

Abstract
A broad portfolio of phenotypic diversity in natural organisms can buffer against exploitation and increase species persistence in disturbed ecosystems. The study of genomic variation that accounts for ecological and evolutionary adaptation can represent a powerful approach to extend understanding of phenotypic variation in nature. Here we present a chromosome-level reference genome assembly for Chinook salmon (Oncorhynchus tshawytscha; 2.36 Gb) that enabled association mapping of life-history variation and phenotypic traits for this species. Whole-genome re-sequencing of populations with distinct life-history traits provided evidence that divergent selection was extensive throughout the genome within and among phylogenetic lineages, indicating that a broad portfolio of phenotypic diversity exists in this species that is related to local adaptation and life-history variation. Association mapping with millions of genome-wide SNPs revealed that a genomic region of major effect on chromosome 28 was associated with phenotypes for premature and mature arrival to spawning grounds and was consistent across three distinct phylogenetic lineages. Our results demonstrate how genomic resources can enlighten the genetic basis of known phenotypes in exploited species and assist in clarifying phenotypic variation that may be difficult to observe in naturally occurring organisms.
Keywords: evolution, ecology, genomics
1. Introduction
Natural selection may maintain phenotypic variation within populations across temporal and spatial scales, and generate distinct phenotypic diversity among populations across a species' range. This portfolio of diversity within species has been demonstrated to contribute to long-term resilience of species and ecosystems and has become the focus of efforts to protect species within critical areas [1]. In aquatic species such as salmonids, phenotypic variation is abundant and considered to represent evolutionary adaptation to highly variable environments [2]. Recent efforts have begun to elucidate the genomic basis for phenotypic traits in wild populations [3,4] with the availability of salmonid genome assemblies [5,6].
The Chinook salmon (Oncorhynchus tshawytscha) is an anadromous fish species with considerable ecological, economic and social value, and has been a cultural icon that has sustained native people of western North America for millennia [7]. This species has experienced dramatic long-term declines in abundance due to anthropogenic impacts but Chinook salmon (Oncorhynchus tshawytscha) have retained extensive phenotypic variation across their native range from the west coast of North America to the Kamchatka peninsula in north eastern Asia [8]. This includes diverse phenology traits that are heritable such as timing of adult migration and sexual maturation [9]. Phenotypic variation for these traits in Chinook salmon can be segregating within populations or distinct among populations and represent patterns of parallel or divergent evolution depending on phylogenetic lineage [10]. Distinct phylogenetic lineages of Chinook salmon include those in the centre of the species range in North America known as coastal, interior ocean-type and interior stream-type [11]. Phenotypic variation for phenology traits exists within and among these lineages. The coastal lineage is known to have both sub-yearling and yearling juvenile outmigration [12], and adult migratory run-timing occurs across multiple seasons related to premature or mature state of sexual maturity when entering freshwater (e.g. spring-run premature versus fall-run mature) [9,10]. By contrast, the two interior lineages exhibit more narrow life-history traits that are highly distinct from each other [10,12,13]. The interior ocean-type lineage is most genetically similar to the coastal lineage [10,14], with life-history traits that include sub-yearling juvenile outmigration [12], and summer to fall spawning migration [12,15] with both premature and mature entry to freshwater, respectively [9]. The interior stream-type lineage is highly distinct from the other lineages with yearling juvenile outmigration [12], spring to summer spawning migration [12,15] and adults exclusively enter freshwater as premature adults [9]. However, understanding of genomic architecture for differing life-history traits is limited due to lack of genomic resources for this species and challenges of characterizing phenotypic variation in migratory aquatic species.
In this study, we present a novel chromosome-level genome assembly for Chinook salmon and examine the genetic basis for life-history traits across multiple phylogenetic lineages of this species. We address the following questions: (i) is there evidence for candidate genes of major effect associated with phenology traits across multiple lineages of this species? (ii) Are maturation phenotypes consistent with genomic variation in interior lineages in addition to coastal populations previously studied [16]? (iii) Do signals of adaptation suggest that selection acts on populations within lineages in addition to expected adaptive divergence among lineages?
2. Methods
(a). Collection of tissues for genome assembly
Tissues were collected from a wild, sexually mature male Chinook salmon to obtain DNA for genome assembly and RNA for transcriptome assembly. This fish was collected at a weir on Johnson Creek, Idaho, USA (latitude 44.899, longitude −115.492) and was euthanized post-spawning. Chinook salmon in this population represent an ecotype known to be part of the recognized Snake River spring/summer-run evolutionarily significant unit (ESU) and are considered interior Columbia River stream-type phylogenetic lineage [10,11]. Tissues collected included caudal fin clips stored in 100% ethanol, blood stored in sterile haematology tubes on ice, and eight organ tissues stored in RNAlater and flash-frozen in liquid nitrogen (muscle, stomach, gill, liver, kidney, brain, heart, intestine).
(b). Genome assembly and annotation
The 2.36 Gb reference genome of a single wild diploid male (electronic supplemental material, figure S1) was assembled with various sequence libraries that included TruSeq synthetic long reads (long read contig N50 of 4.95 Kb), bacterial artificial clones (BAC) and mate-pair libraries (ranging from 1–40 Kb; electronic supplementary material, Methods File 1). Contigs and scaffolds were assembled de novo with multiple pipelines and scaffolds were error-corrected and re-scaffolded with BioNano optical maps (electronic supplementary material, Methods File 1 contains details regarding bioinformatic steps involved in genome and transcriptome assembly). Corrected scaffolds were ordered into chromosomes using available genetic maps [17,18] and aligned against an existing reference genome of a related species, rainbow trout (O. mykiss; electronic supplemental material, figure S2). Final gap-filling produced the final genome assembly that was annotated with deeply sequenced transcriptomes from eight tissues collected from the same individual fish. Transcriptome sequences were integrated with reference models to predict gene models having homology with curated Swissprot proteins (electronic supplementary material, Methods File 1). A BUSCO (benchmarking universal single-copy orthologues) [19] analysis was completed to estimate the completeness of the genome assembly (electronic supplementary material, Methods File 1). Determination of large collinear homeologous regions (greater than 1 Mb) was performed by a self-alignment of the masked Chinook chromosome sequences (electronic supplementary material, Methods File 1).
(c). Population samples and traits for whole-genome re-sequencing
Whole-genome re-sequencing of pooled sample collections was completed with 14 pools representing O. tshawytscha populations from three phylogenetic lineages that exhibit contrasting maturation and run-timing phenotypes (figure 1a–d; electronic supplemental material, table S1). Specifically, four distinct populations were included to represent each of the three lineages known as coastal, interior ocean-type and interior stream-type [11]. Two additional libraries were prepared from one population (Johnson Creek) to represent two phenotypic groups of females (early versus late arrival to spawning grounds; electronic supplemental material, table S1). Each of the other 12 pools consisted of at least 46 individuals (mean = 77) with a mix of sexes (electronic supplemental material, table S1).
Figure 1.

Chinook salmon collections and migration phenotypes. (a) Map of 12 collection locations corresponding to sites listed in electronic supplementary material, table S1; square = premature adult migration, circle = mature adult migration. (b) Coastal lineage collections—adult fish passage during migration into freshwater; curve colours correspond to collection locations on the map, dashed line = premature, solid line = mature. (c) Interior stream-type lineage collections—adult fish passage during migration into freshwater; curve colours correspond to collection locations on the map, dashed line = premature. (d) Interior ocean-type lineage collections—adult fish passage during migration into freshwater; curve colours correspond to collection locations on the map, dashed line = premature, solid line = mature.
(d). Pool-seq library preparation
Population pools were prepared and sequenced using a standardized pool-seq protocol following best practices [20]. This pooled approach provides estimated allele frequencies for variants across the genome but not individual genotypes. Library preparation included normalizing individual DNA quantity using picogreen fluorescence on a Tecan M200 (Tecan Trading, AG, Switzerland). To ensure similar contribution of each individual to a pool, individual DNA concentrations were not allowed to deviate more than 20% of the average DNA concentration of all individuals. For a given population, samples were fragmented with NEBNext Ultra dsDNA Fragementase (New England Biolabs, Ipswich, MA, USA), pooled together and filtered using Minelute purification (Qiagen, Venlo, Netherlands). Fragment end repair was performed with NEBNext Ultra End Prep (New England Biolabs, Ipswich, MA, USA) and fragments between 400 and 500 bp were selected for using a 25X AMPure beads solution (Beckman Coulter Inc., Indianapolis, IN, USA). Fragments were then amplified with a NEBNext Ultra Q5 PCR protocol, cleaned with AMPure beads, and finally quantified with SYBR quantitative PCR (Thermofisher Scientific, Waltham, MA, USA). Amplified fragments were normalized and then sequenced with high-output runs on an Illumina NextSeq 500 (Illumina, San Diego, CA, USA) with paired-end 150 bp reads (2 × 150 bp).
(e). Pool-seq bioinformatics and analyses
For each Pool-seq library, raw 150 bp paired-end reads (2 × 150 bp) were processed using the PoolParty pipeline (Micheletti & Narum [21]) that integrates several existing resources into a single pipeline. Briefly, this included multiple steps that started with trimming reads (to a minimum of 50 bp) with a quality score less than 20 using the trim-fastq.pl script part of Popoolation2 [22]. Trimmed reads were then aligned to the O. tshawytscha assembly using bwa mem [23] with default parameters. PCR duplicates were identified and removed using SAMblaster [24]. SAMtools view module [25] was used to sort BAM files, which were then combined using the SAMtools mpileup module that extracts SNP and coverage information for each pool. To remove any false-positive SNPs that often occur around insertion–deletions (indels), we used the identify-genomic-indel-regions.pl and filter-sync-by-gtf.pl scripts from PoPoolation2 and eliminated SNPs within 5 bp of indel regions from further analysis [22]. We only retained variant positions with a minimum of 15× depth of coverage and a maximum of 250× depth of coverage which eliminated regions that may be paralogues (high coverage) or regions that are probably represented by a small number of individuals (low coverage). Additionally, the minimum minor allele count for variant sites was set to 0.05 to drop rare alleles and to avoid overestimating heterozygous positions as recommended [22].
Filtered allele frequency data were then used to calculate the fixation index (FST) between collections using a sliding window of 5000 bp with a step size of 50 bp. In concert with FST, we statistically determined genomic regions with significant differentiation using a local score technique [26]. The local score uses single SNP Fisher's exact test p-values to determine genomic regions of differentiation while reducing false positives by incorporating linkage disequilibrium. In short, the local score has a score function related to −log10, which is related to window size. While other windowed approaches rely on combining p-values within a fixed window size, the local score uses the local maximum of Lindley processes, which is a function of statistically significant p-values in proximity to determine window size iteratively. The window size is ultimately influenced by a tuning parameter (ξ) which implies a p-value threshold on a log10 scale. We performed local score analyses that iteratively determined ξ based on the mean of log10 p-values for each comparison [26] and displayed significant regions (Bonferroni-corrected α = 0.05) in the form of Manhattan plots using the R package qqman [27].
To test for consistent differences in allele frequencies across biological replicates, we implemented a Cochran–Mantel–Haenszel (CMH) test that computes significance between groups of interest [22]. The CMH test identified consistent differences in allele frequency changes that occurred between pairs of premature and mature groups from each of the three phylogenetic lineages. We deemed genomic regions significant if they were consistently differentiated in both local score analyses (analogous to a Bonferroni-corrected α = 0.05), and the CMH test (Bonferroni-corrected α = 0.05). Any regions deemed significant were then investigated for variant annotations using SnpEff [28]. SnpEff predicts non-synonymous SNPs (nsSNPs) using frame information from a genome assembly annotation file. Significant regions were annotated following gene models identified in the gff3 file that was generated to support annotation of the genome assembly. Gene ontology enrichment (Panther over-representation test) was completed with the Panther database (http://geneontology.org/) for significant annotated genes that were significant between lineages.
Filtered allele frequency data was also used to inform neutral population structure (genome-wide SNPs) in contrast to the genetic relationship of collections by maturation phenotype (SNPs from a region of major effect on Ots28). Using variant sites in common across the genome (7 324 591 SNPs excluding extremely high FST markers > 0.90), we calculated Nei's standard genetic distance between all population comparisons and created a consensus neighbour-joining tree based on 10 000 bootstraps in the R package ape [29]. We then performed the same analyses using only SNPs from the divergent region of Ots28 (580 SNPs spanning from GREB1 L through ROCK1) to evaluate relationships of collections based on maturation status.
To examine SNP allele frequencies from GREB1 L across the North American range of Chinook salmon, previous RAD (restriction site-associated DNA) data from 53 collection sites [11] (electronic supplemental material, table S2) were aligned to the Chinook salmon reference genome using BWA-mem [23]. Only RAD loci that had a MAPQ score of 10 and SNPS that fell within the putative maturation region of chromosome 28 were retained. Genotypes were produced using the reference-based STACKS pipeline [30] and only one SNP at Ots28 position 11 033 590 showed sufficient variation and was thus retained for estimating genotype frequencies for fish collected from each site.
3. Results and discussion
The final reference assembly resulted in a 2.36 Gb genome with 72.2% (1.70 Gb) of the de novo assembly anchored to 34 chromosomes, with contig N50 of 19.1 Kb, scaffold N50 of 153.3 Kb (prior to chromosome placement) and anchored chromosome N50 of 45.4 Mb (electronic supplemental material, table S3; table S4). A total of 184 duplicated blocks were identified, representing 72% (1.128 Gb) of the un-gapped anchored chromosome sequence (electronic supplemental material, figure S3) with an average sequence similarity of 89.8%. Following criteria established by Lien et al. [6], 37.4% of the identified duplicated regions in Chinook salmon can be assigned to a state of delayed re-diploidization (similarity 90–95%) and 4.9% to a state of retained residual tetrasomy (similarity ≥95%). The assembly was annotated with deeply sequenced transcriptomes from eight tissues collected from the same individual fish. Analysis with BUSCO revealed that 87.3% of 4584 Actinopterygii genes were found as complete genes in the assembly (NCBI: GCA_002831465), with 4.0% fragmented and 8.7% missing genes. The annotated genome and transcriptome assemblies were submitted to NCBI (accession numbers: PRJNA402052; PIPH010000000; GGDU00000000).
Whole-genome re-sequencing of populations with variable life histories (figure 1) provided evidence that divergent selection was extensive throughout the genome among populations with differing phenology and reproductive traits. Variation across greater than 19 million SNPs (19 627 832 variant sites across all populations) throughout the genome revealed that a broad portfolio of life-history and phenotypic diversity exists in this species, both within and among phylogenetic lineages. Several candidate regions for divergent selection were evident across many chromosomes among phylogenetic lineages (electronic supplemental material, figure S4a–c), and between populations within lineages (electronic supplemental material, figure S5a–c), indicating evidence for adaption to local environments through multiple evolutionary processes [31]. Several candidate regions for divergent selection among phylogenetic lineages occurred across many chromosomes. This included large numbers of annotated genes within (Dryad table 1 [32]) and among lineages (Dryad table 2 [32]) with significant enrichment of multiple pathways (Dryad table 3 [32]).
Genome-wide association mapping with millions of SNPs provided evidence for association of major effect genes on chromosome 28 with maturation phenotypes, but both the underlying genomic region and the maturation phenotypes were found to be more complex than previously understood. Recent studies in multiple Pacific salmonid species suggest that the function of GREB1 L is conserved for sexual maturation status, which drives the seasonal migration timing of adults returning to spawn in coastal streams [16,32]. However, in addition to GREB1 L we found multiple genes on Ots28 associated with the trait of sexual maturation status including strong evidence from CMH tests for an adjacent candidate gene ROCK1 (Rho-associated protein kinase 1) and intergenic SNPs (figure 2a–e; electronic supplemental material, table S5). Both GREB1 L and ROCK1 have been characterized as oestrogen receptors [33,34] involved with biological pathways including embryo development in zebrafish (Danio rerio [35]). In the 203 Kb region of Ots28, the majority of the 580 SNPs were located in introns (84.5%), with only 2.7% in exons, and the remaining variants upstream of or downstream from these two genes. One non-synonymous SNP was found in coding sequences of both GREB1 L and ROCK1 (figure 2e), but neither SNP was significant. Several significant intergenic SNPs were found 3′ of GREB1 L and 5′ of ROCK1 that could be involved in gene regulation. The significant association of many SNPs across the 203 Kb region of these two genes may indicate epistasis, but non-coding variation may also suggest cis-regulatory effects on phenotypic expression of this trait.
Figure 2.

Circos Manhattan plot for premature and mature collections of Chinook salmon. (a) Sequence coverage (black outer ring) for each chromosome. (b) Significant divergence between premature (spring-run) and mature (fall-run) migrating Chinook salmon in the Cowlitz River of the coastal lineage. (c) Significant divergence between premature (Methow River summer-run) and mature (Priest Rapids fall-run) migrating Chinook salmon within the interior ocean-type lineage. (d) Significant divergence for Chinook salmon returning to Johnson Creek (interior stream-type) that enter freshwater premature (spring/summer-run), but the final ascent to spawning grounds is bimodal with early premature and late mature females. (e) Annotation of the 203 Kb region on Ots28 between 11.022 and 11.225 Mb (GREB1 L, ROCK1, and intergenic regions) with significance based on CMH tests. Significant genes are labelled and corresponding details are in electronic supplementary material, table S5. Timing of ascent to spawning grounds for premature (dashed line, orange) and mature (solid line, blue) collection pairs are shown within the ring for each lineage (b–d). Purple dots show two non-synonymous SNPs.
Candidate genomic regions on chromosome 28 were significant for premature versus mature migration traits within all three lineages that span from coastal to inland streams ranging across 1200 km of the Columbia River drainage. A population near the coast (less than 250 km inland) that exhibits both premature (spring-run) and mature (fall-run) migration within the same river (Cowlitz River) provided strong evidence that GREB1 L was a primary gene underlying this trait. However, this coastal lineage had the widest region of significance on Ots28 (390 Kb, ranging from positions 10.907–11.297 Mb) that included seven total candidates (MIB1, ABHD3, GREB1 L, ROCK1, USP14, THOC1 and AQPA) along with three candidate regions from other chromosomes (figure 2b; electronic supplemental material, table S5). Within a pair of collections from the interior ocean-type lineage, early premature migrating adults (Methow River summer-run) were highly distinct from late mature migrating fish (Priest Rapids fall-run) at GREB1 L (figure 2c), but this was the only significant candidate gene in a narrower region of Ots28 (57 Kb, ranging from positions 11.022–11.079 Mb; electronic supplemental material, table S5). Four additional candidate genes were present on chromosomes Ots10, Ots15 and Ots33 (electronic supplemental material, table S5) but at lower significance than Ots28.
Surprisingly, diverse phenotypes for sexual maturation were also found to be associated with variation at chromosome 28 in the interior stream-type lineage. This lineage consists solely of premature migrating adults when they enter freshwater (figure 2d) but exhibit extended upstream migration distance of over 1200 km to reach inland spawning grounds. Close monitoring of one population in Johnson Creek, Idaho during multiple stages of migration revealed a bi-modal pattern of maturation during the final ascent to spawning grounds (figure 3). Specifically, early and late return peaks predominantly consisted of premature and mature individuals, respectively. Disruptive selection due to high water temperatures in summer months may explain the bimodal migration in this population as has been shown in other salmonids (figure 3) [36]. Mapping genomic divergence between early and late ascending groups across approximately 6 million SNPs (6 146 606 SNPs in common) revealed that the significant region on Ots28 (128 Kb, ranging from positions 11.093–11.221 Mb) did not include GREB1 L, but rather an adjacent candidate gene ROCK1 (figure 2d; electronic supplemental material, table S5). Two additional regions of the genome had similar significance for this trait in the Johnson Creek population including at Ots11 (TMX1) and Ots34 (NPR1) in this population (figure 2d; electronic supplemental material, table S5). These additional regions of divergence may reflect that the maturation trait is more highly polygenic in this population, or they may represent additional unresolved phenotypes among the early and late ascending groups.
Figure 3.
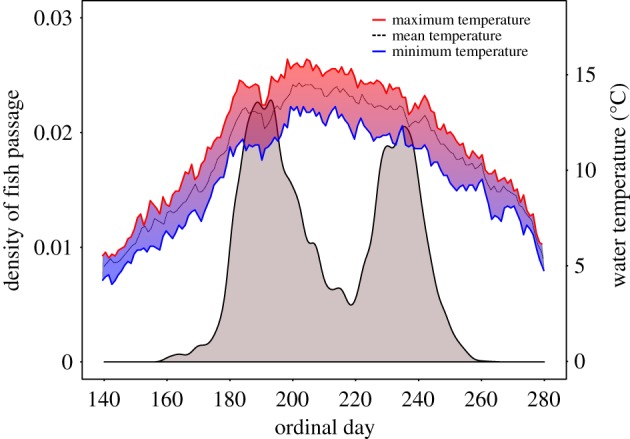
Environmental driver of disruptive selection. Bimodal ascent to spawning grounds in fish passage density of Chinook salmon versus daily water temperatures in Johnson Creek, ID, USA. Fish passage and water temperature data were collected and summarized over multiple years between 2004 and 2015 and shown by day of year (ordinal day). (Online version in colour.)
Specific candidate genes among lineages may suggest complexity in the genetic basis for specific maturation phenotypes, but there was clear evidence from CMH tests that 203 Kb of Ots28 was conserved as a region of major effect for maturation across lineages (positions between 11.022 and 11.225 Mb of Ots28 including GREB1 L and ROCK1; figure 2e). This was also evident from genetic relationships of collections based on 580 SNPs in this region of Ots28 that clustered by maturation phenotype regardless of phylogenetic lineage (figure 4a,b), which contrasted with monophyletic clustering of populations by major lineage as seen with genome-wide SNPs (7.3 million SNPs after removing high FST markers). The genomic basis for these phenology traits are of critical biological importance as standing variation may limit phenological responses to climate change in salmonids, which can have ecological ramifications for many aquatic and terrestrial species [37,38].
Figure 4.

Neighbour-joining trees of Nei's genetic distance (DA). (a) Genome-wide distance estimate representing putatively neutral genetic structure for all collections by phylogenetic lineage (7 324 591 SNPs). (b) Distance estimate from candidate region on chromosome Ots28 (580 SNPs spanning from GREB1 L through ROCK1) representing relationship of collections with premature and mature phenotypes. Collection numbers correspond to site list in electronic supplementary material, table S1 and locations in figure 1. ‘Mixed’ indicates cryptic ascent timing phenotypes that were combined in one collection that was then split into early premature and late mature groups. (Online version in colour.)
Consistent results for genomic divergence at chromosome Ots28 across lineages of Chinook salmon in this study suggested that the genomic basis for maturation traits might be conserved throughout the species range. We re-examined previously published sequence data of 53 Chinook salmon populations [11] and found that a diagnostic marker from candidate gene GREB1 L (Ots28, SNP position 11 033 626 bp) was associated with maturation-related run-timing throughout the North American range (figure 5; electronic supplemental material, table S2). This result supports the hypothesis that variation at chromosome 28 contains a broadly conserved genomic region under selection for migratory maturation phenotypes in Chinook salmon.
Figure 5.

SNP genotype frequencies at candidate gene (GREB1 L) for premature versus mature Chinook salmon across North America. (a) Northern distribution of mature (red), premature (green) and heterozygous (yellow) genotypes. (b) Southern distribution of mature (red), premature (green) and heterozygous (yellow) genotypes. Outside ring colours indicate run-timing phenotypes. Details for each collection site are in electronic supplementary material, table S2.
Assembly of salmonid genomes has been challenging due to duplication events in their evolutionary history that have resulted in regions with residual tetrasomy [39,40]. However, sequencing and linkage mapping with haploid individuals has helped to identify many homeologous regions [6,18,41,42]. While the individual for the current Chinook salmon genome assembly was diploid, we used existing resources such as linkage maps [18] and a recent genome assembly for rainbow trout (GenBank accession MSJN01000000) to help place homeologue to mapped positions. Recent studies indicate that homeologues may be an important source of adaptive variation in salmonids and should be accounted for when possible in genome scans [43–45]. Our study suggests that adaptive variation is extensive throughout the Chinook salmon genome and includes homeologous genes in addition to those that have reverted to a diploid state. However, alignment of re-sequencing data was not possible to homeologues that retain very high sequence similarity that represent approximately 4.9% of duplicated regions in the assembly. Thus, it is likely that our study under-represents signals of adaptive variation in duplicated regions with very high sequence similarity (e.g. greater than 95%).
Throughout this study, evidence for divergent selection was pervasive both within and among phylogenetic lineages and supports a diverse portfolio of phenotypic variation in Chinook salmon. While selective sweeps were expected between lineages with well-documented differences in life histories [10], there were also large numbers of candidate genes among populations of the same lineage that provided support for locally adaptive traits that have yet to be determined. Undetermined phenotypes could represent unresolved traits related to development, environmental tolerance, immune response, phenology and reproduction. However, phenotypic variation is often difficult to characterize in natural organisms at both short- and long-term temporal scales due to occupation of areas that are challenging for biologists to access (rugged mountains or deep seas), animal activity that is not readily visible to humans (nocturnal or underwater) and behavioural tactics to avoid humans.
Unusual phenotypes and physiological capabilities continue to be discovered in natural organisms such as naked mole rats (Heterocephalus glaber) that can tolerate oxygen deprivation for extended periods of time in underground tunnels [46], or the first species of fish that exhibits whole-body endothermy (opah, Lampris guttatus [47]). In other cases, extreme phenotypic variation has been well characterized within species [48], but little is known about selection gradients, genetic basis or environmental cues that influence quantitative traits [49]. In our study, genomic variation revealed that anadromous salmonids migrating long distances retain unexpected variation in maturation phenotypes related to environmental drivers of selection. Thus, applications with advanced genomic resources may provide clues to detect cryptic phenotypes within natural populations of exploited species where maintenance of a broad portfolio of diversity is necessary for long-term persistence [50].
4. Conclusion
Here we examined the genomic basis of phenotypic diversity in natural populations of Chinook salmon through association mapping of adult migration and maturation traits to a novel genome assembly for this species. This study provides a chromosome-level reference genome assembly for this species, while whole-genome re-sequencing of Chinook salmon collections with divergent life histories enabled tests for consistent association of specific genomic regions associated with phenotypic traits. This genome-level approach pinpointed multiple major effect genes on chromosome 28 beyond the single GREB1 L candidate previously identified [16]. Results also demonstrated consistent association of maturation phenotypes with this genomic region across three distinct phylogenetic lineages. Genotypes of a candidate SNP from GREB1 L also indicated that phenotypic variation for maturation is highly diverse across the species' range in North America. Finally, several candidate regions for divergent selection were evident within and among phylogenetic lineages, which suggests that local adaptation is abundant in natural populations of this species. Our study demonstrates how genomic resources can enlighten the genetic basis of phenotypic diversity in impacted species that require a broad portfolio of variation to persist in contemporary and evolutionary time frames.
Supplementary Material
Supplementary Material
Acknowledgements
Nate Campbell assisted with tissue collection, library preparation and sequencing strategies. Stephanie Harmon prepared libraries for whole-genome re-sequencing of populations. Ben Hecht assisted with library preparation and coordination of sequencing data collection. Mike Miller assisted with tissue collection and strategy for BAC library preparation for genome assembly. Scott Monsma assisted with strategy and preparation of long mate-pair libraries. Melanie Oakes assisted with optical mapping. Craig Rabe provided water temperature data and assisted with tissue samples. Matt Rockwell and Brian Steffy assisted with sequencing of synthetic long-read libraries. Keith Stormo assisted with strategy and preparation of BAC libraries.
Ethics
Trapping and sampling was authorized by permit from the National Marine Fisheries Service under the Endangered Species Act.
Data accessibility
Genome and transcriptome assemblies and sequencing data are available on NCBI under project PRJNA402052 (genome assembly PIPH000000000, and transcriptome assembly GGDU00000000). The gene annotation file for genome assembly and tables 1 -3 listing significant candidate genes within and among lineages, and gene ontology enrichment are available from the Dryad Digital Repository at https://doi.org/10.5061/dryad.dr1qs08 [32].
Authors' contributions
S.R.N. designed the study, collected tissue samples, coordinated sequencing, assisted with data analyses and wrote the manuscript. A.D.G. completed bioinformatics including assembly of genome and transcriptomes, annotated the assembly, assisted with analyses and assisted with writing the manuscript. S.J.M. completed bioinformatics for re-sequencing data, analysed population allele frequencies and assisted with writing the manuscript. A.M. assisted with genome-sequencing strategy, genome assembly and writing the manuscript.
Competing interests
We declare we have no competing interests.
Funding
Funding was provided by Bonneville Power Administration (200890700), Center for Mathematical Modeling (Basal project AFB 170001 and doctoral folio 21140124) and National Laboratory of High Performance Computing of Chile (ECM-02).
References
- 1.Schindler DE, Hilborn R, Chasco B, Boatright CP, Quinn TP, Rogers LA, Webster MS. 2010. Population diversity and the portfolio effect in an exploited species. Nature 465, 609–613. ( 10.1038/nature09060) [DOI] [PubMed] [Google Scholar]
- 2.Fraser DJ, Weir LK, Bernatchez L, Hansen MM, Taylor EB. 2011. Extent and scale of local adaptation in salmonid fishes: review and meta-analysis. Heredity 106, 404–420. ( 10.1038/hdy.2010.167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayllon F, et al. 2015. The vgll3 locus controls age at maturity in wild and domesticated Atlantic salmon (Salmo salar L.) males. PLoS Genet. 11, e1005628 ( 10.1371/journal.pgen.1005628) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barson NJ, et al. 2015. Sex-dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature 528, 405–408. ( 10.1038/nature16062) [DOI] [PubMed] [Google Scholar]
- 5.Davidson WS, Koop BF, Jones SJ, Iturra P, Vidal R, Maass A, Jonassen I, Lien S, Omholt SW. 2010. Sequencing the genome of the Atlantic salmon (Salmo salar). Genome Biol. 11, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lien S, et al. 2016. The Atlantic salmon genome provides insights into rediploidization. Nature 533, 200–205. ( 10.1038/nature17164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnsen DB. 2009. Salmon, science, and reciprocity on the Northwest Coast. Ecol. Soc. 14, 43 ( 10.5751/ES-03107-140243) [DOI] [Google Scholar]
- 8.Quinn TP. 2005. The behavior and ecology of pacific salmon and trout. Seattle, WA: University of Washington Press. [Google Scholar]
- 9.Quinn TP, McGinnity P, Reed TE. 2015. The paradox of ‘premature migration’ by adult anadromous salmonid fishes: patterns and hypotheses. Can. J. Fish. Aquat. Sci. 73, 1015–1030. ( 10.1139/cjfas-2015-0345) [DOI] [Google Scholar]
- 10.Waples RS, Teel DJ, Myers JM, Marshall AR. 2004. Life-history divergence in Chinook salmon: historic contingency and parallel evolution. Evolution 58, 386–403. ( 10.1111/j.0014-3820.2004.tb01654.x) [DOI] [PubMed] [Google Scholar]
- 11.Hecht BC, Matala AP, Hess JE, Narum SR. 2015. Environmental adaptation in Chinook salmon (Oncorhynchus tshawytscha) throughout their North American range. Mol. Ecol. 24, 5573–5595. ( 10.1111/mec.13409) [DOI] [PubMed] [Google Scholar]
- 12.Healey MC. 1991. Life history of Chinook salmon (Oncorhynchus tshawytscha). In Pacific salmon life histories (eds Groot C, Margolis L), pp. 311–395. Vancouver, Canada: University of British Columbia Press. [Google Scholar]
- 13.Brannon EL, Powell MS, Quinn TP, Talbot A. 2004. Population structure of Columbia River Basin Chinook salmon and steelhead trout. Rev. Fish. Sci. 12, 99–232. ( 10.1080/10641260490280313) [DOI] [Google Scholar]
- 14.Narum SR, et al. 2008. Differentiating salmon populations at broad and fine geographic scales with microsatellites and SNPs. Mol. Ecol. 17, 3464–3477. [DOI] [PubMed] [Google Scholar]
- 15.Hess JE, Whiteaker JM, Fryer JK, Narum SR. 2014. Monitoring stock specific abundance, run-timing, and straying of Chinook salmon in the Columbia River using genetic stock identification. N. Am. J. Fish. Manage. 34, 184–201. ( 10.1080/02755947.2013.862192) [DOI] [Google Scholar]
- 16.Prince DJ, O'Rourke SM, Thompson TQ, Ali OA, Lyman HS, Saglam IK, Hotaling TJ, Spidle AP, Miller MR. 2017. The evolutionary basis of premature migration in Pacific salmon highlights the utility of genomics for informing conservation. Sci. Adv. 3, e1603198 ( 10.1126/sciadv.1603198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brieuc MS, Waters CD, Seeb JE, Naish KA. 2014. A dense linkage map for Chinook salmon (Oncorhynchus tshawytscha) reveals variable chromosomal divergence after an ancestral whole genome duplication event. G3-Genes Genom. Genet. 4, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKinney GJ, et al. 2016. An integrated linkage map reveals candidate genes underlying adaptive variation in Chinook salmon (Oncorhynchus tshawytscha). Mol. Ecol. Res. 16, 769–783. ( 10.1111/1755-0998.12479) [DOI] [PubMed] [Google Scholar]
- 19.Waterhouse RM, Seppey M, SimãO FA, Manni Mè, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. ( 10.1093/molbev/msx319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schlötterer C, Tobler R, Kofler R, Nolte V. 2014. Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 15, 749–763. ( 10.1038/nrg3803) [DOI] [PubMed] [Google Scholar]
- 21.Micheletti SM, Narum SR. 2018. Utility of pooled sequencing for association mapping in non-model organisms. Mol. Ecol. Resour. 18, 825–837. [DOI] [PubMed] [Google Scholar]
- 22.Kofler R, Pandy RV, Schlotterer C. 2011. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-seq). Bioinformatics 27, 3435–3436. ( 10.1093/bioinformatics/btr589) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 1303.3997.
- 24.Faust GG, Hall IM. 2014. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. ( 10.1093/bioinformatics/btu314) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. ( 10.1093/bioinformatics/btr509) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fariello MI, et al. 2017. Accounting for linkage disequilibrium in genome scans for selection without individual genotypes: the local score approach. Mol. Ecol. 26, 3700–3714. ( 10.1111/mec.14141) [DOI] [PubMed] [Google Scholar]
- 27.Turner SD. 2014. QQman: an R package for visualizing GWAS results using Q-Q and Manhattan plots. biorXiv. ( 10.1101/005165) [DOI]
- 28.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. ( 10.4161/fly.19695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. ( 10.1093/bioinformatics/btg412) [DOI] [PubMed] [Google Scholar]
- 30.Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. 2013. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140. ( 10.1111/mec.12354) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones FC, et al. 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61. ( 10.1038/nature10944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hess JE, Zendt JS, Matala AR, Narum SR. 2016. Genetic basis of adult migration timing in anadromous steelhead discovered through multivariate association testing Proc. R. Soc. B 283, 20153064 ( 10.1098/rspb.2015.3064) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohammed H, et al. 2013. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 3, 342–349. ( 10.1016/j.celrep.2013.01.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, Duan H, Zhang Y, Sun FQ. 2016. Abnormal activation of RhoA/ROCK1 signaling in junctional zone smooth muscle cells of patients with adenomyosis. Reprod. Sci. 23, 333–341. ( 10.1177/1933719115602764) [DOI] [PubMed] [Google Scholar]
- 35.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. 2016. Thomas. PANTHER version 11: expanded annotation data from Gene Ontology and reactome pathways, and data analysis tool enhancements. Nucl. Acids Res. 45, D183–D189. ( 10.1093/nar/gkw1138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodgson S, Quinn TP. 2002. The timing of adult sockeye salmon migration into freshwater: adaptations by populations to prevailing thermal regimes. Can. J. Zool. 80, 542–555. ( 10.1139/z02-030) [DOI] [Google Scholar]
- 37.Gende SM, Edwards RT, Willson MF, Wipfli MS. 2002. Pacific salmon in aquatic and terrestrial ecosystems. BioScience 52, 917–928. ( 10.1641/0006-3568(2002)052%5B0917:PSIAAT%5D2.0.CO;2) [DOI] [Google Scholar]
- 38.Kovach RP, Joyce JE, Echave JD, Lindberg MS, Tallmon DA. 2015. Earlier migration timing, decreasing phenotypic variation, and biocomplexity in multiple salmonid species. PLoS ONE 8, e53807 ( 10.1371/journal.pone.0053807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allendorf FW, Thorgaard GH. 1984. Tetraploidy and the evolution of salmonid fishes. In Evolutionary genetics of fishes (ed. Turner BJ.), pp. 1–53. New York, NY: Plenum Press. [Google Scholar]
- 40.Macqueen DJ, Johnston IA. 2014. A well-constrained estimate for the timing of the salmonid whole genome duplication reveals major decoupling from species diversification. Proc. R. Soc. B 281, 20132881 ( 10.1098/rspb.2013.2881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berthelot C, et al. 2014. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 5, 3657 ( 10.1038/ncomms4657) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christensen KA, Leong JS, Sakhrani D, Biagi CA, Minkley DR, Withler RE, Rondeau EB, Koop BF, Devlin RH. 2018. Chinook salmon (Oncorhynchus tshawytscha) genome and transcriptome. PLoS ONE 13, e0195461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbey J, et al. 2016. Accuracy of assignment of Atlantic salmon (Salmo salar L.) to rivers and regions in Scotland and northeast England based on single nucleotide polymorphism (SNP) markers. PLoS ONE 11, e0164327 ( 10.1371/journal.pone.0164327) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Limborg MT, Larson WA, Seeb L, Seeb JE. 2017. Screening of duplicated loci reveals hidden divergence patterns in a complex salmonid genome. Mol. Ecol. 26, 4509–4522. ( 10.1111/mec.14201) [DOI] [PubMed] [Google Scholar]
- 45.Waples RK, Seeb JE, Seeb LW. 2017. Congruent population structure across paralogous and nonparalogous loci in Salish Sea chum salmon (Oncorhynchus keta). Mol. Ecol. 26, 4131–4144. ( 10.1111/mec.14163) [DOI] [PubMed] [Google Scholar]
- 46.Park TJ, et al. 2017. Fructose-driven glycolysis supports anoxia resistance in the naked mole rat. Science 356, 307–311. ( 10.1126/science.aab3896) [DOI] [PubMed] [Google Scholar]
- 47.Wegner NC, Snodgrass OE, Dewar H, Hyde JR. 2015. Whole-body endothermy in a mesopelagic fish, the opah, Lampris guttatus. Science 348, 786–789. ( 10.1126/science.aaa8902) [DOI] [PubMed] [Google Scholar]
- 48.Lamichhaney S, et al. 2016. Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nat. Genet . 48, 84–88. ( 10.1038/ng.3430) [DOI] [PubMed] [Google Scholar]
- 49.Kingsolver JG, Hoekstra HE, Hoekstra JM, Berrigan D, Vignieri SN, Hill CE, Hoang A, Gibert P, Beerli P. 2001. The strength of phenotypic selection in natural populations. Am. Nat. 157, 245–261. ( 10.1086/319193) [DOI] [PubMed] [Google Scholar]
- 50.Norberg J, Swaney DP, Dushoff J, Lin J, Casagrandi R, Levin SA. 2001. Phenotypic diversity and ecosystem functioning in changing environments: a theoretical framework. Proc. Natl Acad. Sci. USA 98, 11 376–11 381. ( 10.1073/pnas.171315998) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome and transcriptome assemblies and sequencing data are available on NCBI under project PRJNA402052 (genome assembly PIPH000000000, and transcriptome assembly GGDU00000000). The gene annotation file for genome assembly and tables 1 -3 listing significant candidate genes within and among lineages, and gene ontology enrichment are available from the Dryad Digital Repository at https://doi.org/10.5061/dryad.dr1qs08 [32].