate that AMPAR-mediated synaptic transmission depends on both dynein and kinesin superfamily proteins (Kim and Lisman, 2001). Moreover, increase in AMPAR number at the PM during long-term potentiation (LTP) directly depends on their secretory transport (Broutman and Baudry, 2001, Esteves da Silva et al., 2015).
In C. elegans, motor-mediated transport is the major mechanism for delivery, removal, and redistribution of GLR-1 glutamate receptors (Hoerndli et al., 2015). The microtubule-dependent motor UNC-116 (homolog of mammalian KIF5) drives the delivery and the removal of AMPARs, while UNC-43 (homolog of mammalian Ca2+/calmodulin-dependent protein kinase II [CaMKII]) plays an essential role in modulating the transport of AMPARs between the cell body and the insertion or removal of synaptic AMPARs (Hoerndli et al., 2015). In vertebrates, AMPARs interact with the microtubule motor KIF5 through GRIP1 (Setou et al., 2002). Other motors have also been implicated in AMPAR transport, such as Myosin-VI, an actin-dependent motor protein (Wu et al., 2002). In hippocampal neuronal cultures, it was reported that GFP-GluA2-containing vesicles transport bidirectionally in a dendritic endosomal fraction and in synaptic spines (Esteves da Silva et al., 2015, Evans et al., 2017).
The intracellular transport of vesicles containing fluorescently tagged AMPARs and its regulation by synaptic activity have proven difficult to resolve by live-cell imaging, because at steady state, the fluorescent signal from an abundance of AMPARs expressed in the ER (Greger et al., 2007) and at the PM of dendrites obscures detection of transport vesicle movement. To overcome this limitation, we used ARIAD, an ER release synchronization system (Rivera et al., 2000), combined with spinning disk confocal microscopy and photo-bleach to reveal the dendritic transport of newly synthesized AMPARs in cultured rat hippocampal neurons. Here we report the characteristics of AMPAR-containing transport vesicles under basal conditions and how vesicular transport is regulated during synaptic activity.
Results
Controlled Release of Newly Synthesized GluA1-Containing AMPARs from the ER
To monitor transport of GFP-tagged AMPARs released synchronously from the ER, ARIAD technology (Rivera et al., 2000) was used, which allowed GFP-GluA1 secretion to be temporally controlled with a cell-permeant, small-molecule drug (AP-21998) termed Ariad ligand (AL) (Figure 1A). ARIAD cDNA codes for a signal peptide sequence, followed by four conditional aggregation domains (CADs), a furin cleavage site, and the protein of interest (Figure 1B). In this system, ER retention of fusion proteins is driven by the CADs that interact in a ligand-dependent manner and gradually accumulate in the ER (Figure 1A). Their simultaneous release upon addition of AL allows expressed proteins to progress through the secretory pathway in a synchronous manner, which is particularly adapted to monitor intracellular transport. Important features of this system include (1) no or low basal secretion and (2) a rapid and high level of secretion in response to the addition of AL.
Figure 1.

Temporal Control of AMPAR from the Endoplasmic Reticulum to the PM
(A) Natural secretion (green arrow) versus controlled secretion (yellow arrow) of AMPAR from the ER to the PM. Addition of the ligand allows proteins to be exported through the secretory pathway until the PM.
(B) ARIAD vector and strategy allowing export of GluA1. The retention machinery is cut by the endogenous protease Furin in the Golgi.
(C) Images of hippocampal neurons expressing ARIAD/GFP-GluA1. Individual and merged labeling are shown at different incubation times with the AL (t = 0–30 min). Calreticulin and GM130 antibodies were used to stain ER and Golgi, and colocalization with GluA1 was performed using line scan analysis. Results are plotted as GluA1 (green), calreticulin (blue), and GM130 (red) in function of the length of the dotted arrow drawn across the somatic region in the zoomed image. Correspondence of the peak for each line is indicative of the colocalization of the proteins.
(D) Images illustrating total GFP-GluA1 (green) and live labeling of GFP-GluA1 with an anti-GFP antibody (red) at different incubation times with the AL (30–120 min) and the related quantification of the kinetic (see also Figure S1D). The yellow window delineates the acquisition time of the videos of AMPAR intracellular transport.
Mean ± SEM of 4 independent experiments (n > 20 cells).
To study the transport of GluA1-containing AMPARs, GFP-GluA1 was expressed in ARIAD (Figure 1B). Furin, a trans-Golgi resident protease that processes protein precursors before their secretion, cleaved the CAD from GluA1, allowing its progression through the GA to the PM (Figure 1A). To establish the kinetics of GluA1-containing AMPAR transport from the ER to the GA, we triggered secretion of GFP-GluA1 by incubations with AL from 0 to 30 min (Figure 1C). Colocalization of GFP-GluA1 with immunostaining for endogenous calreticulin (ER marker) and GM130 (GA marker) were compared by line scan profiles. Before addition of AL (t = 0), GFP-GluA1-containing AMPARs were entirely localized in the ER. The staining profile shifted 15 min after the addition of AL, appearing in perinuclear structures corresponding to the GA. GFP-GluA1-containing AMPARs were detected in the GA within 30 min of induction.
To assess the kinetics of GluA1 insertion at the PM (Figures 1D and S1), neurons transfected with GFP-GluA1 in ARIAD were treated with AL and live immunostaining for GFP was used to detect GFP-GluA1 on the cell surface. At time zero, before AL addition, no surface labeling for GFP was observed (Figure S1A). Thirty minutes after addition of AL, surface labeling for GFP was detected, on both the dendritic shaft and the synaptic spines, and continued to intensify until reaching a plateau 1 hr after incubation with the AL (Figures 1D and S1D). Surface detection of GluA1 occurred at the cell body and in proximal and in distal dendrites with similar kinetics (Figures S1B and S1C). Induction of GluA1 export did not modify endogenous synaptic GluA2 content (mean ± SEM of GluA2 synaptic content at t = 0 min: 244.5 ± 18.95 fluorescence a.u., n = 200; at t = 45 min: 242.9 ± 14.17 fluorescence a.u., n = 160; p = 0.95). Based on the kinetics of GFP-GluA1 detection at the cell surface, the optimal time window to monitor AMPAR intracellular transport in subsequent live-cell imaging experiments was estimated to be between 20 and 60 min after AL addition.
Characterization of GluA1 Intracellular Transport under Basal Conditions
To investigate the impact of the ARIAD system on GluA1 vesicular trafficking per se, neurons were transfected with either TdTomato-GluA1 in ARIAD (ARIAD/TdT-GluA1) or TdTomato-GluA1 in pRK5 vector (TdT-GluA1) lacking the aggregation control system of secretion (Figures 2B–2D). After 7–8 days of expression, intracellular transport of GluA1 was monitored by videomicroscopy after its release from the ER. Before recording, we photo-bleached a cell-body proximal dendritic segment to erase the background signal due to the presence of tagged receptors in the ER (for ARIAD/TdT-GluA1) or the ER and PM (for TdT-GluA1) (Figures 2A and 2B, dotted areas). This allowed clear visualization and tracking of GluA1-containing vesicles in the dendritic shaft as they invaded the photo-bleached area (Video S1). To analyze GluA1 transport, kymographs were generated from video recordings, as a representation of the distance traveled by vesicles over time (Figure 2B). An ImageJ macro was designed that allowed filtering, isolation of vesicles, and denoising of the images. Vesicle trajectories were labeled green for vesicle movements away from the cell body (anterograde, i.e., OUT), red for vesicle movements toward the cell body (retrograde, i.e., IN), and blue for static vesicles (PAUSE).
Figure 2.

Characterization of GluA1-Containing AMPAR Intracellular Transport in Basal Condition
(A) Protocol to monitor AMPAR vesicular transport in the proximal neuronal dendritic shaft. After incubation with AL, positions of transfected neurons are registered. GluA1 vesicular transport becomes visible after photo-bleaching (FRAP) of the region of interest (ROI) in the proximal dendrite. Transport is recorded by streaming one image every 100 ms during 1 min.
(B) Images of neurons expressing TdT-GluA1 in pRK5 (top panel) or in ARIAD (bottom panel), with raw and annotated kymographs (right panels). Trajectories are shown with a color code (green for outward movements, red for inward movements, and blue for pausing vesicles). Dotted lines in the crude image delineate the ROI from which kymographs were generated.
(C) Background analyses of the ROI after FRAP of neurons in the two conditions. Results are expressed as the ratio of the background measured with ImageJ at the end of the acquisition (frame 550) over the one measured just after the FRAP (frame 20).
(D) Mean vesicle number passing through a 20 μm2 bleached ROI during 1 min of recording.
(E) Image of neurons expressing ARIAD/TdT-GluA1 (left panel) and the zoomed images of its ROI (right panels) at different time points (t = 4–20 s). Colored arrows indicate outward (green), inward (red), or static (blue) trajectories of GluA1-containing vesicles.
(F) Percentage of time spent in each state by a vesicle containing ARIAD/TdT-GluA1.
(G) Velocities of ARIAD/TdT-GluA1 vesicles.
(H) Mean frequency distribution of instantaneous speeds of mobile GluA1 vesicles (>500 values).
For (C) and (D), mean ± SEM of 2 independent experiments (n = 20/18 cells). For (F) to (H), mean ± SEM of 8 independent experiments (n = 76 cells). See also Figures S2A–S2C and Tables S1 and S2.
TdTomato tagged GluA1 was expressed in the Ariad vector in cultured hippocampal neurons and imaged 30′ after addition of the AL. Time stamp in seconds.
Kymographs obtained from videos recorded with GluA1 in ARIAD have less background than when GluA1 was expressed in pRK5 (Figures 2B and S2A). We calculated the overall background in the region of interest (ROI) just after the fluorescence recovery after photo-bleaching (FRAP) (image 20) or at the end of the video (image 550) (Figure 2C). When GluA1 was expressed in ARIAD, the background at frame 550 was 2 (±1.6) times higher than at frame 20, whereas this ratio was 5.2 (±3.2) times higher in pRK5. This is probably because of a recovery of a GluA1 signal at the PM when expressed in pRK5, which is absent from ARIAD. This difference in background allowed us to detect around 2 times more vesicles when GluA1 was under the ARIAD system compared to pRK5 (GluA1 in pRK5, 6.5 ± 3.7 vesicles/20 μm2/min; GluA1 in ARIAD, 11.5 ± 4.3 vesicles/20 μm2/min) (Figure 2D). We also quantified the intensity of signal to noise by tracing line scans every 10 s on the images of the Figure 2B (Figure S2A). When GluA1 is expressed in ARIAD, vesicles were more easily detected than in pRK5. Moreover, after 40 s, the TdT-GluA1 signal-to-noise ratio was still high in the ARIAD condition, whereas vesicles could hardly be detected in pRK5. There was no significant difference between the speeds of TdT-GluA1 expressed in ARIAD or those in pRK5 (Figure S2B).
The efficiency of the analysis plugin to automatically detect vesicles and calculate their speeds was tested. We expressed ARIAD/TdT-GluA1 and manually tracked ten GluA1-containing vesicles. The distribution of the mean instantaneous speeds obtained manually was similar to that of the ones obtained using the automatized plugin (Figure S2C). Altogether, the ARIAD system did not change the overall characteristics of GluA1 transport compared to a standard expression system and presented several advantages. It synchronized the transport vesicles to exclusively track newly synthesized receptors and allowed a specific population of Golgi-derived vesicles to be imaged. Due to the low background in the videos, numerous vesicles were detected and analyzed.
With imaging and analysis conditions established, GluA1 intracellular transport in basal conditions expressing ARIAD/TdT-GluA1 was characterized (Figures 2E–2H). The mean percentage of time spent by individual vesicles in each state was calculated (Figures 2E and 2F). Vesicles spent significantly more time moving anterogradely (OUT, 37% ± 1.44%) than retrogradely (IN, 27% ± 1.13%). In addition, they spent around one-third of their time in a static state (PAUSE, 36% ± 1.56%) (Figure 2F). The detection of vesicles in pause highly depends on the time elapsed between the FRAP and the start of the acquisition (fraction of vesicles in pause as a function of elapsed time: <5 s, 35% ± 2.20%; 300 s, 62% ± 2.95%) (Figure S2D). Here we used <5 s, the acquisition starts right after photo-bleaching the ROI, and no static vesicles remain at the beginning of the video, because they are all initially bleached. If a delay is introduced between photo-bleaching and beginning of the recording, the percentage of vesicles in pause increases, because pausing events accumulate during this short period.
No significant difference in the vesicle velocity between OUT and IN directions was detected (OUT, 1.46 ± 0.04 μm.s−1; IN, 1.37 ± 0.04 μm.s−1) (Figure 2G). Similarly, the mean time spent by vesicles in one direction before changing direction was not significantly different between OUT and IN directions (OUT, 2.74 ± 0.25 s; IN, 2.48 ± 0.28 s) (Figure S2E). However, the mean distance traveled by vesicles going OUT was higher than that of vesicles going IN (OUT, 3.21 ± 0.46 μm; IN, 2.54 ± 0.32 μm) (Figure S2F). We found no differences between the frequency distribution of the instantaneous speed of GluA1-containing vesicles moving OUT and that of vesicles moving IN, both defined by two main populations at 0.5 and 1.6 μm.s−1 (Figure 2H). Finally, for a given vesicle, the number of transitions that go from the OUT or IN state to the PAUSE state is higher than the transitions from one moving state immediately to the opposite direction (Figure S2G), meaning that vesicles preferentially stop before inverting their direction.
The GluA1 transport properties in proximal versus secondary branch dendrites were also compared (Figures S2H and S2I). Vesicles spent slightly less time outward in secondary versus primary dendrites, while pause time and inward movements were similar. The mean speed was higher in secondary versus primary dendrites.
Properties of GluA1 Intracellular Transport upon Calcium Chelation
To decipher whether intracellular transport of GluA1 is regulated by neuronal activity, we adjusted our recording strategy and simultaneously recorded intracellular transport and synaptic activity with the calcium sensor GCaMP6f (Figure S3A).
First, the effect of lowering intracellular Ca2+ levels on GluA1 intracellular transport was analyzed (Figure 3) using bis–N,N,N',N'-tetraacetic acid-AM (BAPTA-AM), a cell-permeant calcium chelator. Neurons cotransfected with ARIAD/TdT-GluA1 and GCaMP6f were treated with AL, and 20 μM BAPTA-AM was added 5 min before recording a series of alternating frames in the green and red channels to image Ca2+ levels and GluA1 transport, respectively (Figures 3A and S3A). Neurons incubated with BAPTA-AM exhibited less variation in Ca2+ levels than did control cells (Figures 3B and S3B). The mean frequency distribution of Ca2+ levels was shifted to lower ΔF/F with BAPTA-AM (Figure S3C).
Figure 3.

Decreasing Calcium Level Increases Vesicle Speeds
(A) Images of neurons coexpressing ARIAD/TdT-GluA1 and GCaMP6f (left panels) and their associated kymographs (right panels) in control condition (Ctrl, top panels) and with 20 μM of BAPTA-AM (BAPTA, bottom panels).
(B) Variations of GCaMP6f mean fluorescence (ΔF/F) over time in Ctrl and BAPTA-treated neurons (see also Figures S3B and S3C).
(C) Mean outward (green), inward (red), and static (blue) vesicle states over time in Ctrl and BAPTA-treated neurons.
(D) Mean vesicle number passing through a 20 μm2 dendritic-bleached ROI during 1 min of recording.
(E) Mean outward (green) and inward (red) velocities of GluA1-containing vesicles in Ctrl and BAPTA-treated neurons.
(F) Mean frequency distribution of the outward (green) and inward (red) instantaneous speeds of mobile GluA1-containing vesicles in Ctrl and BAPTA-treated neurons (>500 values).
Mean ± SEM of 4 independent experiments (n = 43 cells for Ctrl, n = 28 cells for BAPTA). See also Figures S3A and S3B and Tables S1 and S2.
We then compared the characteristics of GluA1 transport in both conditions (control [Ctrl] versus BAPTA) (Figures 3C–3F). Vesicles spent the same percentage of time in each state: OUT (Ctrl, 38% ± 2.04%; BAPTA, 43% ± 2.58%), IN (Ctrl, 27% ± 1.24%; BAPTA, 28% ± 2.47%), and PAUSE (Ctrl, 35% ± 2.20%; BAPTA, 29% ± 2.25%) (Figure 3C). As described earlier in the basal condition, the time spent by vesicles in the OUT state remained higher than in the IN state in the presence of BAPTA. Similarly, the mean vesicle density was not changed by chelating Ca2+ (Ctrl, 8.8 ± 0.79; BAPTA, 8.5 ± 0.75) (Figure 3D). In contrast, velocity was increased by about 20% in the mean of both OUT and IN speeds in the presence of BAPTA-AM (Ctrl versus BAPTA: OUT, 1.52 ± 0.05 μm.s−1 versus 1.77 ± 0.06 μm.s−1; IN, 1.43 ± 0.05 μm.s−1 versus 1.72 ± 0.07 μm.s−1) (Figure 3E). The frequency distributions of both OUT (green) and IN (red) instantaneous speeds displayed a significant decrease in the vesicle population moving slowly (≤1.5 μm.s−1) in favor of an increase in those moving quickly (>1.5 μm.s−1), with a new population at 3.5 μm.s−1 (Figure 3F).
Effect of Glutamate-Mediated Increase in Intracellular Calcium on AMPAR Transport
The effects of increased Ca2+ concentration on GluA1 transport were tested by glutamate uncaging to trigger Ca2+ entry into the cell (Figure 4). One-photon uncaging and 2-color imaging were performed on neurons coexpressing ARIAD/TdT-GluA1 with GCaMP6f (Figures 4A and S4A). After 21 s of baseline recording (before), we applied 10 series at 0.2 Hz of uncaging laser pulses on about 10 spines identified as protrusions from the dendrites over the last 26 s of the video (during) and in the presence of 0.5 mM caged 4-Methoxy-7-nitroindolinyl-caged-L-glutamate (MNI-Glu) or the absence of MNI-Glu (Ctrl). The GCaMP6f dendritic signal increased during the uncaging pulses in the presence, but not in the absence, of MNI-Glu. GCaMP6f kymographs (Figure 4B) and time course variations of the GCaMP6f mean fluorescence (Figures 4C and S4B) revealed that waves of Ca2+ were synchronized with the ten uncaging pulses. Glutamate uncaging led to a shift in the frequency distribution of Ca2+ to the higher ΔF/F in neurons exposed to MNI-Glu compared to Ctrl (Figure S4C).
Figure 4.

Glutamate Uncaging Increases Intracellular Calcium and Decreases Vesicle Speeds
(A) Images of control (Ctrl) and MNI-caged-L-glutamate (MNI-Glu)-exposed dendrite from neurons expressing GCaMP6f showing an increase of the Ca2+ fluorescence intensity after release of glutamate. Purple circles indicate the regions of laser pulses.
(B) GCaMP6f crude kymographs in the absence (Ctrl) or presence of MNI-Glu in the media before and during the uncaging protocol after the purple line.
(C) Variation of GCaMP6f fluorescence (ΔF/F) over time in Ctrl and MNI-Glu media before and after uncaging (purple line). See also Figures S4B and S4C.
(D) Images of neurons coexpressing ARIAD/TdT-GluA1 and GCaMP6f and their associated GluA1 kymographs in Ctrl and MNI-Glu conditions.
(E) Mean outward (green) and inward (red) velocities of GluA1-containing vesicles in Ctrl and MNI-Glu media.
(F) Mean frequency distribution of the outward (green) and inward (red) instantaneous speeds of GluA1-containing vesicles in both conditions before and during uncaging (>200 values).
(G) Correlation between outward and inward instantaneous speeds and changes in the GCaMP6f fluorescence (ΔF/F) in MNI-Glu-incubated neurons during uncaging. Each dot represents one vesicle at a specific time point. See also Figure S4D for Ctrl.
(H) Mean frequency of the time spent by vesicles in pause in Ctrl and MNI-Glu media before and during uncaging.
Mean ± SEM of 6 independent experiments (n = 42 cells for Ctrl, n = 37 cells for MNI-Glu). See also Figure S4 and Tables S1 and S2.
Next, the effects of glutamate-mediated increase in intracellular Ca2+ concentrations on GluA1 transport were investigated (Video S2). From the GluA1 kymographs (Figure 4D), we extracted the different parameters related to intracellular transport before and during uncaging of MNI-Glu (Figures 4E–4H, S4D, and S4E). Both OUT (green) and IN (red) mean velocities were decreased after glutamate release (OUT: 1.24 ± 0.06 μm.s−1 [Ctrl_before], 1.14 ± 0.06 μm.s−1 [Ctrl_during], 1.17 ± 0.05 μm.s−1 [MNI-Glu_before], 0.88 ± 0.04 μm.s−1 [MNI-Glu_during]; IN: 1.22 ± 0.07 μm.s−1 [Ctrl_before], 1.05 ± 0.05 μm.s−1 [Ctrl_during], 1.11 ± 0.07 μm.s−1 [MNI-Glu_before], 0.78 ± 0.04 μm.s−1 [MNI-Glu_during]) (Figure 4E). The frequency distribution of the instantaneous speed of individual vesicles displayed a moderate but significant decrease in the population of rapidly moving vesicles for both directions (Figure 4F). In addition, at the level of individual vesicles, the instantaneous speed (IN and OUT) was negatively correlated to the intracellular Ca2+ levels in uncaged MNI-Glu (Figure 4G), but not in Ctrl (Figure S4D). Finally, a robust and significant increase in the frequency of static vesicles during glutamate uncaging was observed (Ctrl_before, 49% ± 2.67%; Ctrl_during, 58% ± 2.50%; MNI-Glu_before, 49% ± 2.09%; MNI-Glu_during, 69% ± 2.50%) (Figure 4H). The uncaging protocol alone had a small effect on several transport parameters (mean speed IN, Figure 4E; time in pause, Figure 4H; vesicle number, Figure S4E), albeit a weaker one than in the presence of MNI-Glu. This is likely an off-target effect of blue light illumination.
TdTomato tagged GluA1 was expressed in the Ariad vector in cultured hippocampal neurons and imaged 30′ after addition of the AL. MNI-caged-L-glutamate was uncaged at the time of appearance of the stars, leading to vesicle arrest. Time stamp in seconds.
Altogether, these results indicate that variations in intracellular Ca2+ concentrations modify GluA1 intracellular transport properties: low Ca2+ activates trafficking, and inversely, high Ca2+ slows and even stops vesicles. Because increases in intracellular Ca2+ levels are hallmarks of various forms of synaptic plasticity, we next studied the impact of chemical long-term potentiation (cLTP) on GluA1 vesicular trafficking.
AMPAR Intracellular Transport during the Induction of cLTP
We applied a classical cLTP induction protocol on 17-day-old hippocampal neurons coexpressing ARIAD/TdT-GluA1 and GCaMP6f. cLTP was divided into two phases: (1) the induction phase of LTP (stim-cLTP), defined as 5 min of chemical stimulation with no magnesium, 30 μM bicuculline, and 200 μM glycine, and (2) the cLTP, defined as the 20–60 min period following the induction. We first focused our interest on stim-cLTP (Figure 5). For these experiments, we slightly modified the protocol of acquisition by first photo-bleaching 2–3 neurons in Tyrode’s solution (Figure S5A) and then applying the cLTP solution and starting the recording. As mentioned earlier, increased duration between FRAP and video acquisition led to an increase in the initial percentage of vesicles in pause compared to previous experiments (Figures 5C and S2D).
Figure 5.

cLTP Induction Decreases Vesicle Speeds and Increases Static Vesicle Frequency
(A) Images of 16-day neurons coexpressing ARIAD/TdT-GluA1 and GCaMP6f (left panels) and their associated kymographs (right panels) in basal condition (Ctrl, top panels) or during stim-cLTP (bottom panels).
(B) Variations of GCaMP6f fluorescence over time in basal condition (Ctrl) or during stim-cLTP. See also Figures S5B and S5C.
(C) Mean of the percentage of time spent in each state—outward (green), inward (red), pause (blue)—by a vesicle in both conditions. See also Figure S2C.
(D) Mean outward (green) and inward (red) velocities of GluA1-containing vesicles.
(E) Mean frequency distribution of the outward (green) and inward (red) instantaneous speeds of GluA1-containing vesicles (>200 values).
(F) Correlation between outward and inward instantaneous speeds and changes in the GCaMP6f fluorescence during stim-cLTP. See also Figure S5D for Ctrl.
(G) Images of neurons expressing ARIAD/GFP-GluA1 and their related quantifications. Staining has been performed on live cells with an anti-GFP antibody and showed the time course externalization of newly synthesized GFP-GluA1 analyzed between 5 and 20 min in basal conditions (Ctrl) or after stim-cLTP (cLTP).
For (B) to (F), mean ± SEM of 4 independent experiments (n = 22 cells). For (G), mean ± SEM of 3 independent experiments (n > 30 cells). See also Tables S1 and S2.
Induction of cLTP strongly increased the GCaMP6f signal in the entire dendrite, inducing waves of Ca2+ with a high intensity compared to the basal condition (Ctrl) (Figures 5A and 5B). This increase in Ca2+ was observed in each cell and led to a shift of the frequency distribution of Ca2+ to the higher ΔF/F compared to the Ctrl (Figures S5B and S5C).
We next investigated the effects of the stim-cLTP on GluA1 transport and found a strong increase in pause frequency during stim-cLTP compared to the basal state, to the detriment of both outward and inward movements (Ctrl versus stim-cLTP: OUT, 18% ± 1.78% versus 9% ± 1.48%; IN, 20% ± 2.04% versus 7% ± 1.65% (IN); PAUSE, 62% ± 2.94% versus 83% ± 2.69%) (Figure 5C). Both IN and OUT vesicle velocities were highly decreased during the stimulation compared to Ctrl (Ctrl versus stim-cLTP: OUT, 1.21 ± 0.07 μm.s−1 versus 0.83 ± 0.06 μm.s−1; IN, 1.27 ± 0.06 μm.s−1 versus 0.83 ± 0.08 μm.s−1) (Figure 5D). The relative frequency of slowly moving OUT and IN vesicles (≤1.0 μm.s−1) increased in favor of a decrease in the rapidly moving ones (>1.0 μm.s−1) (Figure 5E). At the level of individual vesicles, the instantaneous speeds (OUT and IN) were negatively correlated to intracellular Ca2+ levels in chemically stimulated neurons (Figure 5F), but not in Ctrl (Figure S5D). Vesicle number slightly decreased in neurons during stim-cLTP (Ctrl versus stim-cLTP: 9.0 ± 0.84 vesicles/20 μm2/min versus 6.2 ± 0.65 vesicles/20 μm2/min; ∗p = 0.0121) (data not shown).
cLTP is associated with an increase in AMPAR exocytosis (Huganir and Nicoll, 2013, Makino and Malinow, 2009) that has been linked to release of receptors from recycling pools (Park et al., 2006). Our system gave us the opportunity to investigate whether cLTP also triggers exocytosis of newly synthesized GluA1-containing AMPARs. To do so, the externalization of newly synthesized GluA1 in cells expressing ARIAD/GFP-GluA1 was monitored by live immunolabeling for GFP after stim-cLTP or Ctrl (Figure 5G). Quantification of the GFP staining revealed a significant rise in GluA1 surface labeling as early as 10 min after cLTP induction, which continued until the end of the monitoring (Ctrl versus cLTP: 15 min, 7.61 ± 0.90 versus 10.11 ± 0.79; 20 min, 6.88 ± 0.79 versus 10.00 ± 0.73).
Altogether, these results confirm that a protocol known to increase intracellular Ca2+ through opening of N-methyl-D-aspartate (NMDA) receptors significantly slowed GluA1 transport, decreasing both IN and OUT vesicle velocities up to their immobilization. Following this Ca2+ entry, a massive exocytosis of newly synthesized GluA1 occurred, suggesting that arrest of intracellular transport of newly synthesized GluA1 followed by their exocytosis could participate to the increase in synaptic transmission during the early phase of cLTP.
AMPAR Intracellular Transport after cLTP Induction
We next analyzed the state of GluA1 intracellular transport in the longer term, i.e., 15–60 min after the application of the induction protocol (called cLTP) (Figure 6). To this aim, we characterized Ca2+ activity and GluA1 intracellular transport at least 15 min after stim-cLTP (Figures 6A and S6A). We observed moderate changes in intracellular Ca2+ levels in neurons challenged by the cLTP stimulation, with some high Ca2+ rise events in some individual cells (Figures 6A–6C and S6B).
Figure 6.

cLTP Potentiates GluA1-Containing AMPAR Intracellular Transport
(A) Images of 16-day neurons expressing ARIAD/TdT-GluA1 and GCaMP6f (left panels) and their associated kymographs (right panels) in basal condition (Ctrl, top panels) and neurons recorded during cLTP (cLTP, bottom panels).
(B) Variations of the GCaMP6f fluorescence over time in basal condition (Ctrl) or after during cLTP (cLTP).
(C) Mean outward (green), inward (red), and static (blue) vesicle states over time.
(D) Mean vesicle number passing through a 20 μm2 bleached ROI during 1 min of recording.
(E) Mean outward (green) and inward (red) velocities of GluA1 vesicles.
(F) Mean frequency distribution of the outward (green) and inward (red) instantaneous speeds of mobile vesicles in Ctrl or during cLTP (>900 values).
Mean ± SEM of 4 independent experiments (n = 33 cells for Ctrl, n = 36 cells for cLTP). See also Figure S6 and Tables S1 and S2.
During cLTP, the time spent by the vesicles in each state returned toward basal levels compared with the stim-LTP phase (Ctrl versus cLTP: OUT, 37% ± 2.02% versus 38% ± 1.76%; IN, 28% ± 2.06% versus 31% ± 1.52%; PAUSE, 36% ± 2.21% versus 31% ± 2.15%) (Figure 6C). However, both the number of vesicles (Ctrl versus cLTP: 8.7 ± 1.02 vesicles/20 μm2/min versus 14.9 ± 1.14 vesicles/20 μm2/min) (Figure 6D) and their OUT and IN speeds (Ctrl versus cLTP: OUT, 1.39 ± 0.06 μm.s−1 versus 1.63 ± 0.04 μm.s−1; IN, 1.30 ± 0.06 μm.s−1 versus 1.60 ± 0.05 μm.s−1) were strongly increased (Figure 6E). These changes are clearly visible through the numerous vesicle traces and their slopes on the kymographs generated from potentiated neurons (cLTP) (Figure 6A; Video S3). In agreement with these results, frequency distributions of the outward and inward instantaneous velocities displayed a significant decrease in the population of vesicles moving slowly (<1.5 μm.s−1) to the benefit of an increase in those traveling quickly (>1.5 μm.s−1), which represented approximately 30% of the total population upon cLTP (OUT, 27.01%; IN, 29.72%) (Figure 6F).
TdTomato tagged GluA1 was expressed in the Ariad vector in cultured hippocampal neurons and imaged (in red) 30′ after addition of the AL and 20′ after application of the c-LTP protocol. Note the large increase in vesicle density. In green homer1c-GFP to label synapses. Time stamp in seconds.
Altogether, application of the cLTP protocol has two distinct effects on GluA1 vesicular transport over time: (1) a slowdown of transport up to the immobilization of vesicles during stim-cLTP when intracellular Ca2+ is massively increasing and (2) an increase in vesicle number and velocities during the later phase, when some waves of Ca2+ can still be detected.
AMPAR Intracellular Transport and Phosphorylation of GluA1
LTP-inducing stimuli trigger rapid insertion of AMPARs at synapses through a cascade of CaMKII-dependent phosphorylations. Considering the correlation between Ca2+ activity and GluA1 intracellular transport that we observed, we investigated the transport of phosphomimetic (S/D) and phosphodeficient (S/A) GluA1 mutants (Figures 7 and S7A).
Figure 7.

Phosphorylation of GluA1 Influences the Distances and Directions of Vesicle Travel
(A) Images of 14-day-old neurons expressing phosphodeficient (S831A-S845A) or phosphomimetic (S831D-S845D) TdT-GluA1 mutants and their associated kymographs.
(B) Mean outward (green), inward (red), and static (blue) vesicle states over time for the two GluA1 phosphorylation (phospho) mutants.
(C) Mean outward (green) and inward (red) velocities of vesicles in neurons expressing GluA1 phospho mutants.
(D) Mean vesicle number passing through a 20 μm2 bleached ROI during 1 min of recording in neurons expressing phospho mutants of GluA1.
(E) Epifluorescence images of neurons expressing ARIAD/GFP-GluA1 mutants SS/AA (top panel) and SS/DD (bottom panel) and incubated for 40 or 50 min with the AL. Surface receptors are labeled in red, and total receptors—i.e., surface and intracellular—are in green.
(F) Kinetics of GluA1 phospho mutant externalization after incubation with AL. Results were normalized according to the dendritic area in which the extracellular GluA1 fluorescence intensity has been quantified.
(G) Quantification of the extracellular fluorescence intensity of the GluA1 phospho mutants (SS/AA and SS/DD) expressed as the ratio of the signal measured in the distal over the proximal dendrites.
Mean ± SEM of 4 independent experiments (n > 20 cells). See also Figure S7 and Tables S1 and S2.
We first analyzed the two GluA1 mutants: S831A/D and S845A/D (Figures S7B and S7C). The single mutation at position 831 or 845 did not significantly change the mean velocities of OUT (S845A, 1.8 ± 0.08 μm.s−1; S845D, 1.9 ± 0.09 μm.s−1; S831A, 1.7 ± 0.07 μm.s−1; S831D, 1.6 ± 0.08 μm.s−1) and IN (S845A, 1.7 ± 0.09 μm.s−1; S845D, 1.6 ± 0.08 μm.s−1; S831A, 1.5 ± 0.07 μm.s−1; S831D, 1.4 ± 0.06 μm.s−1) or the number of vesicles for each condition (Figures S7B and S7C). Both S831A and S831D mutants behaved like wild-type (WT) GluA1 regarding the time spent by vesicles in OUT and IN directions (Figures S7B and S7C; for comparison, see Figure 2F), with a preference for the OUT movement (S/A, 39% ± 0.02%; S/D, 33% ± 0.02%) compared to the IN movement (S/A, 30% ± 0.02%; S/D, 26% ± 0.02%) (PAUSE: S/A, 31% ± 0.02%; S/D, 41% ± 0.02%). However, the S831D mutant seemed to be more static (PAUSE) than the S831A mutant (Figure S7B). In contrast, changing S845 from A to D had a drastic effect on the percentage of time spent in each state (Figure S7C). Whereas the S845A mutant went equally in the two directions OUT and IN, the S845D mutant massively went in the OUT direction (S/A, 33% ± 0.03%; S/D, 41% ± 0.02%) compared with IN (S/A, 31% ± 0.02%; S/D, 28% ± 0.02%) and PAUSE (S/A, 36% ± 0.03%; S/D, 31% ± 0.02%). In summary, it seems that these two single mutations do not affect the speeds of the vesicles but change the OUT and IN movements for S845 or the pauses for S831.
We thus analyzed the effect of double mutation (S831A-S845A or S831D-S845D) for transport (Figure 7). As exemplified in the kymographs, the properties of transport of these two mutants are markedly different (Figure 7A). The SS/DD mutant spent more time in the OUT direction, to the detriment of the pausing time, while this was the reverse for the SS/AA mutant: OUT (SS/AA, 33% ± 0.02%; SS/DD, 43% ± 0.02%), IN (SS/AA, 27% ± 0.02%; SS/DD, 28% ± 0.02%), and PAUSE (SS/AA, 40% ± 0.02%; SS/DD, 29% ± 0.02%) (Figure 7B). The DD mutant traveled OUT about 15% faster than the AA mutant, while the IN speeds were comparable: OUT (SS/AA, 1.56 ± 0.08 μm.s−1; SS/DD, 1.81 ± 0.08 μm.s−1) and IN (SS/AA, 1.44 ± 0.07 μm.s−1; SS/DD, 1.49 ± 0.06 μm.s−1) (Figure 7C). In addition, the SS/DD mutant had more vesicles than the SS/AA one (OUT: SS/AA, 4.5 ± 0.37 vesicles/20 μm2/min; SS/DD, 7.9 ± 0.69 vesicles/20 μm2/min) (Figure 7D).
Because the SS/DD mutant is going outward more than the SS/AA one, we wondered whether we could see a difference in their externalization. We expressed the two proteins and performed immunochemistry on live cells following induction of the transport by AL (Figures 7E and 7F). As soon as 40 min after addition of AL, the SS/DD mutant was sent farther from the cell body than the SS/AA mutant. The quantification of the distal versus proximal signal indicated a differential externalization of the two mutants (Figure 7G). Whereas the SS/DD mutant was externalized with similar kinetics in all neurons, the non-phosphorylatable protein was more externalized at the proximal part of the dendrite than at the distal one.
Discussion
GluA1 intracellular transport under basal conditions is characteristic of microtubule-based transport, with vesicles alternating within seconds between pauses and periods of retrograde or anterograde movements over several microns. Changes in intracellular Ca2+ were associated with strong modulation of GluA1 transport. Although lowering Ca2+ accelerated vesicle movement, acute rises in Ca2+ decreased vesicle speed and arrested most them. Application of a cLTP protocol induced a biphasic modulation of vesicular movement. During the induction phase of cLTP (0–5 min), vesicles transporting GluA1 were slowed and often arrested, likely allowing exocytosis of AMPARs (5–20 min). In contrast, during the second phase of cLTP (after 20 min), the number of vesicles transporting GluA1, as well as their anterograde speed, increased. In accordance with the known change in the phosphorylation state of GluA1 after cLTP induction, we found that the SS/DD mutant of GluA1 (S831-S845) traveled with a higher speed of outward displacement and had a higher rate of exocytosis at the tip of the dendrites compared to the corresponding non-phosphorylatable GluA1. Altogether, these results reveal the characteristics of GluA1-containing AMPAR intracellular transport and its regulation by synaptic activity, introducing a new level of regulation of GluA1 trafficking during LTP in hippocampal neurons.
AMPAR Distribution and Transport within the Different Compartments in Basal Conditions
Synaptic AMPARs represent a small fraction of the total pool of AMPARs, with large amounts of receptors present in the extrasynaptic PM and in intracellular pools, such as recycling endosomes, the GA, and the ER (Henley et al., 2011, Shepherd and Huganir, 2007). AMPARs are in dynamic equilibrium between these compartments, and different mechanisms have been proposed for their delivery to synapses: local synthesis (Hanus and Schuman, 2013) or motor-dependent transport of receptors synthesized at the cell body (Greger et al., 2007, Kim and Lisman, 2001, Setou et al., 2002), exocytosis (Gerges et al., 2006, Yudowski et al., 2007), and lateral diffusion (Choquet and Triller, 2013). The contribution of each of these mechanisms in the delivery of AMPARs to synapses is still under debate.
AMPAR transport along the biosynthetic pathway, although crucial, is one of the least characterized trafficking processes. AMPARs can be synthesized in the soma and locally in dendrites, although probably at a lower level (Bowen et al., 2017, Ju et al., 2004). AMPARs can be exocytosed all along the dendrite (Anggono and Huganir, 2012, Yudowski et al., 2007), and it is thus likely that they are transported by a vesicular mechanism before membrane delivery. Inhibition of dynein or kinesin substantially reduces synaptic AMPAR-mediated responses (Kim and Lisman, 2001). Following ER exit, GluA1-containing AMPARs undergo spatially restricted entry into the secretory pathway and accumulate in recycling endosomes at or near spines before reaching the PM (Bowen et al., 2017). Moreover, GluA1 traffics through the secretory pathway faster than GluA2 (Evans et al., 2017, Greger et al., 2007). Finally, when expressed with GluA1, GluA2 can be detected in the dendritic shaft in moving vesicles going in both directions, likely following the endosomal transport routes that are largely microtubule based (Esteves da Silva et al., 2015).
We found that under basal conditions, newly synthesized GluA1 is transported both antero- and retrogradely, which is likely important to rapidly distribute AMPARs all along the dendrite. This transport in both directions and the pause time have been previously observed in mammalian neurons (Ju et al., 2004) and C. elegans neurons (Hoerndli et al., 2015) and may indicate that the same motors are powering the antero- and retrograde movements. A previous slower speed of 1–2 μm.min−1 (50–150 times slower) was reported for vesicles transporting GluA1 or GluA2 (Ju et al., 2004), likely due to limitations in the imaging used. Accordingly, GluA2-containing vesicles have been detected at a speed of 1 μm.s−1, close to the one we found for GluA1 (Esteves da Silva et al., 2015).
We found two main populations of vesicles in terms of speed centered, respectively, on 0.5 and 1.6 μm.s−1, with a small proportion moving at 3.5 μm.s−1. In C. elegans, KIF5 (UNC-116) microtubule-dependent motor transport is the major mechanism for the delivery of GLR-1 at synapses (Hoerndli et al., 2015). The calculated velocity was 1.6 μm.s−1 in both directions, comparable with our results. However, it seems that the profile of speed is less complex in C. elegans than in hippocampal neurons.
AMPAR Transport during Synaptic Plasticity
A major result of our study is to unravel the strong activity-dependent regulation of GluA1-containing AMPAR transport. Activation of 5 to 10 synapses with a glutamate uncaging protocol was necessary to trigger Ca2+ elevations inside dendrites efficient to modulate transport. These waves of Ca2+ induced a massive reduction of the vesicle speed and increased the time in pause. This phenomenon could be the prerequisite for the detachment of vesicles from microtubules and the exocytosis of receptor-containing vesicles. We sometimes observed a disappearance of the vesicle signal after pausing, although we could not directly visualize exocytosis. In agreement with this hypothesis, de novo GluA1-containing AMPARs are heavily externalized at the PM in all parts of the dendritic shaft right after induction of cLTP.
In the late phase of LTP (20–60 min after induction), some waves of Ca2+ could still be detected; these were rare and of smaller amplitude than in the early phase but higher than in Ctrl cells. During this phase, the number of vesicles and their speed strongly increased, particularly through a rise in the population of vesicles moving around 3.5 μm.s−1. This increase in mobility could be due to a change in the motor driving this population of vesicles, but this cannot explain by itself the increase in vesicle density that could originate from an increased production of vesicles from the Golgi. Altogether, this phenomenon could allow refurnishing of the intracellular pool of receptors emptied by prior externalization.
In C. elegans, UNC-43/CaMKII controls synaptic strength by regulating motor-driven AMPAR transport, and the kinase activity was required for AMPAR transport (Hoerndli et al., 2015). UNC-43/CaMKII has either a direct or an indirect role in the loading of AMPAR cargo onto UNC-116/KIF5 kinesin motors, which is critical for the synaptic delivery of AMPARs. In vertebrates, GluA1 can be phosphorylated by protein kinase A (PKA), protein kinase C (PKC), and CaMKII specifically on Ser831 and Ser845 (Huganir and Nicoll, 2013). This plays important roles during synaptic plasticity, in which phosphorylated GluA1 represents around 50% of the total protein after induction of LTP (Diering et al., 2016). We found that the SS/DD mutant has more vesicles traveling and is expressed at the tip of the dendrites compared with the null mutant. This suggests that Ca2+ could control GluA1 transport partly by regulating its phosphorylation. This mechanism could allow the replenishment of AMPAR at the tip of dendrites far from the soma.
Relevance of Transport to Address Receptors or Distribute Receptors over Long Distances
Altogether, our data reveal that the delivery of newly synthesized GluA1-containing AMPARs to the cell surface is a highly regulated mechanism. GluA1 cargos distributed along the length of a neuronal dendrite can repopulate synapses with AMPARs. This provides a high degree of regulation for the delivery of newly synthesized receptors from the ER to the PM. Intracellular AMPAR transport is quantitatively and qualitatively different from AMPAR Brownian diffusion observed at the PM. Vesicular transport corresponds to fast, directed, linear movements around 1.5 μm.s−1, whereas when the receptors have reached the PM, they diffuse randomly around 0.05–0.2 μm2.s−1 (Choquet and Triller, 2013). Random surface diffusion is efficient to rapidly distribute receptors over short distances (of a few microns), while directed intracellular transport is efficient over long distances. These changes in the modality of receptor movements allow different steps of regulation during the journey of AMPARs.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-calreticulin | ABR | Cat# PA3-900 |
| Mouse anti-GM130 | BD Transduction Lab. | Cat# 610822 |
| Mouse anti-GFP | Roche | Cat# 11814460001; RRID: AB_390913 |
| Alexa 647 Goat anti-rabbit IgG(H+L) | Thermo Fisher Scientific | Cat# A21244; RRID: AB_2535812 |
| Alexa 568 Goat anti-mouse IgG(H+L) | Thermo Fisher Scientific | Cat# A11004; RRID: AB_2534072 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D/D Solubilizer: FM AP21998-AL | Clontech Laboratories | Cat# 635054 |
| MNI-Glu | TOCRIS | Cat# 1490 |
| Bicuculline | TOCRIS | Cat# 2503 |
| Glycine | TOCRIS | Cat# 0219 |
| BAPTA-AM | Sigma | Cat# A1076 |
| Experimental Models: Cell Lines | ||
| Rat: embryonic day 12-17 hippocampal primary neuron culture | N/A | N/A |
| Recombinant DNA | ||
| Plasmid: TdT-GluA1/ARIAD | This paper | N/A |
| Plasmid: GFP-GluA1/ARIAD | This paper | N/A |
| Plasmid: TdT-GluA1 | This paper | N/A |
| Plasmid: GCaMP6f | H. Bito’s lab | N/A |
| Plasmid: TdT-GluA1 S831A-S845A/ARIAD | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| KymoToolset Plugin | This paper | fabrice.cordelieres@gmail.com |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Françoise Coussen (fcoussen@u-bordeaux.fr).
Experimental Model and Subject Details
All experiments were performed on primary hippocampal neurons cultures from Sprague-Dawley rat E18 embryos of either sex. All the animals were used according to the guidelines of the University of Bordeaux/CNRS Animal Care and Use Committee.
Method Details
Molecular Biology
WT and mutated GluA1 (NCBI Reference Sequence: NM_031608.1) were cloned in the ARIAD system following the below procedure: the Apa1 site localized in position 3231 of the original pC4S1-FM4-FCS-hGH vector (Gift from ARIAD Pharmaceuticals, Inc.) was mutated to erase the restriction site. In replacement of the original Spe1 - Furin cleavage site – hGH – BamH1, a DNA coding for the Spe1 (nuc: 2078) - Furin cleavage site (nuc: 2081-2104) - Age1 (nuc: 2105) – GFP (nuc: 2105-2826) - Nhe1 (nuc: 2828) - Apa1 (nuc:2834) - Hind3 (nuc: 2840) - BamH1 (nuc: 2846) was subcloned. This vector was then used either to introduce the different cDNAs coding for GluA subunits by subcloning into the Nhe1/Apa1/Hind3/BamH1 polycloning site or to change GFP tag by subcloning the different Tag DNAs, such as TdTomato or HA in the Age1/Nhe1 restriction sites. To reduce expression toxicity in neurons, GCaMP6f and ARIAD-GluA1 WT or mutated were finally subcloned into the eukaryotic expression vector pBI TET-on, which give access to the tetracycline-regulated expression system. The pTRE-GCaMP6f construct we used to monitored calcium activity in cultured rat hippocampal neurons was a gift from Haruhiko Bito’s lab.
Cell Culture and Transfection Procedure
Dissociated hippocampal neurons from E18 Sprague-Dawley rats embryos were prepared as described previously at a density of 300,000 cells per 60-mm dish on poly-L-lysine pre-coated coverslips. Neuron cultures were maintained at 36.5°C, 5% CO2 in Neurobasal medium supplemented with 2mM L-glutamine and 1X NeuroCult SM1 Neuronal supplement (STEMCELL technologies), during 6 days before adding progressively BrainPhys medium supplemented with 1X NeuroCult SM1 Neuronal supplement.
Neurons from 8-10 days in vitro were transfected with the different cDNA following the calcium phosphate procedure. Doxycycline (0.2 mg/ml) was used to induce protein expression for 36-48 hr. Experiments were performed 3 to 7 days after transfection (12-17 days in vitro), depending on the neuron maturation degree required.
Immunocytochemistry
For intracellular labeling, neurons were fixed at 12-14 DIV in 4% PFA (4% paraformaldehyde, 4% sucrose) for 10 min at room temperature either before AL addition (time point = 0) or 15 or 30 min after addition of 1μM AL AP21998 to release expressed GluA1 from retention in the ER. Fixed coverslips were rinsed in PBS and then quenched with 50mM NH4Cl for 10 min followed by additional PBS rinses. Cells were permeabilized with 0.2% Triton X-100 (Sigma T9284) for 3 min and then rinsed with PBS. Non-specific binding was blocked by 45 min incubation with PBS containing 2% BSA (Sigma, A9647). Polyclonal anti-calreticulin (1:500) antibody and monoclonal anti-GM130 (1:2000) were used as specific markers of the ER and the Golgi compartments respectively. Both of them were diluted in this blocking solution and incubated with the coverslips for 60 min after which coverslips were rinsed and blocked again for 30 min prior to incubation with a goat anti-rabbit or a goat anti-mouse respectively as secondary antibodies.
For extracellular labeling, after addition of 1 μM of AP-21998 AL in the cell culture medium, neurons were incubated for additional time at 36.5°C before being fixed with PFA. Extracellular labeling of GFP-tagged receptors was performed with a monoclonal anti-GFP antibody (1/2000) for 15 min at room temperature following by a goat anti-mouse Alexa-568. Coverslips were mounted with ProLong Gold Antifade Mountant (LifeTechnologies).
Image Analysis and Quantification
Fluorescence images of intra- and extracellular stained proteins were collected on a wild field upright Leica DM5000 epifluorescence microscopy with a LED light source (Leica Microsystems, Germany), using a 63x oil objective and a Greyscale CCD camera Coolsnap HQ2. Extracellular labeling of GFP-GluA1 has been quantified with MATLAB software following the below procedure: surfaces of interest were drawn by hand (Surface) and number of red pixels (extracellular GluA1) was quantified. Images were also processed using an “in-house” developed ImageJ macro. Briefly, quantifications were performed by first asking the user to draw multiple sets of regions of interest: one in the background (devoid of any structure), and as many ROIs as required in the cell body and the proximal or distal dendritic shaft. For each of them, maximum intensity and area were retrieved. In a later step, the maximum intensity retrieved from the background area, to which 3 times its standard deviation was added, is subtracted to the full image. Results are expressed as the mean intensity fluorescence at the PM.
Colocalization images were analyzed with the Metamorph software. A line of 5 μm was drawn and pixel intensity for each color was determined on the line.
Videomicroscopy
Videomicroscopy was performed on inverted Leica DMI6000B, equipped with a spinning-disk confocal system (Yokogawa CSU-X1, beam lines: 408 nm, 491 nm, 561 nm) fitted with an EMCCD camera (Photometrics Quantem 512), using a HCX PL Apo 100X 1.4 NA oil immersion objective. The setup was driven by the Metamorph software (Molecular Devices, Sunnyvale, USA) and place under appropriate environmental control (Life Imaging Services).
2 colors imaging: Neurons expressing TdTomato-GluA1 and the calcium sensor GCaMP6f were imaged between 15 to 17 days at 37°C in a Ludin chamber filled with 1ml of normal Tyrode solution (20 mM HEPES, 3.5 mM KCl, 150 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 25mM D-glucose). GluA1 exit from the ER using 1μM of AP21998 AL in Tyrode. The following acquisition sequence is performed: (1) Using low-magnification objective, position of co-transfected cells (4-10 cells / coverslip) are recorded; (2) If applicable, drugs are applied to modify calcium activity for 5min; (3) Photobleaching is performed in a user-defined ROI placed over the dendritic shaft (∼60μm2, using 561nm laser) to enhance visualization of AMPAR-containing vesicles within this region; (4) Both calcium activity and vesicular trafficking are recorded (115 time points, 200 msec time interval); (5) Analysis is performed using an in-house developed plugin to the ImageJ software (“KymoToolSet”).
LTP was chemically induced by incubating neurons in a magnesium-free Tyrode solution (stim-media: 20 mM HEPES, 3.5 mM KCl, 150 mM NaCl, 2 mM CaCl2, 25mM D-glucose, 30 μM bicuculline and 200 μM glycine). (1) For experiments performed during cLTP stimulation, positions of co-transfected cells and dendritic regions of interest are first registered during 15 min in Tyrode solution containing 1μM of AP21998. After photo-bleaching of the dendritic regions of interest, neurons are transferred in the stim-media and videos are taken during the 5 min of induction (2 to 3 cells / coverslip). (2) For experiments performed after cLTP induction, positions of co-transfected cells are first registered during 15 min in normal Tyrode solution containing 1μM AP21998. Neurons are then incubated 5 min in the stim-media, before return in normal Tyrode for an additional 15 min. Same imaging and analysis procedure as above is used on a dendritic ROI for a maximum 30min, so cells spent 1 hr maximum with the AL.
Uncaging experiments: Same imaging and analysis procedure as above is used with the following variations and additional steps: (1) positions recording is performed in normal Tyrode containing 1μM of AP21998; (1b) MNI-Glu is added (final concentration: 0.5 mM); (2b) ROIs in close vicinity to 5-10 spines are drawn for later glutamate uncaging; (4) Both calcium activity and vesicular trafficking are recorded: a-(Before stimulation) 50 time points, 426 msec time interval; b-(During stimulation) 10 repetitions of 5 time points followed by 405nm laser uncaging.
Quantification and Statistical Analysis
Graphpad Prism version 7.0 was used for statistical analysis. All statistical analysis details are listed in the Table S1 for main figures and Table S2 for supplemental figures. Comparison of the means of two independent samples has been made with two-tailed t test. Unpaired t test has been used to test the statistical difference of the means of two independent samples whether paired t test has been used to compare the means of two matched pair’s samples. To compare the means of more than two independent samples, one-way ANOVA with Bonferroni’s multiple comparisons test has been used. Asterisks notify the following significance levels: p < 0.05 (∗), p < 0.01 (∗∗), p < 0.001 (∗∗∗) and p < 0.0001 (∗∗∗∗).
Acknowledgments
pGP-CMV-GCaMP6f was a gift from Douglas Kim (Addgene plasmid no. 40755). We thank the ARIAD Company for the gift of the ARIAD vector. We thank the Bordeaux Imaging Center, part of the national infrastructure France BioImaging (ANR-10INBS-04-0), and appreciate the help of S. Marais and C. Poujol. We thank the IINS cell biology core facilities (LABEX BRAIN [ANR-10-LABX-43]). This work was supported by funding from the Ministère de l’Enseignement et de la Recherche (ANR-PAINT), the Centre National de la Recherche Scientifique, the Conseil Régional de la Nouvelle Aquitaine, and ERC Grant ADOS (339541) to D.C.
Author Contributions
F.C. and D.C. were the main supervisors of the project and secured funding. E.H., F.C., and D.C. conceived the study and wrote the original draft of the manuscript. E.H. designed, performed, and analyzed all intracellular transport experiments for Figures 2, 3, 4, 5, 6, and S2–S6 and prepared all figures of the manuscript. F.C. performed and analyzed most of the immunocytochemistry experiments, as well as the intracellular transport experiments for Figures 7 and S7. J.D.P. performed the immunocytochemistry experiments and the related quantifications for Figure 1C. All authors contributed to the preparation of the manuscript. F.P.C. developed the software required to automatize the acquisitions and the following transport analyses.
Declaration of Interests
The authors declare no competing interests.
Published: July 24, 2018
Footnotes
Supplemental Information includes seven figures, two tables, and three videos and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.095.
Supplemental Information
References
- Anggono V., Huganir R.L. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen A.B., Bourke A.M., Hiester B.G., Hanus C., Kennedy M.J. Golgi-independent secretory trafficking through recycling endosomes in neuronal dendrites and spines. eLife. 2017;6:e27362. doi: 10.7554/eLife.27362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutman G., Baudry M. Involvement of the secretory pathway for AMPA receptors in NMDA-induced potentiation in hippocampus. J. Neurosci. 2001;21:27–34. doi: 10.1523/JNEUROSCI.21-01-00027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet D., Triller A. The dynamic synapse. Neuron. 2013;80:691–703. doi: 10.1016/j.neuron.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Diering G.H., Heo S., Hussain N.K., Liu B., Huganir R.L. Extensive phosphorylation of AMPA receptors in neurons. Proc. Natl. Acad. Sci. USA. 2016;113:E4920–E4927. doi: 10.1073/pnas.1610631113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves da Silva M., Adrian M., Schätzle P., Lipka J., Watanabe T., Cho S., Futai K., Wierenga C.J., Kapitein L.C., Hoogenraad C.C. Positioning of AMPA receptor-containing endosomes regulates synapse architecture. Cell Rep. 2015;13:933–943. doi: 10.1016/j.celrep.2015.09.062. [DOI] [PubMed] [Google Scholar]
- Evans A.J., Gurung S., Wilkinson K.A., Stephens D.J., Henley J.M. Assembly, secretory pathway trafficking, and surface delivery of kainate receptors is regulated by neuronal activity. Cell Rep. 2017;19:2613–2626. doi: 10.1016/j.celrep.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerges N.Z., Backos D.S., Rupasinghe C.N., Spaller M.R., Esteban J.A. Dual role of the exocyst in AMPA receptor targeting and insertion into the postsynaptic membrane. EMBO J. 2006;25:1623–1634. doi: 10.1038/sj.emboj.7601065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger I.H., Ziff E.B., Penn A.C. Molecular determinants of AMPA receptor subunit assembly. Trends Neurosci. 2007;30:407–416. doi: 10.1016/j.tins.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Hanus C., Schuman E.M. Proteostasis in complex dendrites. Nat. Rev. Neurosci. 2013;14:638–648. doi: 10.1038/nrn3546. [DOI] [PubMed] [Google Scholar]
- Hanus C., Geptin H., Tushev G., Garg S., Alvarez-Castelao B., Sambandan S., Kochen L., Hafner A.S., Langer J.D., Schuman E.M. Unconventional secretory processing diversifies neuronal ion channel properties. eLife. 2016;5:e20609. doi: 10.7554/eLife.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley J.M., Wilkinson K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016;17:337–350. doi: 10.1038/nrn.2016.37. [DOI] [PubMed] [Google Scholar]
- Henley J.M., Barker E.A., Glebov O.O. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34:258–268. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N., Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Hoerndli F.J., Wang R., Mellem J.E., Kallarackal A., Brockie P.J., Thacker C., Madsen D.M., Maricq A.V. Neuronal activity and CaMKII regulate kinesin-mediated transport of synaptic AMPARs. Neuron. 2015;86:457–474. doi: 10.1016/j.neuron.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir R.L., Nicoll R.A. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W., Morishita W., Tsui J., Gaietta G., Deerinck T.J., Adams S.R., Garner C.C., Tsien R.Y., Ellisman M.H., Malenka R.C. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- Kapitein L.C., Schlager M.A., Kuijpers M., Wulf P.S., van Spronsen M., MacKintosh F.C., Hoogenraad C.C. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 2010;20:290–299. doi: 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
- Kim C.H., Lisman J.E. A labile component of AMPA receptor-mediated synaptic transmission is dependent on microtubule motors, actin, and N-ethylmaleimide-sensitive factor. J. Neurosci. 2001;21:4188–4194. doi: 10.1523/JNEUROSCI.21-12-04188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H., Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M., Salgado J.M., Ostroff L., Helton T.D., Robinson C.G., Harris K.M., Ehlers M.D. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera V.M., Wang X., Wardwell S., Courage N.L., Volchuk A., Keenan T., Holt D.A., Gilman M., Orci L., Cerasoli F., Jr. Regulation of protein secretion through controlled aggregation in the endoplasmic reticulum. Science. 2000;287:826–830. doi: 10.1126/science.287.5454.826. [DOI] [PubMed] [Google Scholar]
- Setou M., Seog D.H., Tanaka Y., Kanai Y., Takei Y., Kawagishi M., Hirokawa N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- Shepherd J.D., Huganir R.L. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Wang Z., Edwards J.G., Riley N., Provance D.W., Jr., Karcher R., Li X.D., Davison I.G., Ikebe M., Mercer J.A., Kauer J.A., Ehlers M.D. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Nash J.E., Zamorano P., Garner C.C. Interaction of SAP97 with minus-end-directed actin motor myosin VI. Implications for AMPA receptor trafficking. J. Biol. Chem. 2002;277:30928–30934. doi: 10.1074/jbc.M203735200. [DOI] [PubMed] [Google Scholar]
- Yudowski G.A., Puthenveedu M.A., Leonoudakis D., Panicker S., Thorn K.S., Beattie E.C., von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J. Neurosci. 2007;27:11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TdTomato tagged GluA1 was expressed in the Ariad vector in cultured hippocampal neurons and imaged 30′ after addition of the AL. Time stamp in seconds.
TdTomato tagged GluA1 was expressed in the Ariad vector in cultured hippocampal neurons and imaged 30′ after addition of the AL. MNI-caged-L-glutamate was uncaged at the time of appearance of the stars, leading to vesicle arrest. Time stamp in seconds.
TdTomato tagged GluA1 was expressed in the Ariad vector in cultured hippocampal neurons and imaged (in red) 30′ after addition of the AL and 20′ after application of the c-LTP protocol. Note the large increase in vesicle density. In green homer1c-GFP to label synapses. Time stamp in seconds.