

Abstract
Injury, inflammation, and nerve damage initiate a wide variety of cellular and molecular processes that culminate in hyperexcitation of sensory nerves, which underlies chronic inflammatory and neuropathic pain. Using behavioral readouts of pain hypersensitivity induced by angiotensin II (Ang II) injection into mouse hindpaws, our study shows that activation of the type 2 Ang II receptor (AT2R) and the cell-damage-sensing ion channel TRPA1 are required for peripheral mechanical pain sensitization induced by Ang II in male and female mice. However, we show that AT2R is not expressed in mouse and human dorsal root ganglia (DRG) sensory neurons. Instead, expression/activation of AT2R on peripheral/skin macrophages (MΦs) constitutes a critical trigger of mouse and human DRG sensory neuron excitation. Ang II-induced peripheral mechanical pain hypersensitivity can be attenuated by chemogenetic depletion of peripheral MΦs. Furthermore, AT2R activation in MΦs triggers production of reactive oxygen/nitrogen species, which trans-activate TRPA1 on mouse and human DRG sensory neurons via cysteine modification of the channel. Our study thus identifies a translatable immune cell-to-sensory neuron signaling crosstalk underlying peripheral nociceptor sensitization. This form of cell-to-cell signaling represents a critical peripheral mechanism for chronic pain and thus identifies multiple druggable analgesic targets.
SIGNIFICANCE STATEMENT Pain is a widespread health problem that is undermanaged by currently available analgesics. Findings from a recent clinical trial on a type II angiotensin II receptor (AT2R) antagonist showed effective analgesia for neuropathic pain. AT2R antagonists have been shown to reduce neuropathy-, inflammation- and bone cancer-associated pain in rodents. We report that activation of AT2R in macrophages (MΦs) that infiltrate the site of injury, but not in sensory neurons, triggers an intercellular redox communication with sensory neurons via activation of the cell damage/pain-sensing ion channel TRPA1. This MΦ-to-sensory neuron crosstalk results in peripheral pain sensitization. Our findings provide an evidence-based mechanism underlying the analgesic action of AT2R antagonists, which could accelerate the development of efficacious non-opioid analgesic drugs for multiple pain conditions.
Keywords: angiotensin II, AT2R, neuroimmune interaction, oxidative stress, pain, TRPA1
Introduction
Acute and chronic pain conditions arise from dysregulation of sensory neuron function and are often associated with diverse pathological states, including trauma, cancer, infectious diseases, and neuropathy. Most chronic pain is undermanaged by currently available analgesics (Yekkirala et al., 2017). Decades of rodent pain model-based research has yielded mechanistic knowledge of pain/nociceptive neuron function in specific pain states in the PNS and CNS (Patapoutian et al., 2009; Mickle et al., 2016; Yekkirala et al., 2017). However, poor clinical translation of basic mechanistic findings has called into question the validity of such models for specific human pain conditions (Woolf and Mannion, 1999; Moore et al., 2014; Yekkirala et al., 2017). Only a few targets identified from discovery research studies, such as neutralizing antibodies for nerve growth factor (NGF) and calcitonin gene-related peptide (CGRP), have shown effectiveness in osteoarthritis (Miller et al., 2017) and migraine (Deen et al., 2017), respectively. Small-molecule antagonists for the majority of nociceptive targets, sensory neuron receptors, ion channels, and signaling second messengers identified from mechanism-based studies in rodents, have not yet provided successful translation into clinical developments (Mickle et al., 2016; Yekkirala et al., 2017). Interestingly, an antagonist of the angiotensin II (Ang II) type 2 receptor (AT2R) EMA401 has demonstrated pain relief in a phase II clinical trial involving patients with neuropathic pain associated with postherpetic neuralgia (PHN) (Rice et al., 2014). This provides an opportunity to “back-translate” the site and mechanism of action and cellular targets underlying the analgesic effect of AT2R antagonism, as well as involvement of angiotensin signaling in pain sensitization.
The role of Ang II in regulating blood pressure, via its action on the Ang II type 1 (AT1R) receptor has been well documented; however, the role of AT2R has remained largely undefined (de Gasparo et al., 2000). Expression of AT2R in rodent brain has been shown to contribute to regulation of drinking behavior and locomotor activity (Hein et al., 1995; de Gasparo et al., 2000; de Kloet et al., 2016). The role of AT2R antagonism in rodent models of inflammatory, neuropathic, and bone cancer pain has been shown more recently (Chakrabarty et al., 2013; Muralidharan et al., 2014; Smith et al., 2016). Mechanistically, a Gαs-coupled AT2R signaling cascade in sensory neurons was suggested to elicit peripheral pain sensitization (Danser and Anand, 2014; Anand et al., 2015). In contrast, Gαi/o-coupled AT2R signaling has also been suggested to operate in sensory neurons. This signaling was elicited by an ulcerative bacterial toxin, leading to analgesia in mice (Marion et al., 2014). However, a follow-up in vitro study failed to reverse the toxin's effect on sensory neurons with a specific AT2R antagonist (Anand et al., 2016). More recently, it was reported that AT2R antagonists indirectly increased levels of the Ang II fragment Ang1–7, which activates the Mas1 receptor to elicit anti-nociceptive effects in a rodent model of bone cancer pain (Forte et al., 2016). These observations raise questions regarding the mechanism of action and/or the cellular target(s) of AT2R antagonists. Therefore, establishing the mechanistic underpinnings of angiotensin signaling in pain sensitization is essential for formulating further therapeutic developments.
Our study shows that Ang II acutely induces tactile and cold, but not heat pain hypersensitivity in mice, similar to that associated with neuropathy. By combining pharmacological and genetic manipulations, we show the requirement of AT2R and the mechanical/cell-damage-sensing receptor TRPA1 in Ang II-induced pain hypersensitivity. However, our in-depth investigation found no evidence of AT2R expression in mouse or human sensory neurons and Ang II did not directly influence sensory neuron function. Instead, our study shows the critical role of peripheral/skin macrophages (MΦs) in the development of Ang II-induced pain hypersensitivity. Furthermore, we identify Ang II-AT2R-mediated reactive oxygen/nitrogen species (ROS/RNS) production in MΦs as the vital trigger for TRPA1 activation on sensory neurons. Our findings comprehensively define the role of angiotensin signaling and MΦ-to-sensory neuron redox communication in peripheral pain sensitization.
Materials and Methods
Mice.
All experiments involving the use of mice and the procedures followed therein were approved by Institutional Animal Studies Committees of Washington University in St. Louis and The University of Iowa, in strict accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. Every effort was made to minimize the number of mice used and their suffering. Mice were maintained on a 12:12 light:dark cycle (06:00 to 18:00 h) with access to food and water ad libitum. Eight- to 14-week-old male and female mice were used for all experiments. All mice were bred and maintained in-house at the University Animal Facility to obtain the required number of mice of specific genotype, gender, and age. C57BL/6J (157 males and 118 females; The Jackson Laboratory, catalog #000664, RRID:IMSR_JAX:000664), C57BL/6J-Agtr1a-KO (9 males and 10 females; The Jackson Laboratory, catalog #002682, RRID:MGI:3619339), FVB/NJ (21 males and 16 females; The Jackson Laboratory, catalog #001800, RRID:MGI:3028967), B6/129PF2/J (7 males and 6 females; The Jackson Laboratory, catalog #100903, RRID:IMSR_JAX:100903), B6/129PF2/J-Trpa1-KO (9 males and 9 females; The Jackson Laboratory, catalog #006401, RRID:IMSR_JAX:006401), C57BL/6J-Trpv1-KO (8 males and 7 females; The Jackson Laboratory, catalog #003770, RRID:IMSR_JAX:003770), and Macrophage Fas-Induced Apoptosis (MaFIA; 22 males and 17 females; The Jackson Laboratory, catalog #005070, RRID:IMSR_JAX:005070) mice were purchased from The Jackson Laboratory and subsequently bred in the above-mentioned institutional animal facilities. The FVB/NJ-Agtr2-KO mouse line (23 males and 19 females) was generated by Victor J. Dzau and Richard E. Pratt (Hein et al., 1995). The C57BL/6J-Trpv4-KO mouse line (7 males and 7 females) was generated and generously provided by Dr. Wolfgang Liedtke (Liedtke and Friedman, 2003). The AT2R-eGFP reporter mouse line (MMRRC catalog #030278-UCD) was generated by Dr. Nathaniel Heintz under the GENSAT project and backcrossed to C57BL/6J for several generations to yield C57BL/6J-Agtr2GFP. Eleven male and 7 female C57BL/6J-Agtr2GFP mice were used in this study. The MaFIA mouse expresses eGFP and a mutant human FK506-binding protein 1A under the control of the Csf1r promoter. This enables selective, inducible depletion of MΦs with administration of a synthetic homodimerizer, AP20187, also known as B/B homodimerizer (Burnett et al., 2006). To induce MΦ depletion, MaFIA mice received 5 daily injections of B/B homodimerizer (2 mg/kg, i.p.) or vehicle (PBS + 10% v/v PEG-400 + 1.7% v/v Tween 80). This treatment regimen is sufficient to reduce Iba1 immunoreactivity in the skin by ∼85%, as reported previously (Shutov et al., 2016). Specific routes of individual drug injections are provided in figures and figure legends. Intraplantar injections were performed as described previously (Loo et al., 2012; Mickle et al., 2015b). Mice were manually restrained with the aid of a cloth such that the plantar surface of one hindpaw was exposed. A 10 μl volume was injected into the plantar surface of the hindpaw via a 33-gauge stainless steel needle coupled to a Hamilton syringe. Intrathecal injection was performed by lumbar puncture as described previously (Karim et al., 2001) using a Hamilton syringe and a 30-gauge needle to deliver a volume of 5 μl. Mice were continuously monitored after injection. Experimenters were blinded to mouse sham/surgery conditions, saline/drug injection types, and injection laterality, as well as to mouse sex and genotypes, during the experiments, data recordings, and analyses. With no sex-specific differences in mouse behaviors and MΦ angiotensin signaling from our preliminary findings, all subsequent experimental groups used both sexes of mice. Please refer to Table 1 for details on mouse sex-distributed individual group numbers for all experiments conducted in this study.
Table 1.
Sex-distributed mouse numbers in all figures
| Figure(s) | Group(s) | Male mice (no.) | Female mice (no.) |
|---|---|---|---|
| 1A,B | B6 FOR all saline and Ang II dose groups | 25 (5/group) | 15 (3/group) |
| 1C | B6 for both saline and Ang II groups | 8 (4/group) | 6 (3/group) |
| 1D,E | B6 for both saline and Ang II groups | 6 (3/group) | 8 (4/group) |
| 2A,C | B6 for Losartan | 3 | 3 |
| 2A,C | B6 for Ang II + Losartan | 3 | 4 |
| 2A,C | B6-Agtr1a-KO: saline | 4 | 3 |
| 2A,C | B6-Agtr1a-KO: Ang II | 3 | 4 |
| 2B,C | B6 for PD123319 | 3 | 4 |
| 2B,C | B6 for Ang II + PD123319 | 3 | 4 |
| 2B,C | FVB-WT: saline | 5 | 2 |
| 2B,C | FVB-WT: Ang II | 4 | 3 |
| 2B,C | FVB-Agtr2-KO: saline | 4 | 3 |
| 2B,C | FVB-Agtr2-KO: Ang II | 3 | 4 |
| 2D | FVB-Agtr2-KO: bradykinin | 3 | 4 |
| 3A,C | B6 for AP18 | 3 | 3 |
| 3A,C | B6 for Ang II + AP18 | 3 | 5 |
| 3A,C | B6/129-Trpa1-WT: saline | 3 | 3 |
| 3A,C | B6/129-Trpa1-WT: Ang II | 4 | 3 |
| 3A,C | B6/129-Trpa1-KO: saline | 4 | 5 |
| 3A,C | B6/129-Trpa1-KO: Ang II | 5 | 4 |
| 3B,D | B6 for both groups | 6 (3/group) | 8 (4/group) |
| 3B,D | B6-Trpv1-KO: saline | 3 | 4 |
| 3B,D | B6-Trpv1-KO: Ang II | 5 | 3 |
| 3B,D | B6-Trpv4-KO: saline | 3 | 4 |
| 3B,D | B6-Trpv4-KO: Ang II | 4 | 3 |
| 4 | B6 for all Ca2+ imaging | 5 | 3 |
| 5 | B6 for all current-clamp | 6 | 4 |
| 6A | FVB-Agtr2-WT and KO | 4 (2 each) | 2 (1 each) |
| 6B | B6 | 2 | 2 |
| 6C | B6 | 2 | 1 |
| 6D, 9A–D | B6-Agtr2GFP | 9 | 4 |
| 7A | B6 | 2 | 1 |
| 8A,D,E | B6 | 9 | 7 |
| 8B,D,E | FVB-Agtr2-WT and KO | 6 (3 each) | 4 (2 each) |
| 8D,E | B6-Agtr1a-KO | 2 | 1 |
| 10A | MaFIA | 2 | 2 |
| 10B,C | MaFIA: vehicle | 8 | 5 |
| 10B,C | MaFIA: B/B-HmD | 9 | 6 |
| 10D,E | MaFIA: B/B-HmD-BK | 3 | 4 |
| 11 | B6 for all cellular imaging | 27 | 16 |
| 11G | B6-Agtr1a-KO for all ROS/RNS imaging | 2 | 3 |
| 11G | FVB-Agtr2-WT for all ROS/RNS imaging | 3 | 3 |
| 11G | FVB-Agtr2-KO for all ROS/RNS imaging | 3 | 2 |
| 11G | B6-Agtr2GFP for all ROS/RNS imaging | 2 | 3 |
| 12A | B6 for all six treatment groups | 18 (3/group) | 12 (2/group) |
| 12B | B6 for all three treatment groups | 15 (5/group) | 12 (4/group) |
| 13A–C,F | B6 for all Ca2+ imaging | 17 | 7 |
| 13C,D | FVB-Agtr2-WT and KO | 9 (4 + 5) | 5 (2 + 3) |
Behavioral assessment of mechanical and heat hypersensitivity.
Mechanical and heat sensitivity on mouse hindpaws were assessed as described previously (Loo et al., 2012; Mickle et al., 2015b; Shepherd and Mohapatra, 2018). Animals were acclimated to the testing environment for 30 min on both of the 2 d before testing, as well as on every behavioral testing day. Mice were placed within single-occupancy Plexiglas boxes situated on a wire mesh platform at room temperature (22–23°C). Mechanical sensitivity was measured using 8 von Frey hair filaments of increasing strength (0.04 - 2 g) applied to the plantar surface of the hindpaw, as described previously (Mickle et al., 2015b; Shepherd and Mohapatra, 2018). Beginning with the finest filament (0.04 g), each filament was presented to each hindpaw 5 times. The number of paw withdrawal responses was recorded and used for calculating an area under the curve value for each hindpaw (Mickle et al., 2015b; Shepherd and Mohapatra, 2018), which provides a total measure of paw withdrawal response across the entire testing filament range. For the assessment of heat sensitivity, mice were placed within single-occupancy Plexiglas boxes situated on a glass plate maintained at a constant neutral temperature (30°C). The nociceptive heat sensitivity of each hindpaw was measured by focusing a beam of light (IITC Life Science) on the plantar surface. The latency to paw withdrawal from the heat source was recorded and expressed as paw withdrawal latency (PWL). The light intensity was calibrated to elicit baseline PWL values of 10–14 s. The mean of two recordings from each hindpaw was used for analysis. The latency cutoff was 20 s to avoid potential heat-related tissue injury. If a hindpaw was not withdrawn before cutoff, a PWL value of 20 s was assigned. Based on numerous prior studies showing hindpaw injection of several injury/inflammatory mediators, power analysis was performed to determine the appropriate sample size using the online BioMath software (http://www.biomath.info/). The effective sample size for these experiments was determined to be ≥6 per experimental group. For experiments with acute injection of saline/Ang II ± vehicle/drugs in mice, animals were randomly assigned to individual groups after injections. Our study used PD123319 (also known as EMA200), a first-generation AT2R antagonist and EMA401 (used in clinical trial) represents the [S]-enantiomer of EMA400, a modified EMA200 compound (Blankley et al., 1991; Smith et al., 2013a). EMA401 has superior pharmacokinetic and bioavailability properties, as well as ∼100-fold superior ED50 for attenuating mechanical hypersensitivity in rat experimental neuropathic pain compared with PD123319. Nevertheless, both PD123319 and EMA401 are highly selective for AT2R over AT1R and both of these antagonists have been shown to attenuate pain hypersensitivity in rodent experimental models with increasing doses and without any visible nonspecific effects (Blankley et al., 1991; Smith et al., 2013a,b; 2016). Due to the ease of procurement of PD123319 over EMA401, the former was used in this study.
Primary cell culture.
Mouse DRG neurons were isolated, dissociated, and cultured on coated glass coverslips, as detailed in previous reports (Loo et al., 2012; Mickle et al., 2015b). Isolated DRGs were digested with 2 mg/ml collagenase for 20 min, followed by neutralization, centrifugation, and trituration before further digestion with 1 mg/ml Pronase for 10 min. Dissociated cells were pelleted by centrifugation and resuspended in DMEM supplemented with 10% FBS. After 60 min of incubation at 37°C in a 5% CO2 incubator, the medium was changed to a serum-free culture medium supplemented with 50 ng/ml NGF. Neurons were used within 2–3 d of culturing.
Human DRGs from consented donors (3 females, mean age ∼30 years; 6 males, mean age ∼27 years) were acquired through MidAmerica Transplant Services and prepared as detailed in several recent reports (Davidson et al., 2016; Valtcheva et al., 2016). Briefly, lumbar DRGs were extracted 1.5–3 h postmortem and then dissected to remove adipose and connective tissue layers. After enzymatic digestion and mechanical dissociation of isolated ganglia, resuspended cells were plated onto coated glass coverslips. Cells were maintained in culture at 37°C with 5% CO2 in serum-free medium supplemented with 50 ng/ml NGF. Neurons were used within 5–6 d of culturing in vitro.
Mouse peritoneal MΦs were isolated as described previously (Shutov et al., 2016). Five milliliters of 3% FBS in DPBS was injected with a 25-gauge needle into the peritoneal cavity of euthanized mice, followed by gentle massage of the abdomen to dislodge cells resident in the cavity. The cell suspension was aspirated with a Pasteur pipette, pelleted by centrifugation, and resuspended in RPMI 1640 containing 50 ng/μl recombinant murine granulocyte-macrophage colony stimulating factor (GM-CSF; Goldbio). Cells were plated onto 35 mm tissue culture dishes for biochemical experiments or poly-l-lysine-coated glass coverslips for live-cell imaging within 2–3 d of culturing in vitro. For experiments with coculturing of peritoneal mouse MΦs or J774A.1 cells and mouse DRG neurons or U937 cells and human DRG neurons, differentiated MΦs (∼1.5 × 106 cells) were dissociated by incubating with TrypLE Express dissociation reagent (Invitrogen) for 5 min at 37°C. After centrifugation and resuspension in TNB medium, MΦs were plated on coverslips containing DRG neurons (at 24 h in vitro) at a density of 3.5 × 105 per coverslip. Cocultured coverslips were used in live-cell imaging experiments after a further 24 h in vitro.
Mouse neutrophils were isolated from peritoneal fluid as described previously (Swamydas et al., 2015). Twenty-four hours before euthanasia, mice were injected intraperitoneally with 1 ml of a sterile 9% solution of casein in PBS. After euthanasia, 5 ml of a 0.02% EDTA solution in PBS was injected into the peritoneal cavity. After briefly massaging the abdomen, the fluid was withdrawn using the same needle and syringe. Cells were pelleted by centrifugation and resuspended in 1 ml of DPBS mixed with 9 ml of Percoll gradient solution (Sigma-Aldrich). Centrifugation at 60,000 × g for 20 min separates polymorphonuclear leukocytes into a distinct band within the gradient, which was then isolated, washed, and resuspended in RPMI 1640 supplemented with 10% FBS. Experimenters were blinded to mouse sex and genotypes and to vehicle or drug types and their concentrations during the conduct of experiments, data recordings, and analyses on cell cultures.
Culture of cell lines.
The human monocyte-MΦ cell line U937 (ATCC catalog #CRL-1593, RRID:CVCL_0007) was cultured in RPMI 1640 containing 10% FBS and penicillin/streptomycin. When plating onto coverslips for experimentation, medium was supplemented with 100 ng/ml phorbol 12-myristate 13-acetate and 50 ng/ml recombinant human GM-CSF 24 h before use for differentiation into MΦs. The mouse monocyte/MΦ cell line J774A.1 (ATCC catalog #TIB-67, RRID:CVCL_0358) was cultured in DMEM containing 10% FBS and penicillin/streptomycin in a humidified incubator at 37°C with 5% CO2. When plating onto coverslips for experimentation, medium was supplemented with 50 ng/ml recombinant murine GM-CSF to aid MΦ differentiation 24 h before use. Human embryonic kidney cells stably expressing T-antigen (HEK293T; ATCC catalog #CRL-3216, RRID:CVCL_0063) were cultured in DMEM containing Glutamax, 10% FBS, and penicillin/streptomycin. Cells were transiently cotransfected with plasmids containing cDNAs of eGFP and WT or mutant human TRPA1 in which three key cysteine residues (Cys421, Cys621, and Cys655) are mutated to serine (hTRPA1-3C/S) using Lipofectamine 2000 according to the manufacturer's instructions, as detailed previously (Loo et al., 2012; Mickle et al., 2015b; Shepherd et al., 2018). Transfected cells were used in experiments within 36–48 h. Experimenters were blinded to vehicle or drug types and their concentrations and cDNA transfection groups during the conduct of these experiments, data recordings, and analyses.
Immunohistochemistry.
DRG and spinal cord tissue and plantar punch tissue biopsies were harvested from mice as described previously (Shepherd and Mohapatra, 2012; Shepherd et al., 2012, 2013). Forty-micrometer-thick fixed frozen sections of mouse spinal cord and plantar punch and 25-μm-thick sections of mouse DRGs were collected into 0.1 m phosphate buffer (PB). Fifty-micrometer-thick sections of human skin punch biopsies harvested from the lower leg/ankle region (demographic details given in Table 2) were collected into 0.1 m PB. Tissue sections were incubated with a blocking/permeabilizing solution (10% goat serum in 0.1 m PB + 0.3% Triton X-100) at 4°C for 1 h, followed by incubation with either rabbit anti-Iba1 (MΦ marker), rat anti-mouse Ly6g (neutrophil marker), rat anti-F4/80 and mouse anti-CD68 (MΦ markers), mouse anti-Neurofilament 200 (NF200; myelinated sensory marker), mouse anti-CGRP (nociceptive neuronal marker), or rabbit anti-human PGP9.5 (human sensory neuron marker) antibodies in blocking solution overnight at 4°C. For validation of AT2R antibodies in DRG tissue, sections were incubated with individual anti-AT2R antibodies, along with anti-NF200 or CGRP antibodies, in blocking solution overnight at 4°C. After 3 10 min washes in blocking solution, sections were incubated for 3 h with either goat anti-rabbit IgG-Alexa Fluor 488 or 555 (for detection of Iba1), goat anti-rat IgG-Alexa Fluor 568 (for detection of Ly6g or F4/80), goat anti-mouse IgG-Alexa Fluor 568 (for detection of CD68), goat anti-mouse IgG1-Alexa Fluor 568 (for detection of NF200 and CGRP), goat anti-mouse IgG1-Alexa Fluor 555, goat anti-rabbit IgG-Alexa Fluor 555 and donkey anti-goat IgG-Alexa Fluor 555 (for AT2R), and goat anti-GFP-FITC (for detection of GFP in Agtr2GFP mouse tissue) antibodies. Details of antibody source and dilutions are provided in Tables 3 and 4. The sections were then washed for 10 min in blocking solution, for 10 min in 0.1 m PB and finally for 10 min in 0.05 m PB. Finally, sections were dried on microscope slides and mounted with ProLong Gold anti-fade reagent with DAPI (Invitrogen). Confocal fluorescence images were captured using a Leica TCS-SPE confocal microscope with a 20×/numerical aperture (NA) 0.7 plan apochromat or 40×/NA 1.15 apochromatic oil-immersion objective. Images are a composite of 11 focal planes in a 20 μm z-stack at 2 μm increments. Tissue samples from >3 mice per group for each genotype were used for these experiments. Experimenters were blinded to mouse sex and genotypes, sham/surgery conditions, lateralization, and antibodies used during the conduct of these experiments, image acquisitions, and analyses. Eight human patient skin biopsy samples each for the healthy control, diabetic peripheral neuropathy, and chemotherapy-induced peripheral neuropathy groups were used for these experiments.
Table 2.
Human skin biopsy tissue donor demographic details
| Healthy control group | ||||||||
| Donor ID# | 1 | 113 | 24 | 115 | 133 | 335 | 166 | 158 |
| Age | 65 | 55 | 64 | 52 | 57 | 67 | 42 | 71 |
| Sex | Male | Female | Male | Male | Female | Male | Female | Male |
| Race | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian |
| Chronic disease | None | None | None | None | None | None | None | None |
| Type of Pain | None | None | None | None | None | None | None | None |
| Duration of pain | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Current/recent analgesic treatment | None | None | None | None | None | None | None | None |
| Diabetic polyneuropathy (DPN) group | ||||||||
| Donor ID# | 59 | 273 | 117 | 202 | 250 | 245 | 266 | 65 |
| Age | 62 | 60 | 60 | 64 | 62 | 69 | 48 | 56 |
| Sex | Female | Male | Male | Male | Female | Female | Male | Male |
| Race | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian |
| Type of Neuropathy | DPN | DPN | DPN | DPN | DPN | DPN | DPN | DPN |
| Type of diabetes | Type 1 | Type 1 | Type 1 | Type 1 | Type 1 | Type 1 | Type 1 | Type 1 |
| Type of Pain | Constant (no details available) | Constant (no details available) | Constant (no details available) | Constant (no details available) | Constant (no details available) | Constant (no details available) | Constant (no details available) | Constant (no details available) |
| Duration of pain | ∼4 y | ∼5 y | ∼2 y | ∼7 y | ∼3 y | ∼5 y | ∼3 y | ∼6 y |
| Current/recent analgesics | Yes (no details available) | Yes (no details available) | Yes (no details available) | No | Yes (no details available) | Yes (no details available) | Yes (no details available) | Yes (no details available) |
| Chemotherapy-induced peripheral neuropathy (CIPN) group | ||||||||
| Donor ID# | 1 | 2 | 8 | 9 | 10 | 11 | 12 | 14 |
| Age | 60 | 53 | 71 | 65 | 64 | 54 | 68 | 73 |
| Sex | Male | Female | Male | Male | Female | Male | Female | Male |
| Race | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | African-American | Caucasian |
| Type of Neuropathy | CIPN | CIPN | CIPN | CIPN | CIPN | CIPN | CIPN | CIPN |
| Type of cancer | Colorectal | Colorectal | Esophageal | Colorectal | Esophageal | Colorectal | Breast | Pancreatic |
| Chemothe-erapeutic drug | Oxaliplatin | Oxaliplatin | Oxaliplatin | Oxaliplatin | Oxaliplatin | Oxaliplatin | Docetaxel | Oxaliplatin |
| Type of Pain | Painful cold, electric shocks, pins and needles | Burning, pins and needles, numb | Electric shocks, tingling, numb | Burning, painful cold, tingling | Painful cold, tingling, pins and needles | Burning, tingling, numb | Tingling, pins and needles, numb | Painful cold, electric shocks, tingling |
| Duration of pain | 1–2 y | 6–12 mo | 2–5 y | 6–12 mo | 6–12 mo | 6–12 mo | 2–5 y | 6–12 mo |
| Current/recent analgesics | Pregabalin | Duloxetine, pregabalin | Pregabalin | Pregabalin | Pregabalin | Pregabalin | Pregabalin | Hydrocodone |
Table 3.
Primary antibodies used in this study
| Antigen | Antibody species/type | Dilution | Vendor | Catalog # and RRID |
|---|---|---|---|---|
| Angiotensin II type 2 receptor (AT2R) | Rabbit polyclonal/IgG | 1:500 | Abcam | ab19134 (RRID:AB_2273884) |
| Rabbit polyclonal/IgG | 1:100 | Santa Cruz Biotechnology | sc-9040 (RRID:AB_2225723) | |
| Goat polyclonal/IgG | 1:100 | Santa Cruz | sc-7420 (RRID:AB_633728) | |
| Iba1 | Rabbit polyclonal/IgG | 1:500 | Wako Chemicals USA | 019-19741 (RRID:AB_839504) |
| Mouse Ly6g | Rat monoclonal/IgG2b | 1:200 | Abcam | ab25377 (RRID:AB_470492) |
| NF200 | Mouse monoclonal/IgG1 | 1:250 | Sigma-Aldrich | N 0142 (RRID:AB_477257) |
| ERK1/2 | Rabbit polyclonal/IgG | 1:1000 | Cell Signaling Technology | 9102 (RRID:AB_330744) |
| Phospho- ERK1/2 | Mouse monoclonal/IgG1 | 1:1000 | Cell Signaling Technology | 9106 (RRID:AB_331768) |
| p38 MAPK | Rabbit polyclonal/IgG | 1:1000 | Cell Signaling Technology | 9212 (RRID:AB_330713) |
| Phospho-p38 MAPK | Rabbit monoclonal/IgG | 1:1000 | Cell Signaling Technology | 4511 (RRID:AB_2139682) |
| Mortalin/Grp75 | Mouse monoclonal/IgG1 | 1:1000 | NeuroMab | 75-127 (RRID:AB_2120479) |
| GFP | Goat polyclonal/FITC conjugate | 1:250 | Abcam | ab6662 (RRID:AB_305635) |
| F4/80 | Rat monoclonal/IgG2b | 1:250 | Thermo Fisher Scientific | MA5-16632 (RRID:AB_2538128) |
| CD68 | Mouse monoclonal/IgG1 | 1:200 | Abcam | ab955 (RRID:AB_307338) |
| GFP | Mouse monoclonal/IgG2a | 1:500 | NeuroMab | 73-131 (RRID:AB_10671444) |
| PGP9.5 | Rabbit polyclonal | 1:2000 | Zytomed Systems | 516-3344 |
Table 4.
Secondary antibodies used in this study
| Antigen | Antibody species/type | Dilution | Vendor | Catalog # and RRID |
|---|---|---|---|---|
| Mouse IgG1-Alexa Fluor 488 | Goat polyclonal IgG | 1:1000 | Thermo Fisher Scientific (previously Molecular Probes) | A21121 (RRID:AB_2535764) |
| Rat IgG–Alexa Fluor 568 | Goat polyclonal IgG | 1:1000 | Thermo Fisher Scientific | A11077 (RRID:AB_2534121) |
| Rabbit IgG–Alexa Fluor 488 | Goat polyclonal IgG | 1:1000 | Thermo Fisher Scientific | A11008 (RRID:AB_143165) |
| Rabbit IgG–Alexa Fluor 555 | Goat polyclonal IgG | 1:1000 | Thermo Fisher Scientific | A21428 (RRID:AB_2535849) |
| Goat IgG–Alexa Fluor 488 | Donkey polyclonal IgG | 1:1000 | Thermo Fisher Scientific | A11055 (RRID:AB_2534102) |
| Goat IgG–Alexa Fluor 555 | Donkey polyclonal IgG | 1:1000 | Thermo Fisher Scientific | A21432 (RRID:AB_2535853) |
| Rabbit IgG–Alexa Fluor 488 | Donkey polyclonal IgG | 1:1000 | Thermo Fisher Scientific | A21206 (RRID:AB_2535792) |
| Rabbit IgG–Alexa Fluor 555 | Donkey polyclonal IgG | 1:1000 | Thermo Fisher Scientific | A31572 (RRID:AB_162543) |
| Mouse IgG–HRP | Goat polyclonal IgG | 1:10,000 | Cell Signaling Technology | 7076 (RRID:AB_330924) |
| Rabbit IgG–HRP | Goat polyclonal IgG | 1:2,500 | Cell Signaling Technology | 7074 (RRID:AB_2099233) |
| Goat IgG–HRP | Donkey polyclonal IgG | 1:1000 | Santa Cruz Biotechnology | sc-2020 (RRID:AB_631728) |
ImageJ quantification.
Density of Iba1/PGP9.5 in human skin biopsy sections was quantified using ImageJ as described previously (Karlsson et al., 2015). Threshold RGB intensity was set in a blinded fashion and maintained between images to be compared. Approximately 0.5 mm2 of skin area was captured in each field. The area of the ROI that exhibited fluorescence above threshold was recorded as a percentage value.
Immunocytochemistry.
Mouse DRG neurons and peritoneal MΦs cultured on glass coverslips were immunostained using previously described protocols (Shepherd et al., 2012, 2013). Cells were fixed with 3% paraformaldehyde and subsequently blocked with 4% nonfat milk powder in saline containing 0.1% Triton X-100. Cells were costained with rabbit or goat anti-Iba1 (MΦ marker), along with rabbit anti-Iba1 and mouse anti-GFP antibodies, in blocking solution at room temperature for 1 h. After three 10 min washes in blocking solution, cells were incubated with either donkey anti-rabbit IgG–Alexa Fluor 555 and goat anti-mouse IgG–Alexa Fluor 488 antibodies for 1 h at room temperature. Details of antibody source and dilutions are provided in Tables 3 and 4. The cells were then washed 3 times for 10 min each in blocking solution and mounted onto glass slides with ProLong Gold antifade reagent with DAPI (Invitrogen). Epifluorescence microscopic images were captured by an MRc-5 digital camera connected to an AxioImager microscope using the AxioVision software (Carl Zeiss). Images were taken with a 63× Plan-Apochromat objective (NA 1.4), and transferred to Photoshop software (Adobe Systems) as TIFF files. Batches of cultured DRGs and MΦs from >3 mice were used for these experiments. Experimenters were blinded to mouse genotype and antibodies used during the conduct of these experiments, image acquisitions, and analyses.
Live-cell imaging.
Functional Ca2+ imaging on DRG neurons and MΦs were performed as described previously (Loo et al., 2012; Mickle et al., 2015b; Shepherd et al., 2018). Glass coverslips containing cells were incubated at room temperature for 20 min with the Ca2+-sensitive dye Fura 2-AM (2 μm). The coverslip was placed in the recording chamber mounted on the stage of an inverted Leica DMI6000B microscope and washed for 5 min at room temperature with continuous superfusion of standard extracellular HEPES-buffered HBSS (known hereafter as extracellular imaging buffer) containing the following (in mm): 140 NaCl, 5 KCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 0.4 KH2PO4, 0.6 NaHPO4, 3 NaHCO3, 10 glucose, and 10 HEPES adjusted to pH 7.4 with NaOH and 310 mOsm with sucrose before the experiment began. All drug applications are performed in the extracellular imaging buffer with continuous superfusion at room temperature. Fluorescence was alternately excited at 340 and 380 nm (12 nm band-pass) using a Lambda LS Xenon lamp (Sutter Instruments) and a 10×/NA 0.4 objective. Emitted fluorescence was collected at 510 nm using a Hamamatsu ORCA-100 CCD camera for the entire experimental duration, including the first 5 min wash duration. Pairs of images were sampled at 1 Hz, fluorescence was background subtracted, and the ratio of fluorescence (F340/F380) was calculated.
Fluorescent dye-based imaging of cellular ROS/RNS production were conducted on coverslips containing DRG neurons or MΦs were incubated with 5 μm 2′,7′-dichlorofluorescein diacetate (DCFDA) dye for 30 min. The coverslip was placed in the recording chamber mounted on the stage of an inverted Leica DMI6000B microscope and washed for 5 min at room temperature with continuous superfusion of extracellular imaging buffer before the experiment began. All drug applications are performed in the extracellular imaging buffer with continuous superfusion at room temperature. Fluorescence was excited at 485 nm using a Lambda LS Xenon lamp (Sutter Instruments) and a 10×/NA 0.4 plan apochromat objective. Emitted fluorescence was collected at 530 nm using a Hamamatsu ORCA-100 CCD camera, sampling at 0.33 Hz for the entire experimentaratl duration, including the first 5 min wash duration. Fluorescence was background subtracted and the fold change in F485 versus time t0 (immediately before the drug application time point) was calculated.
To confirm the observed effects in cellular imaging experiments, multiple batches of cultured cells from four to five mice per experimental/treatment group for each genotype were used. Cultured cells from individual genotypes and drug treatment experiments were performed in a randomized fashion and experimenters were blinded to mouse sex and genotypes, as well as to drug types used during the conduct of these experiments, image acquisitions, and analyses.
RT-PCR.
Total RNA was extracted from DRGs and MΦs as described previously (Loo et al., 2012; Mickle et al., 2015b), first using TRIzol reagent (Invitrogen), followed by treatment with DNase-I for 30 min (Qiagen). RNA was then reverse transcribed into cDNA using a SuperScript III RT-PCR kit (Invitrogen) as per the manufacturer's instructions. PCRs were performed using these cDNA samples and Pfu DNA polymerase (Agilent Technologies) with specific primer sets, as detailed in Table 5, and 30 amplification cycles. PCR products were visualized on 2% agarose gels stained with ethidium bromide. DRG tissue, isolated neutrophils, and cultured MΦs from 3 mice, as well as 3 culture batches of J774A.1 and U937 monocyte/MΦ cell lines were used for these experiments. Experimenters were blinded to mouse sex and genotypes, as well as to specific primers used during the conduct of these experiments.
Table 5.
Oligonucleotides used in RT-PCR experiments
| Species and gene | Primer sets | Annealing temperature (°C) | Amplicon size (bp) |
|---|---|---|---|
| Mouse Agtr1a | Forward: 5′ CTGAAGCCAGTACCAGCTCT 3′ | 54.0 | 245 |
| Reverse: 5′ CTGGGGCAGTCATCTTGAAT 3′ | |||
| Human AGTR1 | Forward: 5′ GGCCAGTGTTTTTCTTTTGAATTTAGCAC 3′ | 55.0 | 202 |
| Reverse: 5′ TGAACAATAGCCAGGTATCGATCAATGC 3′ | |||
| Mouse/human Agtr2/AGTR2 | Forward: 5′ GTTCCCCTTGTTTGGTGTAT 3′ | 51.0 | 274 |
| Reverse: 5′ CATCTTCAGGACTTGGTCAC 3′ | |||
| Mouse/human Gapdh/GAPDH | Forward: 5′ TGATGACATCAAGAAGGTGGTGAAG 3′ | 57.0 | 240 |
| Reverse: 5′ TCCTTGGAGGCCATGTAGGCCAT 3′ | |||
| Mouse Ly6g | Forward: 5′ CTTCTCTGATGGATTTTGCGTTG 3′ | 52 | 365 |
| Reverse: 5′ CAACTCAGAGCTTCTATATCTC 3′ | |||
| Mouse/human Trpv1/TRPV1 | Forward: 5′ CTGGACTACCTGGAACACCA 3′ | 53.0 | 128 |
| Reverse: 5′ AACCAGGGCAAAGTTCTTCC 3′ |
RNA deep sequencing.
Deep sequencing was performed on total RNA isolated from independently obtained human DRG tissue at two sites. Human lumbar DRGs from tissue donors without any history of pain-related disease conditions were obtained through MidAmerica Transplant Services. In addition, human lumbar DRGs from tissue donors without or with a history of chronic pain conditions were obtained through AnaBios. Demographic details of human DRG tissue donors are provided in Table 6.
Table 6.
Human DRG donor demographic details for RNAseq
| Site 1: Washington University St. Louis School of Medicine | ||||||
| Donor # | 1 | 2 | 3 | 4 | 5 | |
| Age | 44 | 58 | 27 | 41 | 10 | |
| Sex | Male | Female | Female | Male | Male | |
| Race | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | |
| Type of pain | None in medical record | None in medical record | None in medical record | None | None | |
| Duration of pain | N/A | N/A | N/A | N/A | N/A | |
| Known Analgesics Taken | None | None | None | None | None | |
| Any known disease/cause of death | Cerebrovascular accident/stroke | Cerebrovascular accident/stroke | Head trauma/motor vehicle accident (MVA) | Head trauma | Head trauma | |
| Site 2: University of Texas at Dallas | ||||||
| Donor # | 1 | 2 | 3 | 4 | 5 | 6 |
| Age | 40–50 | 40–50 | 40–50 | 37 | 50 | 47 |
| Sex | Female | Female | Female | Female | Male | Female |
| Race | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian |
| Type of Pain | None in medical record | None in medical record | None in medical record | Diabetic neuropathic, low back and leg pain | Diabetic neuropathic, upper and lower extremity pain | Low back pain from herniated discs |
| Duration of pain | N/A | N/A | N/A | 15 y | 3 y | At least 3 weeks |
| Known Analgesics Taken | None | None | None | Acetaminophen/hydrocodone, marijuana | Gabapentin, acetaminophen/hydrocodone | OTC pain medicines |
| Any known disease | None in medical record | None in medical record | None in medical record | Diabetes | Diabetes | Chronic bronchitis |
N/A, Not applicable.
For RNAseq experiments performed at the Genome Technology Access Center of the Washington University in St. Louis, total RNA from frozen DRG tissue cut into small pieces was extracted using TRIzol reagent (Life Technologies) and subsequently purified using the RNeasy microkit (Qiagen). Total RNA with RIN value of >7 were used for further analysis. Library preparation was performed at the Genome Technology Access Center of the Washington University in St. Louis. Briefly, 30 ng of total RNA was reverse transcribed and amplified using the SMARTer Ultra low input RNA for Illumina Sequencing HV kit (Clontech). After cDNA preparation and shearing using an ultrasonicator (Covaris), the libraries were prepared using VeraSeq Ultra DNA Polymerase for 12 cycles.
For next-generation sequencing and analysis, single end (50 bp) sequencing was performed on the Illumina HiSeq2500 platform following the manufacturer's protocol at the Genome Technology Access Center of the Washington University in St. Louis. The raw, de-multiplexed RNA-seq reads were aligned with STAR version 2.0.4b (https://github.com/alexdobin/STAR/releases) (GRCh37 assembly). Low-base-quality reads and adaptors were clipped. All known genes with raw counts were enumerated to the matching gtf file from the same reference build from Ensembl with Subread:featureCounts (version 1.4.5) (http://sourceforge.net/projects/subread/) using the Ensembl gene ID as the key. Reads per kilobase of transcript per million mapped read values were generated in R using raw counts. We assessed sequencing performance for total number of aligned reads, uniquely aligned reads, number of genes, and transcripts detected, <1% of ribosomal fraction, and Spearman correlation of >0.9 between samples. Detailed protocols for RNA isolation, library preparation, and RNAseq are available online on the GUDMAP website (http://www.gudmap.org/).
For RNAseq experiments performed at the Genome Sequencing Facility of the University of Texas at Dallas, total RNA from DRG tissues was extracted using TRIzol reagent (Life Technologies) and subsequently purified using an RNeasy microkit (Qiagen), as detailed previously (Ray et al., 2018). After library preparation, Poly-A+ RNA was sequenced using a 75-bp paired-end library on an Illumina Sequencer by ActiveMotif. The sequenced reads were mapped and mRNA abundance quantified using the Tophat-Cufflinks pipeline (PMCID: PMC3334321). Quantification of mRNA abundance was performed using the Cuffdiff tool in the Tophat-Cufflinks toolkit using the “classic” normalization mode, as detailed previously (Ray et al., 2018). The relative abundances were converted from fragments per kilobase per million (FPKM) reported mapped fragments by Cuffdiff to transcripts per million (TPM) by normalizing each gene FPKM by the sum of all gene FPKMs for the sample and multiplying by 1 million.
Reference genomes and transcriptomes used for human RNAseq mapping were NCBI hg19 and Gencode v14, respectively (PMCID: PMC3431492). Data from RNAseq experiments for the normal, nonpain donors were submitted to dbGaP with the accession number phs001158.v1.p1. The data from pain donor samples are in the process of being submitted to dbGaP.
Western blotting.
Total protein lysates of cultured DRG neurons and MΦs were prepared as described previously (Loo et al., 2012; Mickle et al., 2015b). Proteins in lysates were separated in 10% SDS-PAGE gels and transferred onto nitrocellulose membrane (GE Healthcare). For DRG and MΦ lysates, membranes were blocked with a 4% solution of nonfat powdered milk in saline and then probed with rabbit anti-ERK1/2, mouse anti-phospho-ERK1/2 and/or rabbit anti-p38 MAPK and anti-pp38 MAPK antibodies, along with loading control (Grp75) antibodies. For lysates prepared from Agtr2-WT and Agtr2-KO MΦs, membranes were blocked with a 4% solution of nonfat powdered milk in saline and then probed with rabbit anti-AT2R or goat ant-AT2R and/or rabbit anti-Iba1 or goat anti-Iba1 antibodies (as detailed in Tables 3 and 4). Primary antibody binding was detected on the membranes with donkey anti-rabbit IgG-HRP or donkey anti-rabbit IgG-HRP or goat anti-mouse IgG-HRP secondary antibody, followed by enhanced ECL-Plus reagent incubation (PerkinElmer). Protein bands were visualized and densitometric quantification of band intensity was performed using the Odyssey Fc Imaging System (Li-Cor), as detailed previously (Gupte et al., 2016). Isolated neutrophils, cultured DRG neurons, and MΦs from three mice and three culture batches of J774A.1 and U937 monocyte/MΦ cell lines were used for these experiments. Experimenters were blinded to mouse sex and genotypes, cell line types, and antibodies used during the conduct of these experiments, image acquisitions, and analyses.
Bioluminescence imaging of ROS/RNS species in vivo.
Fifty-five minutes after intraplantar Ang II injection into one hindpaw, mice were injected intraperitoneally with the fluorescent ROS/RNS indicator dye L-012 (50 mg/kg), as described previously (Kielland et al., 2009). After induction of anesthesia with isoflurane, mice were placed on the stage of an IVIS 100 in vivo imaging system (PerkinElmer) and a 5 min exposure of the plantar surface of both hindpaws was taken. Luminescence signal intensities were then quantified using Living Image 2.60.1 software (PerkinElmer). Based on prior studies showing the magnitude of L-012 luminescence in vivo with oxidative stress conditions, power analysis was performed to determine the appropriate sample size using the online BioMath software. The effective sample size for these experiments was calculated to be ≥5 per experimental group. Animals were randomly assigned to individual groups to receive saline/Ang II ± vehicle/drug injections. Experimenters were blinded to mouse injection lateralization and to drug type and concentrations used during the conduct of these experiments, image acquisitions, and analyses.
Electrophysiology.
Mouse and human DRG neurons were superfused at room temperature with external recording solution containing the following (in mm): 145 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 7 glucose, and 10 HEPES adjusted to pH 7.4 with NaOH and 300–310 mOsm. Borosilicate fire-polished glass pipettes (Sutter Instruments) were filled with internal solution containing the following (in mm): 120 K-gluconate, 5 NaCl, 2 MgCl2, 0.1 CaCl2, 1.1 EGTA, 4 Na2ATP, 0.4 Na2GTP, 15 phosphocreatine, and 10 HEPES pH adjusted to 7.3 with KOH and 291 mOsm. Pipettes had open tip resistance values of 3–5 MΩ. Cells were visualized using IR-DIC microscopy on an Olympus BX51 microscope. To confirm the observed effects in cellular electrophysiology experiments, multiple batches of cultured cells from multiple animals and human donors per experimental/treatment group were used. Experiments involving drug treatment of cultured cells were performed in a randomized fashion.
Recordings were made with Patchmaster software controlling a HEKA EPC10 amplifier. After gigaseal formation and stable whole-cell access, membrane excitability was determined in current-clamp configuration at the resting membrane potential of each cell (average: −62 mV for mouse, −64 mV for human DRG neurons). Series resistance values were 4–8 MΩ, and cells were discarded if these values changed by >20%. Excitability thresholds and action potential (AP) firing frequencies were determined from 1 s step or ramp current injections in increments of 1–10 pA (mouse) or 50–100 pA (human). Input resistance was calculated from hyperpolarizing current steps of −20 pA (mouse) or −200 pA (human). Ang II (1 μm), or vehicle diluted in external solution was superfused continuously at room temperature for 5 min after excitability protocols, which were then repeated. Ang II was only applied once per coverslip. Recordings were filtered at 2.9 kHz, digitized at 20–50 kHz, and analyzed offline using NeuroMatic software and custom-written macros in Igor Pro.
Chemicals and reagents.
A967079 (1E,3E)-1-(4-Fluorophenyl)-2-methyl-1-pentene-3-one oxime), AMG 9810 ((2E)-N-(2,3-Dihydro-1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide), Losartan (2-butyl-4-chloro-1-[[2′-(1H-tetrazol-5-yl)-[1,1′-biphenyl]-4-yl]methyl]-1H-imidazole-5-methanol potassium salt), PD123319 ditri-fluoroacetate (1-[[4-(dimethylamino)-3-methylphenyl]methyl]-5-(diphenylacetyl)-4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridine-6-carboxylic acid ditrifluoroacetate), L-012 (8-amino-5-chloro-2,3-dihydro-7-phenyl-pyrido[3,4-d]pyridazine sodium salt), and bradykinin were purchased from Tocris Bioscience and R&D Systems. Ang II, AP-18 (4-(4-chlorophenyl)-3-methylbut-3-en-2-oxime), CGP42112A (Nα-nicotinoyl-Tyr-(Nα-Cbz-Arg)-Lys-His-Pro-Ile), DCFDA, NGF, LPS, AITC, and N-acetylcysteine were purchased from Sigma-Aldrich. The B/B Homodimerizer (AP20187 or B/B-HmD) was purchased from Clontech. Recombinant murine and human TNF-α were purchased from Peprotech. Recombinant murine and human GM-CSF were purchased from GoldBio, Tocris Bioscience, and R&D Systems. All other chemicals used in this study were purchased from Sigma-Aldrich, Bio-Rad, Roche Applied Science, and Thermo Fisher Scientific.
Experimental design and statistical analysis.
Details of specific experimental design, power analysis, data collection/analysis, and experimenter blinding have been provided under each individual type/techniques throughout the Materials and Methods section. Data are presented as mean ± SEM. For behavioral experiments, two-way ANOVA with Tukey's multiple-comparisons post hoc test was performed. p < 0.05 in each set of data comparisons was considered statistically significant. Biochemical, Ca2+, DCFDA, and L-012 imaging data were analyzed using one-way ANOVA with Tukey's or Bonferroni's multiple-comparisons post hoc test. All analysis was performed using Prism 7.0 software (GraphPad Software).
Data availability.
All of the data presented herein are available without any restriction upon request. The RNAseq data are available through the public database, as mentioned above.
Results
Ang II induces peripheral mechanical pain hypersensitivity and is dependent on AT2R
A recent phase II clinical trial of a peripherally acting AT2R antagonist demonstrated successful alleviation of neuropathic pain associated with PHN (Rice et al., 2014). However, the underlying mechanism remains enigmatic, with a number of studies showing that the mechanism of action may involve AT2R antagonism directly on sensory neurons (for review, see Smith et al., 2016) and other studies finding no direct functional role of sensory neuron AT2R (Forte et al., 2016; Liao et al., 2017; Chakrabarty et al., 2018). We began with an unambiguous verification of the role of Ang II signaling in pain sensitization. Ang II injection into mouse hindpaws dose dependently induced mechanical hypersensitivity without inducing any heat hypersensitivity (Fig. 1A,B). Instead of determining mechanical paw withdrawal threshold, the magnitude of total mechanical sensitivity on mouse hindpaws in response to increasing von Frey filament strength was determined, as detailed earlier in several reports (Banik et al., 2006; Mickle et al., 2015b). Interestingly, intrathecal injection of Ang II did not induce mechanical hypersensitivity (Fig. 1C), as reported previously (Cridland and Henry, 1988), suggesting a peripheral rather than central action of Ang II in the induction of mechanical hypersensitivity. No sex differences in Ang II-induced hindpaw mechanical hypersensitivity were observed (Fig. 1D). In addition, hindpaw Ang II injection failed to induce any heat hypersensitivity in male and female mice (Fig. 1E). We next determined the involvement of specific AT receptor subtypes in this phenomenon. Ang II-induced mechanical hypersensitivity was unaffected by coadministration of an AT1R antagonist (losartan) in WT mice and remained intact in Agtr1a-KO mice (Fig. 2A). However, Ang II-induced mechanical hypersensitivity was completely attenuated by coadministration of an AT2R antagonist (PD123319) in WT mice and was absent upon Ang II injection in Agtr2-KO mice (Fig. 2B). Ang II injection in WT, Agtr1a-KO, and Agtr2-KO mice (with or without losartan or PD123319) did not influence hindpaw heat sensitivity (Fig. 2C). Bradykinin injection into Agtr2-KO mouse hindpaws induced heat (Fig. 2D) and mechanical (Fig. 2E) hypersensitivity, suggesting an absence of any gross deficits in induced pain hypersensitivity in mice lacking functional AT2R. Collectively, these data suggest that Ang II-induced mechanical pain sensitivity operates exclusively via AT2R at a peripheral level, not spinally.
Figure 1.

Ang II induces peripheral mechanical pain hypersensitivity in mice, without any sex-specific differences. A, Intraplantar Ang II injection dose dependently induces mechanical hypersensitivity in the ipsilateral hindpaw of C57BL/6 mice. Data are presented as mean ± SEM (n = 8 per group). B, Increasing doses of intraplantar Ang II injection does not influence heat sensitivity in mouse hindpaws. Data are presented as mean ± SEM (n = 8 per group). C, Intrathecal Ang II (100 pmol) does not induce significant mechanical hypersensitivity. Data are presented as mean ± SEM (n = 7 per group). D, E, Both male and female mice develop hindpaw mechanical hypersensitivity of a similar magnitude (D) in response to Ang II injection (intraplantar) without influence on hindpaw heat sensitivity (E). Data are presented as mean ± SEM (n = 7 per group). *p < 0.05, **p < 0.01, ***p < 0.001, and not significant (ns) versus their respective baseline values, as well as individual time points in saline group (A–C) or contralateral groups (D, E), two-way ANOVA with Tukey's multiple-comparisons post hoc test.
Figure 2.

AT2R, but not AT1R, mediates Ang II-induced mechanical pain hypersensitivity in mice. A, Coadministration of losartan, an AT1R antagonist (10 pmol, ipl), does not influence Ang II-induced (100 pmol, ipl) mechanical hypersensitivity in C57BL/6 (B6) mice. Ang II (100 pmol, ipl) induces mechanical hypersensitivity in ipsilateral hindpaws of B6 and B6-Agtr1a-KO mice to a similar magnitude. Data are presented as mean ± SEM (n = 6 for B6-losartan group and n = 7 each for the remaining groups); **p < 0.01 and ***p < 0.001 versus their respective baseline values, as well as individual time points in B6-losartan and B6-Agtr1a-KO-saline groups. B, Coadministration of PD123319, an AT2R antagonist (10 pmol, ipl), completely attenuates Ang II-induced (100 pmol, ipl) mechanical hypersensitivity in ipsilateral hindpaws of B6 mice. Ang II (100 pmol, ipl) induces mechanical hypersensitivity in in ipsilateral hindpaws of B6 and FVB-Agtr2-WT mice to a similar magnitude, which is absent in FVB-Agtr2-KO mice. Data are presented as mean ± SEM (n = 7 each for B6-PD123319, B6-Ang II+PD123319, and FVB-WT-Saline groups and n = 8 each for FVB-WT-Ang II, FVB-Agtr2-KO-Saline, and FVB-Agtr2-KO-Ang II groups). *p < 0.05, **p < 0.01, and ***p < 0.001 versus their respective baseline values, as well as individual time points in saline injection, for FVB-Agtr2-WT mice; not significant (ns) versus FVB-Agtr2-KO-saline and B6-PD123319 groups. C, Ang II injection (100 pmol, ipl) does not influence mouse hindpaw heat sensitivity in experimental conditions shown in A and B. ns, Not significant versus the respective group baseline values, as well as individual time points in saline-injected relevant KO genotype groups. D, E, Bradykinin injection (10 pmol, ipl) into the hindpaws of FVB-Agtr2-KO mice leads to development of mechanical (D) and heat (E) hypersensitivity, suggesting no deficits in induced cutaneous hypersensitivity due to Agtr2 gene deletion. Data are presented as mean ± SEM (n = 7 per group). **p < 0.01 and ***p < 0.001 versus their respective baseline values, as well as individual time points in contralateral groups, two-way ANOVA with Tukey's multiple-comparisons post hoc test.
Requirement of TRPA1 for Ang II-induced mechanical pain hypersensitivity in mice
To further determine the sensory receptor(s) important for Ang II-induced mechanical hypersensitivity, we directed our attention to critical TRP channels such as TRPA1, TRPV1, and TRPV4, which have been suggested to be involved in pain hypersensitivity associated with injury, inflammation, and neuropathy-like conditions (Kwan et al., 2006; Petrus et al., 2007; Alessandri-Haber et al., 2008; Eid et al., 2008; Chen et al., 2011; Ho et al., 2012; Mickle et al., 2015a, 2016). Ang II-induced mechanical hypersensitivity was completely attenuated by systemic administration of the TRPA1 inhibitor AP-18 (Fig. 3A). In addition, hindpaw injection of Ang II failed to induce mechanical hypersensitivity in Trpa1-KO mice (Fig. 3A). In contrast, hindpaw injection of Ang II elicited mechanical hypersensitivity in Trpv1-KO and Trpv4-KO mice to an extent similar to that observed in WT mice (Fig. 3B). As observed in WT mice, hindpaw Ang II injection did not influence heat sensitivity in Trpa1-KO, Trpv1-KO, or Trpv4-KO mice (Fig. 3C,D). These observations indicate that Ang II-AT2R activation in sensory neurons presumably trans-activates/modulates TRPA1 to elicit mechanical hypersensitivity.
Figure 3.
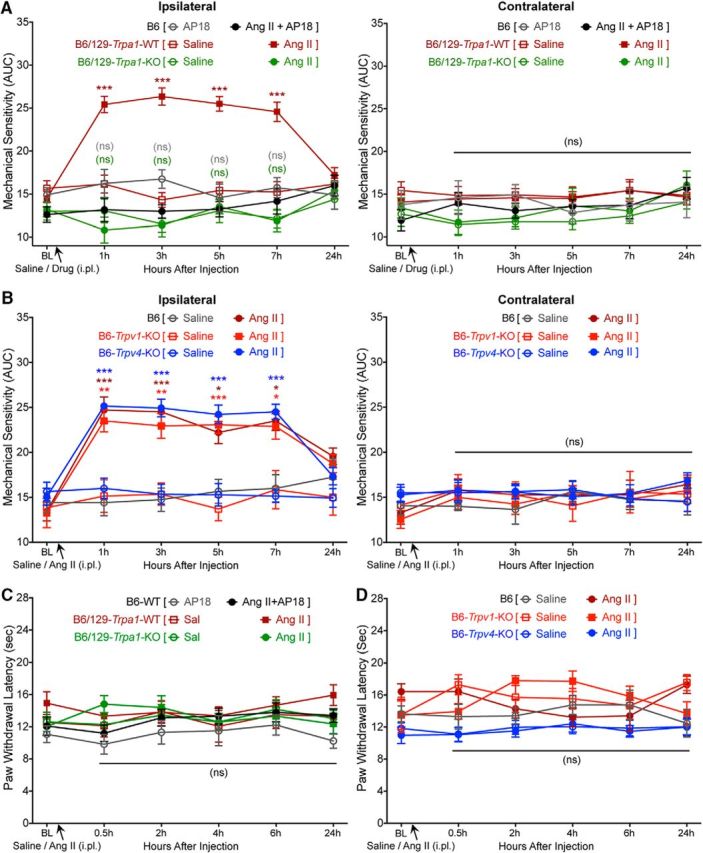
Requirement of TRPA1, but not TRPV1 or TRPV4, for Ang II-induced mechanical pain hypersensitivity in mice. A, Coadministration of AP18, a TRPA1 antagonist (10 nmol, ipl) prevents Ang II-induced (100 pmol, ipl) mechanical hypersensitivity in the ipsilateral hindpaws of B6 mice. Ang II (100 pmol, ipl) elicits mechanical hypersensitivity in B6/129S-Trpa1-WT mice to a similar extent as seen in B6 mice, which is absent in B6/129S-Trpa1-KO mice. Data are presented as mean ± SEM (n = 6 each for B6-AP18 and B6/129-Trpa1-WT-Saline groups, n = 7 for B6/129-Trpa1-WT-Ang II group, n = 8 for B6-Ang II+AP18 group, and n = 9 each for B6/129-Trpa1-KO-Saline and B6/129-Trpa1-KO-Ang II groups). ***p < 0.001 versus their respective baseline values, as well as individual time pointsm in B6/129S-Trpa1-WT-saline group; not significant (ns) versus B6-WT-AP18 and B6/129S-Trpa1-KO-saline groups. B, Ang II (100 pmol, ipl) induces a similar magnitude of mechanical hypersensitivity in the ipsilateral hindpaws of B6, B6-Trpv1-KO, and B6-Trpv4-KO mice. Data are presented as mean ± SEM (n = 7 each for B6-Saline, B6-Ang II, B6-Trpv1-KO-Saline, B6-Trpv4-KO-Saline, and B6-Trpv4-KO-Ang II groups and n = 8 for B6-Trpv1-KO-Ang II group). *p < 0.05, **p < 0.01, and ***p < 0.001 versus their respective baseline values, as well as individual time points for saline injection, in respective mouse genotype groups. C, D, Ang II injection (100 pmol, ipl) does not influence hindpaw heat sensitivity in in the hindpaws of mice used in experimental conditions shown in A and B. ns, Not significant versus the respective group baseline values, as well as individual time points, in saline-injected relevant KO genotype groups, two-way ANOVA with Tukey's multiple-comparisons post hoc test.
Ang II has no direct influence on TRPA1 function and excitability of sensory neurons and lack of AT2R expression in sensory neurons
We next investigated how Ang II/AT2R activation in sensory neurons might influence TRPA1 channel activation and/or modulation, which could lead to the mechanical pain hypersensitivity observed in vivo. Prolonged exposure to Ang II (1 h) did not elevate intracellular Ca2+ ([Ca2+]i) in cultured mouse or human DRG sensory neurons regardless of functional TRPA1 expression (Fig. 4A,B), suggesting that Ang II does not directly activate TRPA1 or other Ca2+-permeable TRP channels in DRG neurons. Previous studies have reported that AT2R activation in mouse DRG neurons modulates TRPV1 function (Anand et al., 2013, 2015; Smith et al., 2016). Intriguingly, in our experiments, Ang II exposure did not influence the function of TRPA1 or TRPV1 channels in mouse or human DRG neurons (Fig. 4C,D). Dual pulse activation of TRPV1 leads to decreased Ca2+ flux in response to second capsaicin application due to Ca2+-dependent channel desensitization (Mohapatra and Nau, 2003, 2005; Mohapatra et al., 2003). Nociceptor-sensitizing mediators that influence TRPV1 channel function significantly enhance the magnitude of Ca2+ flux in response to second capsaicin application (Mickle et al., 2015a). However, Ang II application between two successive application of capsaicin did not significantly alter the magnitude of Ca2+ flux in response to second capsaicin application (Fig. 4D), indicating no influence on TRPV1 channel activation and/or desensitization. Similarly, Ang II application between two successive application of a TRPA1 activator, AITC, did not significantly alter the magnitude of Ca2+ flux in response to second AITC application (Fig. 4C), indicating no influence on TRPA1 channel activation. As a positive control, bradykinin exposure led to significant potentiation of both TRPA1- and TRPV1-mediated Ca2+ flux and prostaglandin E2 (PGE2) exposure led to significant potentiation of TRPV1-mediated Ca2+ flux in both mouse and human DRG neurons (Fig. 4C,D). Furthermore, Ang II exposure did not alter AP firing or membrane excitability properties of cultured mouse and human DRG neurons (Fig. 5A–D). Altogether, these findings suggest that Ang II does not directly influence sensory neuron function or excitability, which prompted us to seek evidence in support of functional AT2R expression in sensory neurons. We first verified AT2R expression in DRG neurons by antibody-based staining, as shown previously (Smith et al., 2013b). Commercially available anti-AT2R antibodies stained DRG tissue from Agtr2-WT and Agtr2-KO mice with similar intensity (Fig. 6A), suggesting possible nonspecific binding of these antibodies, as has been demonstrated earlier in other vascular tissue (Hafko et al., 2013). Furthermore, no amplification of Agtr2 mRNA from mouse and human DRGs could be obtained using species-specific AT2R primer sets in qualitative RT-PCR (Fig. 6B). We next investigated whether functional AT2R signaling could be detected in DRG neurons. In contrast to previous reports (Smith et al., 2013b; Anand et al., 2015), Ang II exposure did not elicit AT2R-dependent ERK1/2 and p38 MAPK phosphorylation in either mouse or human DRG neurons (Fig. 6C,D). All of these observations pointed to a lack of AT2R expression in DRG neurons. To further confirm this assertion, we performed immunostaining of DRG sections from Agtr2GFP reporter mice, in which GFP expression is driven by the Agtr2 promoter. No detectable GFP signal was observed in the entire DRGs of these mice even with anti-GFP antibody-based amplification of GFP fluorescence (Fig. 6E).
Figure 4.

Ang II has no direct influence on sensory neuron TRPA1- and TRPV1-mediated Ca2+ flux. A, Representative traces (from two individual neurons/species) showing that Ang II (1 μm) does not induce [Ca2+]i elevation in cultured mouse and human DRG sensory neurons in culture. AITC (100 μm) was used for the detection of TRPA1+ neurons. B, Quantification of Ang II-induced (1 μm; 1 h) [Ca2+]i elevation in cultured mouse and human DRG neurons. Peak neuronal [Ca2+]i elevation at 5 min intervals are normalized to baseline Ca2+ levels. AITC (100 μm) and 50 mm KCl (K50) are used to detect TRPA1 responsiveness and neurons, respectively. C, D, Ang II (1 μm) fails to potentiate AITC-induced (50 μm; 15 s; C) and capsaicin-induced (50 nm; 15 s; D) [Ca2+]i elevation, as depicted by representative traces from single neuron/species. Potentiation TRPA1- and TRPV1-induced [Ca2+]i elevation are quantified by calculating the ratio of second versus first AITC/Cap-induced Ca2+ flux in cultured mouse/human DRG neurons. As positive controls, bradykinin (100 nm) significantly potentiates AITC- and capsaicin-evoked Ca2+ flux and PGE2 (10 μm) potentiates capsaicin-induced Ca2+ elevation. Data are presented as mean ± SEM in B–D. Numbers shown inside each column in B–D indicate the number of DRG neurons in ≥4 culture batches from ≥4 mice/group or ≥4 human DRG culture batches/group. *p < 0.05, **p < 0.01, ***p < 0.001, and not significant (ns) versus respective baseline/vehicle groups, one-way ANOVA with Bonferroni's post hoc test.
Figure 5.
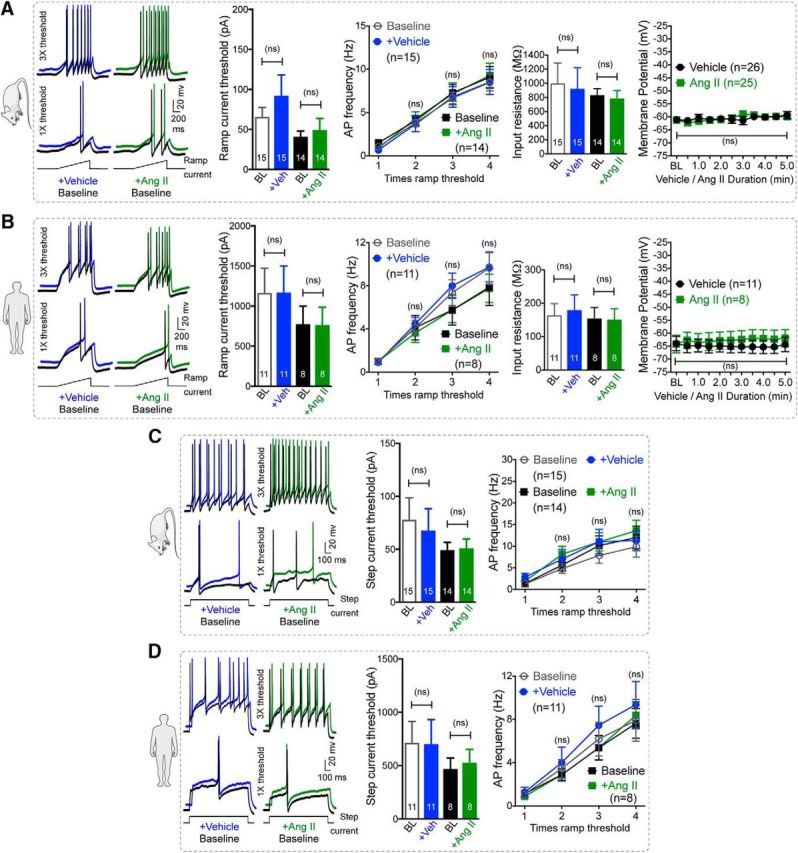
Ang II has no direct influence on sensory neuron membrane potential and excitability. A, B, Ang II (1 μm; 5 min) fails to influence AP firing to ramp current injection and the membrane potential of cultured mouse (A) and human (B) DRG neurons. Data are presented as mean ± SEM. ns, Not significant versus respective vehicle groups, one-way ANOVA with Bonferroni's post hoc test. C, D, Representative AP firing traces to step current injections and graphs for step current thresholds and AP frequency of cultured mouse (C) and human (D) DRG neurons are also unaffected by Ang II exposure (1 μm, 5 min). Data are presented as mean ± SEM. Numbers shown inside each column or on line graphs in all panels indicate the number of DRG neurons in ≥4 culture batches from ≥4 mice/group or ≥4 human DRG culture batches/group. ns, Not significant versus respective vehicle groups, one-way ANOVA with Bonferroni's post hoc test. AP firing traces for vehicle- and Ang II-treated conditions in all panels are offset for distinct visualization from their respective control traces.
Figure 6.
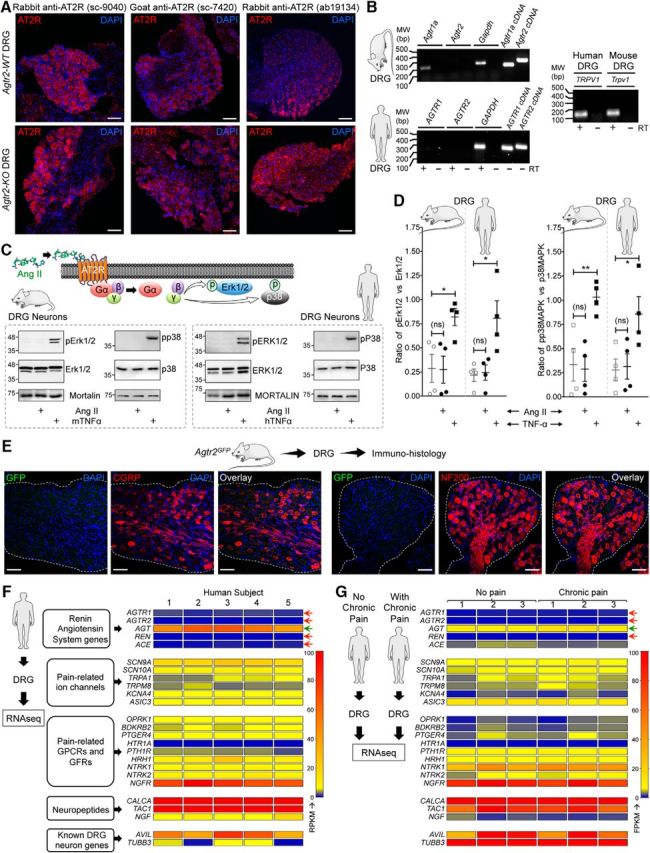
AT2R expression is not detected on mouse and human DRG sensory neurons. A, Representative confocal microscopy images of DRG tissue sections from FVB-Agtr2-WT and FVB-Agtr2-KO mice immunostained with routinely used anti-AT2R antibodies (red). Scale bar, 50 μm. No difference in AT2R signal intensity can be seen in the DRG sections from both mouse genotypes. B, Representative agarose gel electrophoresis images of RT-PCR amplification of AT1R and AT2R genes (Agtr1 and Agtr2, respectively) from the total RNA isolated from mouse and human DRGs. Plasmids containing mouse and human AT1R and AT2R cDNAs are used as a positive control. In addition, TRPV1 amplification is used as a positive control for DRG tissue. Numbers on the left denote DNA molecular weight markers (in base pairs). C, Ang II (1 μm; 30 min) does not induce phosphorylation of ERK1/2 and p38 MAPK, indicative of Ang II/AT2R activation, in cultured mouse and human DRG neurons. TNF-α (10 nm) is used as a positive control for induction of ERK1/2 and p38 MAPK phosphorylation. D, Quantification of the extent of ERK1/2 and p38 MAPK phosphorylation levels in mouse and human DRG neurons in response to Ang II exposure (1 μm; 30 min), as shown in representative immunoblots in Figure 3A. TNF-α (10 nm; 30 min) is used as a positive control for induction of ERK1/2 and p38 MAPK phosphorylation. Data are presented as individual experimental replicates, with mean ± SEM marked therein (n = 4 per group). *p < 0.05, **p < 0.01, and not significant (ns) versus respective comparison groups, one-way ANOVA with Tukey's multiple-comparisons post hoc test. E, The AT2R gene (Agtr2) is not expressed in neurons and non-neuronal cells in mouse DRG, as verified by lack of GFP signal (as a surrogate marker) in DRG sections (L2–L5) from Agtr2GFP reporter mice in which the Agtr2 promoter drives GFP expression. Representative confocal microscopy images of DRG sections stained with anti-GFP antibodies, along with CGRP and NF200 antibodies to mark peptidergic and myelinated sensory neurons. DAPI was used as a nuclear stain. Scale bar, 50 μm. F, G, Heat map showing mRNA expression levels (from RNAseq experiments) of RAS genes compared with critical pain-associated genes in human DRG tissue (F). No alteration in the mRNA levels of RAS genes can be observed in DRGs obtained from humans without or with chronic pain conditions (G). Red arrows indicate no reliable mRNA expression levels and green arrow indicates considerable mRNA expression of RAS genes.
In addition to the lack of sensory neuron GFP expression in the Agtr2GFP mouse, we also verified high-throughput RNAseq data available from published studies and found that Agtr2 mRNA was either absent or its levels were negligible (below baseline noise values) in mouse DRGs (Goswami et al., 2014; Khoury-Hanold and Iwasaki, 2016; Sapio et al., 2016; Yin et al., 2016; Ray et al., 2018). Furthermore, we performed next-generation deep sequencing on total RNA isolated from human DRGs and found no significant expression of AGTR2 mRNA, in contrast to other pain-related channels, receptors, and neuropeptide genes (Fig. 6F). Consistent with this observation, RNAseq datasets for human DRGs generated by an independent research group also showed no significant expression of AGTR2 mRNA from donors without a history of pain or with a medical history of chronic pain conditions (Fig. 6G, Table 6). Collectively, our in-depth analysis argues against any expression of AT2R expression in DRG neurons and/or the existence of direct AT2R-TRPA1 signaling therein. Rather, our data suggest that non-neuronal AT2R presumably underlies Ang II-induced pain hypersensitivity.
MΦ infiltration into the site of skin injury and AT2R expression in skin MΦs
With no indication of sensory neuron expression of AT2R or signaling crosstalk with TRPA1 channels within sensory neurons, we investigated the sites of Ang II injection to obtain histological evidence for the underlying mechanism. A marked increase in MΦ (Iba1 staining) and neutrophil (Ly6G staining) infiltration was observed in the plantar Ang II-injected region of mouse hindpaws, which could be detected both at early (1 h) and late (5 h) time points after injection (Fig. 7A). No such MΦ/neutrophil infiltration was observed in contralateral hindpaw skin or in saline-injected hindpaw skin (Fig. 7A). Similarly, significant increases in MΦ density were observed in leg skin biopsies from human patients with diabetic neuropathy and chemotherapy-induced peripheral neuropathy versus healthy controls, a difference associated with loss of PGP9.5+ nerve fibers (Fig. 7B,C). Because AT2R is required for Ang II-induced mechanical hypersensitivity, we next determined whether MΦs and/or neutrophils express AT2R. No amplification of AT1R or AT2R genes could be obtained from mouse peritoneal polymorphonuclear neutrophils (PMNs), whereas mRNA for both genes could be amplified from mouse peritoneal MΦ RNA (Fig. 8A). This is also consistent with high-throughput mRNA expression data on Agtr2/AGTR2 and other RAS gene expression in mouse and human MΦs available in the NCBI-GEO database (Chang et al., 2008; Gleissner et al., 2010; An et al., 2014; Mauer et al., 2014) (Fig. 8B). To validate functional AT2R expression on MΦs, we next performed biochemical assessment of Ang II-stimulated Erk1/2 phosphorylation, a functional readout of AT2R expression/activation (Gendron et al., 1999; Dhande et al., 2015). Increased Erk1/2 phosphorylation in response to Ang II exposure was observed in WT, Agtr1a-KO, and Agtr2-WT mouse peritoneal MΦs, but not in WT mouse PMNs or Agtr2-KO MΦs (Fig. 8D,E). Furthermore, Ang II-induced Erk1/2 phosphorylation could be mimicked by the AT2R-selective agonist CGP42112A and was blocked by the AT2R antagonist PD123319, but not the AT1R antagonist losartan (Fig. 8D,E), suggesting that functional Ang II/AT2R signaling exists in mouse MΦs.
Figure 7.
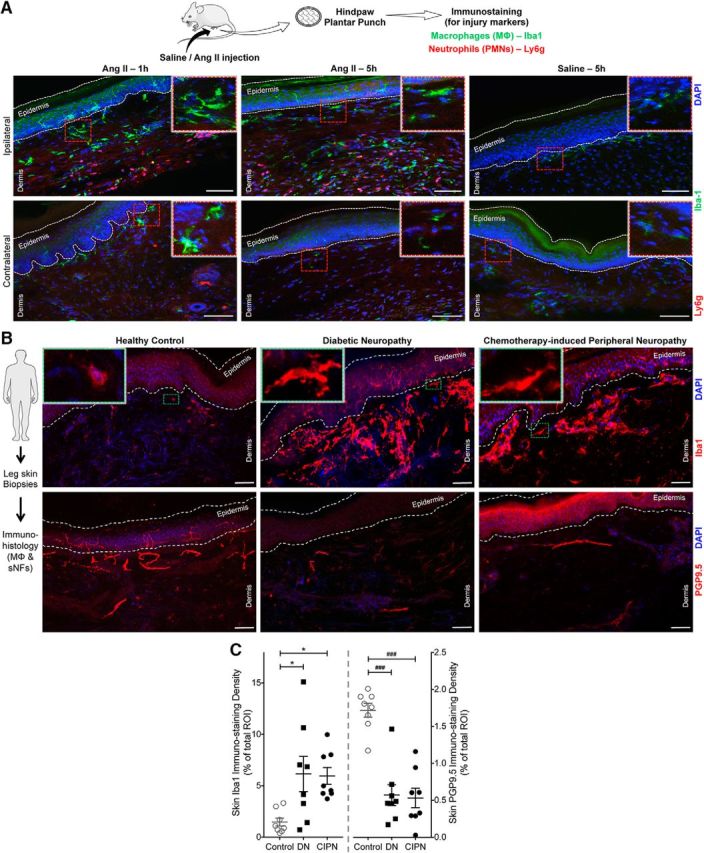
Ang II induces peripheral MΦ infiltration in mouse hindpaw skin and increased MΦ density in skin biopsies from human patients with diabetic and chemotherapy-induced peripheral neuropathy. A, Representative confocal microscopy images of mouse hindpaw plantar punch tissue sections showing hindpaw Ang II injection (100 pmol, ipl) enhances MΦ (green; Iba1) and neutrophil (red, Ly6g; blue, DAPI) infiltration both 1 and 5 h after injection compared with saline injection. Scale bar, 100 μm. Magnified views of area indicated as red dotted rectangular boxes are shown on the right top corner in each image group. B, Representative confocal microscopy images of human plantar punch tissue sections showing increased MΦ density (Iba1) in human leg/ankle skin biopsies from diabetic neuropathy and chemotherapy-induced peripheral neuropathy patients compared with age-matched healthy controls. This is accompanied by a decrease in the density of nociceptive nerve fibers (PGP9.5) in the skin (bottom row images). Green dotted rectangular boxes on the top left corners in top row images represent magnified views of individual MΦs in indicated areas. Scale bar, 100 μm. C, Density of both MΦs and nociceptive fibers in human skin biopsies are quantified and presented as individual experimental replicates, with mean ± SEM marked therein (n = 2 sections each from n = 8 human subjects per group). *p < 0.05, ###p < 0.001, and not significant (ns) versus healthy control groups, one-way ANOVA with Tukey's multiple-comparisons post hoc test.
Figure 8.

AT2R is expressed in MΦs. A, Representative agarose gel electrophoresis images of RT-PCR amplification of AT1R and AT2R genes (Agtr1a and Agtr2, respectively) from total RNA isolated from mouse MΦs and PMNs. Plasmids containing mouse Agtr1a and Agtr2 cDNAs are used as positive controls and Ly6g amplification is used for identification/validation of PMNs. Numbers on the left denote DNA molecular weight markers (in base pairs). B, Heat map showing moderate to high expression levels of RAS genes in monocytes/MΦs from RNA expression data deposited in the NCBI-GEO database. GEO mouse datasets GSE47426 (Mauer et al., 2014) and GSE57468 (An et al., 2014) and human datasets GSE10856 (Chang et al., 2008) and GSE20484 (Gleissner et al., 2010) are analyzed for RAS gene mRNA expression in monocytes/MΦs. C, Representative immunoblots depicting nonspecificity of routinely used anti-AT2R antibodies in peritoneal MΦ lysates from FVB-Agtr2-WT and FVB-Agtr2-KO mice. Anti-Iba1 and anti-mortalin antibodies are used as positive controls for MΦs and housekeeping protein, respectively. Numbers on the left denote protein molecular weight markers (in kilodaltons). D, E, Ang II (100 nm; 30 min) induces Erk1/2 phosphorylation in mouse (B6-WT) peritoneal MΦs, but not in PMNs. The AT2R inhibitor PD123319 (1 μm), but not the AT1R inhibitor losartan (1 μm), attenuates Ang II-induced Erk1/2 phosphorylation in MΦs. Ang II-induced Erk1/2 phosphorylation is absent in MΦs from FVB-Agtr2-KO mice, but intact in FVB-Agtr2-WT mice. The selective AT2R activator CGP42112A (100 nm) and TNF-α (10 nm) are used in mouse MΦs as positive controls for AT2R activation/signaling and Erk1/2 phosphorylation, respectively. LPS (10 nm) is used as a positive control for Erk1/2 phosphorylation in mouse PMNs. Mortalin (Grp75) immunoreactivity is used as loading control and the magnitude of ERK1/2 phosphorylation is quantified in E. Data are presented as individual experimental replicates, with mean ± SEM marked therein (n = 3 per group). *p < 0.05, #p < 0.05, and not significant (ns) versus indicated comparison groups, one-way ANOVA with Tukey's multiple-comparisons post hoc test.
Consistent with these observations, we next verified the expression of Agtr2 on hindpaw skin MΦs by immunohistological staining of hindpaw plantar punch tissue biopsies from Agtr2GFP reporter mice with GFP as a surrogate marker of Agtr2 gene expression. Extensive colocalization of GFP with the MΦ markers F4/80 and CD68 was observed in these sections (Fig. 9A), confirming AT2R expression in skin MΦs. Furthermore, Ang II injection into the hindpaw of Agtr2GFP mice led to increased density of skin MΦs (F4/80+/CD68+) that expressed GFP, suggesting AT2R expression in those infiltrated MΦs (Fig. 9A). Although GFP expression was not detected in any cell type within the lumbar DRGs of Agtr2GFP mice (Fig. 6E), we next investigated whether plantar Ang II injection could lead to infiltration of GFP-positive peripheral MΦs into DRGs and/or induction of GFP expression in DRG microglia/MΦs. Immunostaining of DRG tissue sections from plantar Ang II-injected Agtr2GFP mice did not detect any specific GFP signal (with anti-GFP antibody) in any cell types (Fig. 9B). In addition, it was observed that hindpaw Ang II injection did not visibly alter the density of Iba1-stained cells (microglia/MΦs) in the ipsilateral versus contralateral DRGs (Fig. 9B), suggesting no infiltration/depletion of DRG microglia/MΦs in response to hindpaw Ang II injection. Furthermore, we verified that GFP expression was not detected in the microglia/MΦs present in the primary cultures of dissociated DRGs from Agtr2GFP mice (Fig. 9C), although GFP expression could be detected in cultured peritoneal MΦs from Agtr2GFP mice (Fig. 9D). These observations suggest that increased infiltration of skin MΦs and AT2R expression in these MΦs, but not in the neurons and microglia/MΦs of DRGs, could be associated with Ang II-AT2R-induced peripheral mechanical hypersensitivity.
Figure 9.

AT2R is expressed in skin MΦs, but not in the neurons microglia/MΦs, of DRGs after hindpaw Ang II injection. A, Representative confocal microscopy images of Agtr2GFP mouse hindpaw plantar punch tissue sections showing hindpaw Ang II injection (100 pmol, ipl; 5 h after injection) enhances MΦ density (F4/80 and CD68 staining), which overlaps with GFP signal (green), suggesting AT2R expression therein. Scale bar, 50 μm. Images on the bottom rows represent magnified red dotted rectangular boxes in the respective top row images. B, Representative confocal microscopy images of Agtr2GFP mouse DRG (L2–L5) tissue sections showing hindpaw Ang II injection (100 pmol, ipl; 5 h after injection) does not enhance microglia/MΦ density (red, Iba1 staining) in the ipsilateral DRGs, as well as no detectable GFP signal (green, with anti-GFP antibody staining), suggesting no AT2R expression in neurons and microglia/MΦ. Scale bar, 50 μm. C, Representative microscopy images of cultured-dissociated DRGs from Agtr2GFP mice showing no detectable GFP signal (green, with anti-GFP antibody staining), suggesting no AT2R expression in neurons (red, β3-tubulin staining) and microglia/MΦ (red, Iba1 staining). Scale bar, 25 μm. D, Representative microscopy images of cultured peritoneal monocytes/MΦs from Agtr2GFP mice showing detection of GFP signal (green, with anti-GFP antibody staining) in Iba1-stained (red) MΦs, suggesting AT2R expression. Scale bar, 25 μm.
Peripheral MΦs are required for Ang II-induced mechanical pain hypersensitivity
Because increased MΦ infiltration is observed at the site of Ang II injection and MΦs express functional AT2R, we next verified whether MΦs are critical to Ang II-induced pain hypersensitivity. Chemogenetic depletion of peripheral skin MΦs was achieved with designer drug (B/B-HmD) administration in MaFIA mice (Fig. 10A). Because this designer drug does not penetrate the blood–brain barrier, this method of peripheral MΦ depletion does not influence/deplete MΦs/microglia populations in the DRG and spinal cord (Fig. 10A). Similar to our observations shown in Figure 9, no visible alteration was observed in the density of Iba1-stained cells (microglia/MΦs) in the ipsilateral DRG and spinal cord of vehicle- versus B/B-HmD-administered MaFIA mice upon Ang II injection (Fig. 10A). This again suggests that hindpaw Ang II injection does not lead to any significant infiltration/depletion of microglia/MΦs in the DRG and spinal cord. Hindpaw injection of Ang II failed to elicit any significant mechanical pain hypersensitivity in B/B-HmD-treated mice compared with vehicle-treated mice and did not influence hindpaw heat hypersensitivity (Fig. 10B,C). Peripheral MΦ depletion was not associated with any gross deficits in peripheral pain transduction, as indicated by conservation of bradykinin-induced mechanical and heat hypersensitivity in peripheral MΦ-depleted MaFIA mice (Fig. 10D,E). These observations indicate the critical role of peripheral MΦs, but not of microglia/MΦs, in the DRG and spinal cord of Ang II-induced mechanical pain hypersensitivity in mice.
Figure 10.

Peripheral MΦs are required for Ang II-induced mechanical hypersensitivity on mouse hindpaws. A, Experimental scheme for chemogenetic depletion of skin MΦs in MaFIA mice with the administration of a designer drug (B/B-HmD; 2 mg/kg/d for 5 d). Efficacy of peripheral MΦ depletion in skin, but not DRGs and spinal cord, in these mice were verified with immunostaining. Representative confocal microscopy images of hindpaw plantar punch, DRG (L2–L5) and spinal cord sections from MaFIA mice show depletion of skin MΦs without any alteration in microglia/MΦs in the DRG and spinal cord (red, Iba1 staining) of BB-HmD-treated mice. Scale bar, 50 μm. B, Ang II (100 pmol, ipl) fails to induce mechanical hypersensitivity in the hindpaws of MaFIA mice subjected to chemogenetic depletion of peripheral MΦs (as shown in A). Data are presented as mean ± SEM (n = 6 for Vehicle-Saline group, n = 7 each for Vehicle-Ang II and B/B-HmD-Saline groups, and n = 8 for B/B-HmD-Ang II group). ***p < 0.001 versus vehicle/saline-ipsi group, and ###p < 0.001 versus vehicle/Ang II-ipsi group, two-way ANOVA with Tukey's multiple-comparisons post hoc test. C, Chemogenetic depletion of MΦs and subsequent hindpaw Ang II injection (100 pmol, ipl) in MaFIA mice (B) does not influence hindpaw heat sensitivity. Data are presented as mean ± SEM (n = 7 for B/B-HmD-Saline group and n = 8 for B/B-HmD-Ang II group). D, E, Bradykinin injection (10 nmol, ipl) leads to the development of both mechanical (D) and heat (E) hypersensitivity in MaFIA mice subjected to chemogenetic depletion of peripheral MΦs with B/B-HmD administration (2 mg/kg/d for 5 d). Data are presented as mean ± SEM (n = 7 per group). *p < 0.05 **p < 0.01, and ***p < 0.001 versus respective contralateral groups, two-way ANOVA with Tukey's multiple-comparisons post hoc test.
MΦ AT2R activation leads to increased ROS/RNS production, which underlies Ang II-induced mechanical hypersensitivity
Because MΦs, AT2R, and TRPA1 are all required in Ang II-induced peripheral mechanical hypersensitivity, we next investigated the nature of MΦ angiotensin signaling and its potential crosstalk with sensory neurons. We first performed Ca2+ imaging of cultured mouse peritoneal MΦs, which did not exhibit any [Ca2+]i elevation upon exposure to the TRPA1 activator AITC or TRPV1 activator capsaicin before or after Ang II exposure (Fig. 11A), suggesting no functional TRPA1 expression. In addition, Ang II exposure did not directly induce any [Ca2+]i elevation in cultured mouse peritoneal MΦs (Fig. 11A). We next performed live-cell imaging of mouse peritoneal MΦs, which showed a time- and dose-dependent increase in DCFDA fluorescence emission, an indication of increased ROS/RNS production, when exposed to Ang II and Ang III, but not Ang IV (Fig. 11B,C). These observations are in accordance with the reported selectivity and affinity of Ang III for AT2R, as well as the ∼100-fold lower relative affinity of Ang IV for AT2R (Bosnyak et al., 2011). No mouse sex-specific differences could be detected in the magnitude of Ang II-induced MΦ ROS/RNS production (Fig. 11D). Ang II-induced ROS/RNS production in mouse MΦs could be mimicked by the AT2R-selective agonist CGP42112A and blocked by the AT2R antagonist PD123319, as well as the free radical scavenger n-acetylcysteine (NAC), but not by the AT1R antagonist losartan (Fig. 11E). MΦ ROS/RNS production in response to higher doses of Ang II and Ang III could also be attenuated with coapplication of the AT2R antagonist PD123319 (Fig. 11F), suggesting the specific involvement of AT2R. Ang II elicited ROS/RNS production in MΦs from Agtr1a-KO mice, which could be blocked by PD123319 (Fig. 11G). Ang II and CGP42112A exposure also led to elevated ROS/RNS production in MΦs from Agtr2-WT mice, but not from Agtr2-KO mice (Fig. 11G). We confirmed that similar Ang II/AT2R-dependent ROS/RNS production occurred in GFP-positive MΦs derived from the Agtr2GFP reporter mouse (Fig. 11G). Furthermore, in primary cultures of mouse and human DRG neurons, Ang II exposure did not elicit ROS/RNS production (Fig. 11H), which is consistent with our failure to detect AT2R expression in DRG neurons (Figs. 4, 5, 6, 9). Collectively, these observations suggest that Ang II activation of AT2R in mouse MΦs increases ROS/RNS production, which we next verified in vivo. Hindpaw injection of Ang II increased L-012 luminescence intensity, an indicator of increased redox state, which could be attenuated by coinjection of PD123319 or NAC (Fig. 12A). Furthermore, coinjection of NAC completely attenuated Ang II-induced mechanical hypersensitivity in mouse hindpaws without affecting heat sensitivity (Fig. 12B), indicating that increased local oxidative/nitrosative stress is required for Ang II-induced peripheral mechanical pain hypersensitivity.
Figure 11.

Ang II induces no elevation in Ca2+, but ROS/RNS production in MΦs, in an AT2R-dependent manner. A, Representative traces and quantification of ratiometric Ca2+ imaging of primary C57BL/6J peritoneal MΦs showing no response to the TRPA1 agonist AITC (100 μm, 30 s) before or after application of Ang II (1 μm; 5 min). A terminal pulse of the Ca2+ ionophore ionomycin (Inm; 10 μm) serves as a positive control. Peak Ca2+ signal data (ratio of F340/F380) are plotted on the right column graph with the indicated treatment conditions. In addition, capsaicin (1 μm, 30 s) was used, which failed to induce any Ca2+, before or after Ang II application. B, Representative time-lapse images (left) and quantification traces (right) of cultured mouse peritoneal MΦs showing Ang II-induced (100 nm) increase in ROS/RNS production, as determined by increased intensity of DCFDA redox-sensitive fluorescent dye. Scale bar, 50 μm. C, Ang II and Ang III, but not Ang IV, exposure (15 min) dose dependently induces ROS/RNS production in mouse MΦs. D, Ang II (100 nm; 15 min) induces ROS/RNS production to a similar extent in both male and female mouse MΦs. E, Ang II-induced (100 nm; 15 min) MΦ ROS/RNS production can be attenuated by PD123319 (1 μm) and NAC (3 mm), but not with losartan (1 μm) coapplication. Like Ang II, the AT2R-selective agonist CGP42112A (100 nm; 15 min) also elevates ROS/RNS levels. F, ROS/RNS production in mouse MΦs in response to higher Ang II or Ang II doses (10 μm; 15 min) can be attenuated by PD123319 (1 μm). G, Ang II (100 nm; 15 min) increases ROS/RNS levels in Agtr1a-KO mouse MΦs, which can be attenuated by PD123319 (1 μm) coapplication. Both Ang II and CGP42112A (100 nm each; 15 min) increase ROS/RNS levels in FVB-Agtr2-WT, but not in FVB-Agtr2-KO, mouse MΦs. Furthermore, Ang II and CGP42112A (100 nm each; 15 min) significantly increase ROS/RNS levels in MΦs from B6-Agtr2GFP reporter mice similar to that observed in B6-WT mice. H, DCFDA fluorescence emission in mouse and human DRG neurons is unaffected by Ang II (1 μm; 15 min) and CGP42112A (1 μm; 15 min) exposure, indicating no Ang II-induced ROS/RNS production. Data in all panels are presented as mean ± SEM. Numbers shown inside each column in all panels indicate the number of MΦs and DRG neurons in ≥4 culture batches from ≥4 mice (or human DRG culture batches for H) per group. ***p < 0.001, ###p < 0.001, and not significant (ns) versus respective vehicle (C) and indicated comparison groups (A, B and D, H), one-way ANOVA with Tukey's post hoc test.
Figure 12.
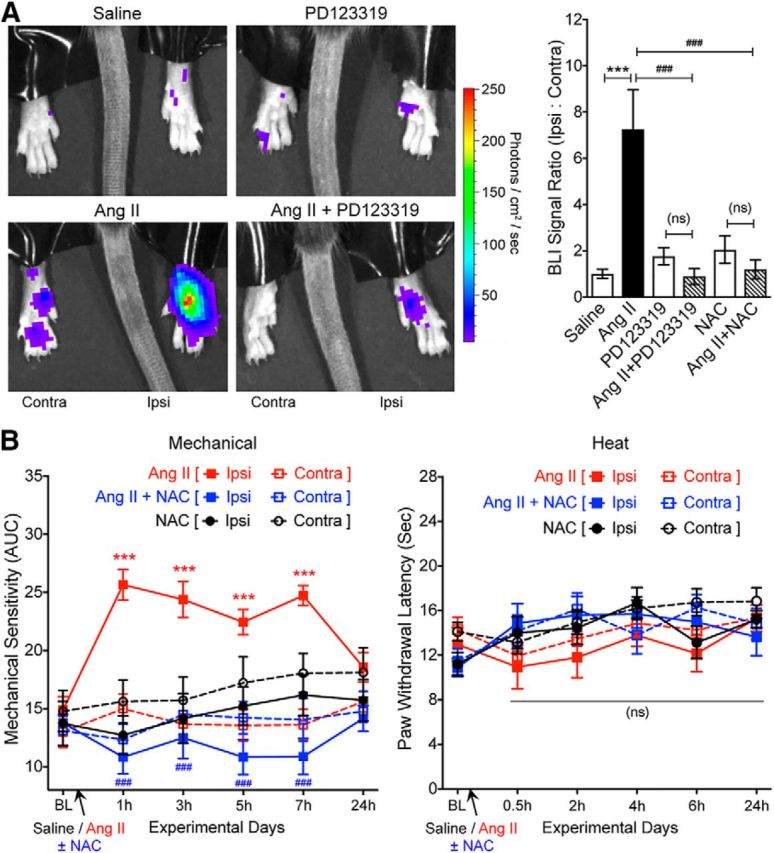
Ang II induces AT2R-dependent ROS/RNS production in mouse hindpaws and attenuation of Ang II-induced hindpaw mechanical hypersensitivity by ROS/RNS scavenging. A, Hindpaw injection of Ang II (100 pmol; 1 h) increases local ROS/RNS production, as determined by increased L-012 redox-sensitive dye luminescence intensity and quantified on the graph (right). Coinjection of PD123319 (10 pmol) and NAC (30 nmol) completely attenuate Ang II-induced ROS/RNS production. Data are presented as mean ± SEM (n = 5 mice per group). ***p < 0.001, ###p < 0.001, and not significant (ns) versus respective comparison groups, one-way ANOVA with Tukey's multiple-comparisons post hoc test. B, Coadministration of NAC (30 nmol, ipl) completely attenuates Ang II-induced (100 pmol, ipl) hindpaw mechanical hypersensitivity in mice. NAC administration does not influence hindpaw heat sensitivity. Data are presented as mean ± SEM (n = 9 per group). ***p < 0.001 versus Ang II/contra, ###p < 0.001 versus Ang II/ipsi groups, and not significant (ns) versus respective comparison groups, two-way ANOVA with Tukey's multiple-comparisons post hoc test.
MΦ AT2R-mediated redox activation of TRPA1 in sensory neurons
Because TRPA1 is a major target for ROS/RNS-mediated sensory neuron excitation (Andersson et al., 2008), we next verified whether Ang II-induced ROS/RNS production in MΦs could trans-activate TRPA1 on sensory neurons using cocultures of mouse DRG neurons and MΦs. Prolonged exposure of Ang II led to a slowly increasing/sustained [Ca2+]i elevation in mouse DRG neurons only upon coculturing with mouse peritoneal MΦs (Fig. 13A,B). We must mention here that, in our cocultures, the DRG neurons were not covered on all sides by MΦs with tight junctions, but rather were present in the vicinity of DRG neurons at a high density. Therefore, the ROIs in these live-cell imaging experiments were reliably restricted to the somata of DRG neurons only. Furthermore, at the end of the recording duration, 50 mm KCl (K50) was applied to confirm the identity of selected ROIs as neurons in these coculture Ca2+-imaging experiments. As observed in Ca2+-imaging experiments on cultured mouse peritoneal MΦs (Fig. 11A), non-neuronal cells adjacent to DRG neurons in cocultures did not exhibit any Ca2+ elevation upon Ang II and K50 applications. Ang II-induced [Ca2+]i elevation in MΦ-cocultured mouse DRG neurons was attenuated with coapplication of the ROS/RNS scavenger NAC (Fig. 13B), suggesting the involvement of ROS/RNS. Furthermore, application of a TRPA1 antagonist A967079 also attenuated the Ang II-induced sustained [Ca2+]i elevation in MΦ cocultured mouse DRG neurons (Fig. 13C), suggesting the involvement of TRPA1. Furthermore, we verified the involvement of AT2R in these experiments. Ang II exposure did not lead to a sustained increase in [Ca2+]i in WT mouse DRG neurons cocultured with peritoneal MΦs from Agtr2-KO mice (Fig. 13C). In contrast, Ang II exposure led to a sustained increase in [Ca2+]i in Agtr2-KO DRG neurons cocultured with Agtr2-WT peritoneal MΦs (Fig. 13C), indicating the critical involvement of MΦ AT2R in this MΦ-to-DRG neuron trans-signaling, without any functional deficit in DRG neurons from Agtr2-KO mice. In support of the latter assertion, we observed significant modulation of TRPA1- and TRPV1-mediated Ca2+ influx by bradykinin in DRG neurons from Agtr2-KO mice (Fig. 13D), as observed in WT mouse DRG neurons (Fig. 4C,D). In addition, we replicated similar Ang II-induced effects on neuronal trans-activation and Ca2+ flux in cocultures of mouse DRG neurons with the mouse MΦ cell line J774A.1, which exhibits functional AT2R expression and Ang II/AT2R-induced ROS/RNS production (Fig. 13E).
Figure 13.

Ang II-induced MΦ ROS/RNS production trans-activates TRPA1 on mouse DRG neurons. A, Representative traces of Ang II-induced (100 nm, 1 h) [Ca2+]i elevation in mouse DRG neurons observed only upon coculturing with mouse MΦs (both B6-WT mice). TRPA1+ neurons are identified by AITC (100 μm) and 50 mm KCl (K50). Area under the curve (AUC) for Ang II-induced [Ca2+]i elevation is subsequently quantified. B, C, Ang II-induced increases in DRG neuron [Ca2+]i elevation in cocultures can be completely attenuated upon coapplication of NAC (3 mm; B) and the TRPA1 antagonist A967079 (1 μm; C). Ang II (100 nm, 1 h) fails to induce [Ca2+]i elevation in FVB-Agtr2-WT DRG neurons cocultured with FVB-Agtr2-KO MΦs; however, increased [Ca2+]i is conserved in FVB-Agtr2-KO DRG neurons cocultured with FVB-Agtr2-WT MΦs (C). Data are presented as mean ± SEM. ***p < 0.001 and not significant (ns) versus respective comparison groups, one-way ANOVA with Tukey's post hoc test. D, Representative traces and quantification of bradykinin (BK; 100 nm; 5 min)-evoked potentiation of AITC-induced (50 μm) and capsaicin-induced (100 nm) Ca2+ flux in Agtr2-KO DRG neurons. Data are presented as mean ± SEM. *p < 0.05 versus control groups, one-way ANOVA with Tukey's post hoc test. E, Top left, Representative agarose gel electrophoresis images of RT-PCR amplification of AT1R and AT2R genes (Agtr1a and Agtr2) from total RNA isolated from the mouse monocyte-MΦ cell line J774A.1. Gapdh amplification is used as a positive control. Numbers on the left denote DNA molecular weight markers (in base pairs). Bottom left, Western blot images demonstrating increased Erk1/2 phosphorylation in J774A.1 cells treated with Ang II (100 nm; 30 min), the AT2R-selective agonist CGP42112A (100 nm; 30 min), or TNF-α (10 nm; 30 min) as a positive control. The Ang II-mediated increase in p-ERK1/2 is inhibited by coapplication of PD123319 (1 μm), but not losartan (1 μm). Numbers on the left denote protein molecular weight markers (in kilodaltons). Right, J774A.1 cells demonstrate AT2R-dependent increased DCFDA fluorescence, which is inhibited by PD123319 (1 μm) or the antioxidant NAC (3 mm), but not losartan (1 μm). Data are presented as mean ± SEM. ***p < 0.001 versus vehicle, ###p < 0.001 versus Ang II. F, Ang II (100 nm, 1 h) induces increase in DRG neuron [Ca2+]i elevation upon coculturing with J774A.1 MΦ cells. Data in B–F are presented as mean ± SEM and numbers shown inside each column in these panels indicate the number of DRG neurons (and MΦs for E) in ≥4 culture batches from ≥4 mice/group. **p < 0.05, ***p < 0.001, ###p < 0.001, and not significant (ns) versus the indicated comparison groups, one-way ANOVA with Tukey's post hoc test.
Consistent with observations in mouse DRG neuron-MΦ cocultures, human DRG neurons were cocultured with the U937 human monocyte-MΦ cell line to verify a similar trans-activation phenomenon. We first verified that U937 MΦs exhibit functional AT2R expression and Ang II-induced ROS/RNS production that is sensitive to PD123319 and NAC, but not to losartan (Fig. 14A). Coculture of human DRG neurons with U937 MΦs exhibited similar Ang II/MΦ-dependent increases in [Ca2+]i in neurons (Fig. 14B). Ang II-induced increases in [Ca2+]i in human DRG neurons cocultured with U937 MΦs was inhibited by coapplication of an AT2R antagonist (PD123319) or TRPA1 antagonist (A967079) (Fig. 14B). ROS/RNS are thought to activate TRPA1 by covalent modification of cysteine residues in the N terminus of the channel (Macpherson et al., 2007a; Andersson et al., 2008; Takahashi et al., 2008). To test whether this underlies Ang II/ MΦ-dependent increases in [Ca2+]i in coculture, we transiently transfected HEK293 cells with plasmids containing either WT human TRPA1 cDNA or a mutant hTRPA1 cDNA in which three key cysteine residues (Cys421, Cys621, and Cys655) are mutated to serine (hTRPA1–3C/S), rendering the channel insensitive to covalent modification-based activation (Macpherson et al., 2007a). HEK cells transfected with eGFP alone showed no significant increase in [Ca2+]i regardless of the presence of Ang II or coculture with U937 cells (Fig. 14C). hTRPA1-WT-expressing cells exhibit Ang II-dependent increased [Ca2+]i, which, as seen in mouse and human DRG neurons, occurred only under MΦ coculture conditions (Fig. 14C). This effect was absent in TRPA1–3C/S-transfected HEK cells (Fig. 14C), indicating a requirement for covalent modification of cysteine residues in the channel protein downstream of MΦ ROS/RNS production to elicit TRPA1 channel activation and Ca2+ flux.
Figure 14.
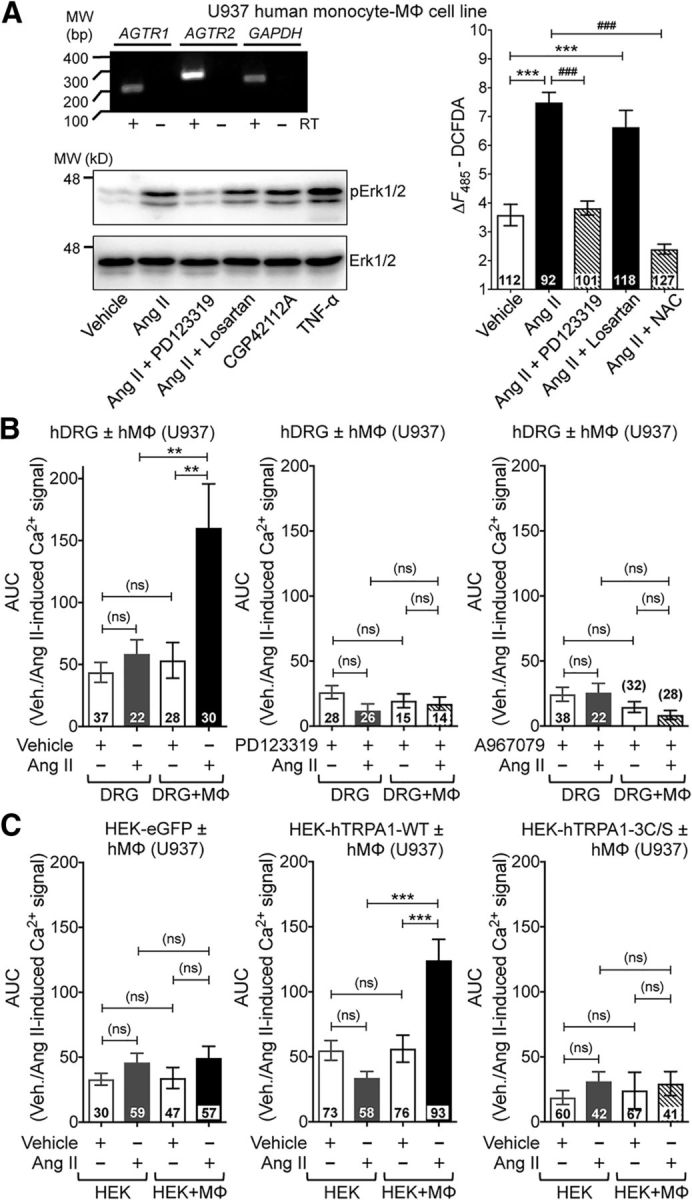
Ang II-induced MΦ ROS/RNS production in U937 human MΦ cells trans-activates TRPA1 on human DRG neurons. A, Top left, Representative agarose gel electrophoresis images of RT-PCR amplification of AT1R and AT2R genes (AGTR1 and AGTR2) from total RNA isolated from the human monocyte-MΦ cell line U937. GAPDH amplification is used as a positive control. Numbers on the left denote DNA molecular weight markers (in base pairs). Bottom left, Western blot images demonstrating increased Erk1/2 phosphorylation in U937 cells treated with Ang II (100 nm; 30 min), the AT2R-selective agonist CGP42112A (100 nm; 30 min), or TNF-α (10 nm; 30 min) as a positive control. The Ang II-mediated increase in p-ERK1/2 is inhibited by coapplication of PD123319 (1 μm), but not losartan (1 μm). Numbers on the left denote protein molecular weight markers (in kilodaltons). Right, U937 cells demonstrate AT2R-dependent increased DCFDA fluorescence, which is inhibited by PD123319 (1 μm) or the antioxidant NAC (3 mm), but not losartan (1 μm). B, Ang II (100 nm, 1 h) induces significant elevation of [Ca2+]i levels in human DRG neurons upon coculturing with the U937 human MΦ cell line, which can be completely attenuated upon coapplication of the AT2R antagonist PD123319 (1 μm) and the TRPA1 antagonist A967079 (1 μm). C, Ang II (100 nm, 1 h) induces a significant elevation in [Ca2+]i levels in HEK293 cells transfected with eGFP+hTRPA1-WT, but not with eGFP alone or eGFP+hTRPA1–3C/S mutant cDNAs, upon coculturing with U937 human MΦ cell line. Data in all panels are presented as mean ± SEM and numbers shown inside each column indicate the number of MΦs (A) and DRG neurons (B, C) in ≥4 human DRG culture batches per group. **p < 0.05, ###p < 0.001, and not significant (ns) versus the indicated comparison groups, one-way ANOVA with Tukey's post hoc test.
Together, these results suggest that Ang II activates MΦ AT2R to elicit ROS/RNS production, which then trans-activates TRPA1 on sensory neurons. This signaling axis is targeted by the AT2R antagonist PD123319, which dampens ROS/RNS production, representing a mechanism by which nociceptor excitation could be attenuated to provide relief from chronic pain conditions observed in experimental rodent models (Chakrabarty et al., 2013, 2018; Smith et al., 2016; Liao et al., 2017) and in humans (Rice et al., 2014).
Discussion
Our findings demonstrate that intercellular crosstalk between peripheral MΦs and sensory neurons, mediated by AT2R-to-TRPA1 redox signaling, constitutes a critical mechanism for peripheral pain sensitization. Analgesic efficacy of the AT2R antagonist EMA401 for neuropathic pain associated with PHN in humans has been shown recently (Rice et al., 2014). Prior studies have suggested that Ang II acts directly on DRG neurons to induce neurite outgrowth and modulation of TRPV1 function via Gαs-coupled AT2R-PKA signaling, resulting in peripheral pain sensitization (Danser and Anand, 2014; Anand et al., 2015). Conversely, activation of Gαi/o-coupled AT2R on sensory neurons by a bacterial mycolactone toxin has been reported to exert analgesic effects in mice (Danser and Anand, 2014; Marion et al., 2014). However, our in-depth investigations show that mouse and human DRG neurons do not express AT2R, nor did we observe any direct influence of Ang II on sensory neuron function. Our study reveals that Ang II induces infiltration of MΦs, and MΦ AT2R activation triggers ROS/RNS production, which subsequently trans-activates TRPA1 channel on sensory neurons. By uncovering how angiotensin signaling drives peripheral pain sensitization, we have identified a mechanism for peripheral pain sensitization and cellular/molecular targets underlying the analgesic effectiveness of AT2R antagonists.
We demonstrate that antagonism of AT2R, but not AT1R, could attenuate local Ang II-induced mechanical hypersensitivity. PD123319 and EMA401 (used in a clinical trial) have 3000- and 10,000-fold selectivity, respectively, for AT2R over AT1R and EMA401 has been shown to have an approximately twofold stronger binding affinity for rodent and human AT2R over PD123319. Furthermore, both these antagonists were highly effective in attenuating pain hypersensitivity in rodent experimental models without any visible nonspecific effects (Blankley et al., 1991; Smith et al., 2013a,b; 2016). Previous studies have shown attenuation of both heat and mechanical hypersensitivity by the same AT2R antagonist in CFA-induced chronic inflammation (Chakrabarty et al., 2013, 2018), experimental neuropathy (Smith et al., 2013b), and bone cancer pain (Muralidharan et al., 2014) models in rodents. At 10 mg/kg dose, PD123319 produces saturating attenuation of mechanical hypersensitivity in these rodent experimental pain conditions without any gross physiological effects, suggesting the target specificity and engagement of AT2R. TRPV1, the critical nociceptive target for heat hypersensitivity (Caterina et al., 2000), has been implicated as the nociceptive target downstream of AT2R activation on sensory neurons (Anand et al., 2013, 2015; Smith et al., 2016). However, our study conclusively shows no involvement of Ang II in heat sensitivity in mice and direct Ang II/AT2R signaling is absent in both mouse and human DRG neurons. Furthermore, Ang II has no influence on TRPV1 channel function and no expression of AT2R was detected in mouse and human DRG neurons, which supports and justifies our observations regarding the lack of Ang II-induced heat hypersensitivity. Furthermore, TRPA1 and TRPV4, but not TRPV1, have been suggested to mediate mechanical hypersensitivity in rodent models of inflammation and experimental neuropathy (Kwan et al., 2006; Petrus et al., 2007; Alessandri-Haber et al., 2008; Eid et al., 2008; Chen et al., 2011; Ho et al., 2012; Mickle et al., 2016). Our study found that only TRPA1, not TRPV1 or TRPV4, is required for Ang II-induced mechanical hypersensitivity. However, we observed no functional evidence of direct TRPA1 modulation by Ang II in mouse or human DRG sensory neurons, nor did we observe, in contrast to prior observations (Marion et al., 2014), any Ang II-induced changes in neuronal membrane potential. This may be explained by the different origin of the cells used, PC12 cells and hippocampal neurons (Marion et al., 2014), versus mouse and human DRG sensory neurons in this study. Instead, our findings suggest that peripheral intercellular signaling is critically involved in pain sensitization via MΦ AT2R-ROS/RNS-mediated trans-activation of TRPA1 on sensory neurons. TRPA1 can be activated by mechanical force and cell damage responses, including ROS/RNS (Macpherson et al., 2007b; Trevisani et al., 2007; Andersson et al., 2008). It must be noted here that Ang II-induced trans-activation of TRPA1-mediated Ca2+ flux in our coculture experiments was relatively slow compared with previous reports on ROS activation of TRPA1 in DRG neurons (Macpherson et al., 2007a; Andersson et al., 2008; Takahashi et al., 2008). Higher concentrations of ROS (H2O2 and/or 4-HNE) were used (high micromolar range) as agonists for TRPA1 activation in prior reports (Macpherson et al., 2007a; Trevisani et al., 2007; Andersson et al., 2008; Takahashi et al., 2008), leading to rapid and robust activation of the channel, whereas, in DRG neuron/MΦ cocultures, the concentration of cellular ROS/RNS levels produced from MΦs in response to Ang II application could be of much lesser extent. In addition, Ang II exposure leads to a slow and steady increase in ROS/RNS in MΦs (Fig. 11B), which would be followed by the secretion and subsequent trans-activation of TRPA1 on DRG neurons (Fig. 13A). Therefore, in MΦ/DRG cocultures, a time-dependent cellular ROS/RNS production in MΦs upon Ang II exposure presumably leads to subthreshold TRPA1 channel activation, which could underlie a slowly increasing/sustained [Ca2+]i elevation in DRG neurons. Furthermore, TRPA1 has been shown to have a U-shaped temperature dependence of channel activation and modulation by H2O2 (Moparthi et al., 2016), which could also influence the time course and/or kinetics of ROS/RNS-mediated Ca2+ flux through the channel in MΦ/DRG cocultures. ROS/RNS are known to activate TRPA1 via covalent modification of Cys residues (Macpherson et al., 2007a; Trevisani et al., 2007). In agreement with these findings, we report that mutation of three critical Cys residues in hTRPA1 was associated with a loss of Ang II-induced sensory neuron Ca2+ flux upon coculture with MΦs. In summary, our study using cocultures of primary MΦs and DRG neurons shows that AT2R on MΦs and TRPA1 on DRG neurons are critical components of a peripheral immunosensory neuron crosstalk, which is conserved in rodents and humans at a cellular level.
Infiltration of a variety of immune cells, including MΦs, at the site of injury and in DRGs have been reported in multiple pathological pain models in rodents (Ristoiu, 2013; Ghasemlou et al., 2015; Trevisan et al., 2016; Chakrabarty et al., 2018). However, hindpaw Ang II injection led to local MΦ infiltration in the skin without any influence on microglia/MΦs in the DRG and spinal cord. Increased peripheral MΦ infiltration and expression of renin–angiotensin system (RAS) components therein, such as angiotensinogen, renin, angiotensin-converting enzyme, and AT2R, have been shown in rodent models of CFA-induced inflammation and vestibulodynia (Chakrabarty et al., 2013, 2018; Liao et al., 2017). Furthermore, increased expression of RAS components, including AT2R, has been shown to accompany the differentiation of MΦs from monocytes (Okamura et al., 1999). Our study shows that Ang II injection into mouse hindpaws leads to local infiltration of neutrophils and MΦs and AT2R is functionally expressed in skin MΦs, but not in neutrophils. In addition, no expression of AT2R is detected on any cell types in DRGs, as supported by RNAseq data and immunostaining for GFP expression in Agtr2GFP mouse DRGs. Furthermore, our study found increased density of GFP-expressing MΦs in the hindpaw skin of Ang II-injected Agtr2GFP mice both at 1 and 5 h after injection, indicative of MΦ infiltration, but not division/expansion of resident MΦs, whereas no alteration in the density of DRG and spinal cord microglia/MΦs and no GFP expression was observed therein. This suggests that infiltrating MΦs at the site of Ang II injection represent critical immune cell components underlying mechanical pain hypersensitivity. Interestingly, intrathecal administration of Ang II in mice did not induce mechanical hypersensitivity. Furthermore, our study shows that chemogenetic depletion of peripheral/skin MΦs, which spares microglia/MΦs in the DRG and spinal cord, completely attenuated Ang II-induced mechanical pain hypersensitivity in mice. This again confirms that peripheral/skin MΦs, but not DRG or spinal cord microglia/MΦs, are an indispensable component for Ang II-induced pain hypersensitivity. MΦ/microglia-derived inflammatory mediators, growth factors, and spinal modulatory signaling have been suggested as predominant nociceptor-sensitizing factors in pain (Kuner, 2010; Old et al., 2014; Trevisan et al., 2016; Weaver et al., 2017). Our findings indicate that MΦ Ang II-AT2R signaling leads to ROS/RNS production that trans-activates TRPA1 on sensory neurons to elicit nociceptor excitation and pain. Further studies are thus warranted to determine any interdependence of MΦ/microglia-derived inflammatory mediators, growth factors, and spinal modulatory signaling with AT2R-ROS/RNS signaling in MΦs in relation to specific pathological pain conditions.
Our study comprehensively shows functional expression of AT2R in MΦs and that its activation by Ang II increases cellular ROS/RNS production that is dependent on AT2R. Furthermore, we directly demonstrate increased local AT2R-dependent ROS/RNS production in vivo upon Ang II injection. Accordingly, the ROS/RNS scavenger NAC completely attenuates Ang II-induced mechanical hypersensitivity. AT2R has been shown to activate NADPH oxidase (NOX) (Park et al., 2013), which plays a critical role in ROS production, as well as cGMP/nitric oxide synthase-mediated production of RNS (Ewert et al., 2003). NOX2 constitutes the predominant MΦ NOX isoform (Lambeth, 2004). Prior studies have shown local elevation of H2O2 levels in injured sciatic nerves in mice and NOX2-deficient mice exhibit diminished mechanical hypersensitivity in response to peripheral nerve injury (Kim et al., 2010; Kallenborn-Gerhardt et al., 2014). Several antioxidants have been proposed as alternative therapeutics for multiple neuropathic and inflammatory pain conditions (Onysko et al., 2015; Carr and McCall, 2017; Drewes et al., 2017). Importantly, our study demonstrates that local MΦ Ang II/AT2R signaling serves as a critical source of oxidative stress. Therefore, AT2R antagonists exert analgesic effects, presumably via blockade of MΦ-derived ROS/RNS production, and thus attenuate pathological excitation of both mouse and human sensory neurons. Furthermore, oxidative and nitrosative stress have also been shown to induce mitochondrial dysfunction and nerve fiber degeneration (Vincent et al., 2011). Because Ang II/AT2R activation on MΦs is the predominant source of ROS/RNS production, it is plausible that AT2R inhibition could also play a neuroprotective role, which may also contribute in part to its analgesic efficacy.
The performance of existing analgesics for diverse chronic pain conditions are suboptimal and the success rate of new-generation analgesic drug development has proven insufficient (Percie du Sert and Rice, 2014; Yekkirala et al., 2017). This necessitates a comprehensive understanding of the pathophysiology and mechanisms underlying distinct chronic pain conditions. The recent success of the AT2R antagonist EMA401 in a phase II clinical trial for treatment of neuropathic pain associated with PHN (Rice et al., 2014) has led us to back-translate the mechanisms underlying angiotensin signaling in pain sensitization. Our findings implicate peripheral MΦ AT2R-mediated oxidative/nitrosative stress as a critical trigger for TRPA1 activation on sensory neurons, which constitutes a mechanism for peripheral nociceptive sensitization in pathological pain states.
Footnotes
The vast majority of this study was supported by funds from the Washington University Pain Center and Washington University School of Medicine, Department of Anesthesiology. Additional funding sources that supported this study are as follows: Pilot and Feasibility Grant from the Washington University Nutrition Obesity Research Center NIH Grant P30DK056341 (A.J.S.), NIH Grant NS069898 (D.P.M.); NIH Grant CA171927 (A.D.M.); Danish Diabetes Academy, supported by the Novo Nordisk Foundation (P.K.); NIH Grants HL125805 (A.D.d.K.); NIH Grants DK102520 and U01DK101039 (S.J.); NIH Grant NS072432 (Y.M.U.); NIH Grant NS065926 (T.J.P.); University of Texas STARS funding (T.J.P. and G.D.); and NIH Grant NS042595 (R.W.G.). We thank Samantha Kelly, Sherri Vogt, and Masato Hoshi for technical assistance; Justin Grobe, Nicole Littlejohn, and Carmen Halabi for help with Agtr2-WT and Agtr2-KO mouse breeding; Eric Tycksen from the Genome Technology Access Center (GTAC), Washington University in St. Louis (NIH-CA91842 and UL1TR000448) for assistance; Andrew Torck and Matthew Neiman (University of Texas, Dallas) for executing the RNA-seq data quantification and analysis; Troels Staehelin and Jens Randel Nyengaard for input and support on human skin biopsy experiments; Wolfgang Liedtke for providing Trpv4-KO mice; Curt D. Sigmund and Justin Grobe for providing Agtr2-KO mice (originally generated by Drs. Victor J. Dzau, and Richard E. Pratt); Sven Eric Jordt and Michael J.M. Fischer for providing human TRPA1 wild-type and mutant cDNAs; Mid-America Transplant, AnaBios, and the families of human DRG donors used in this study; and Joseph Vogel for providing the U937 human MΦ cell line (originally obtained from the ATCC).
The authors declare no competing financial interests.
References
- Alessandri-Haber N, Dina OA, Joseph EK, Reichling DB, Levine JD (2008) Interaction of transient receptor potential vanilloid 4, integrin, and SRC tyrosine kinase in mechanical hyperalgesia. J Neurosci 28:1046–1057. 10.1523/JNEUROSCI.4497-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An D, Kim K, Lu W (2014) Defective entry into mitosis 1 (Dim1) negatively regulates osteoclastogenesis by inhibiting the expression of nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1 (NFATc1). J Biol Chem 289:24366–24373. 10.1074/jbc.M114.563817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand U, Facer P, Yiangou Y, Sinisi M, Fox M, McCarthy T, Bountra C, Korchev YE, Anand P (2013) Angiotensin II type 2 receptor (AT2R) localization and antagonist-mediated inhibition of capsaicin responses and neurite outgrowth in human and rat sensory neurons. Eur J Pain 17:1012–1026. 10.1002/j.1532-2149.2012.00269.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand U, Yiangou Y, Sinisi M, Fox M, MacQuillan A, Quick T, Korchev YE, Bountra C, McCarthy T, Anand P (2015) Mechanisms underlying clinical efficacy of angiotensin II type 2 receptor (AT2R) antagonist EMA401 in neuropathic pain: clinical tissue and in vitro studies. Mol Pain 11:38. 10.1186/s12990-015-0038-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand U, Sinisi M, Fox M, MacQuillan A, Quick T, Korchev Y, Bountra C, McCarthy T, Anand P (2016) Mycolactone-mediated neurite degeneration and functional effects in cultured human and rat DRG neurons: mechanisms underlying hypoalgesia in Buruli ulcer. Mol Pain 12: pii: 1744806916654144. 10.1177/1744806916654144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DA, Gentry C, Moss S, Bevan S (2008) Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci 28:2485–2494. 10.1523/JNEUROSCI.5369-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik RK, Woo YC, Park SS, Brennan TJ (2006) Strain and sex influence on pain sensitivity after plantar incision in the mouse. Anesthesiology 105:1246–1253. 10.1097/00000542-200612000-00025 [DOI] [PubMed] [Google Scholar]
- Blankley CJ, Hodges JC, Klutchko SR, Himmelsbach RJ, Chucholowski A, Connolly CJ, Neergaard SJ, Van Nieuwenhze MS, Sebastian A, Quin J 3rd (1991) Synthesis and structure-activity relationships of a novel series of non-peptide angiotensin II receptor binding inhibitors specific for the AT2 subtype. J Med Chem 34:3248–3260. 10.1021/jm00115a014 [DOI] [PubMed] [Google Scholar]
- Bosnyak S, Jones ES, Christopoulos A, Aguilar MI, Thomas WG, Widdop RE (2011) Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci 121:297–303. 10.1042/CS20110036 [DOI] [PubMed] [Google Scholar]
- Burnett SH, Beus BJ, Avdiushko R, Qualls J, Kaplan AM, Cohen DA (2006) Development of peritoneal adhesions in macrophage depleted mice. J Surg Res 131:296–301. 10.1016/j.jss.2005.08.026 [DOI] [PubMed] [Google Scholar]
- Carr AC, McCall C (2017) The role of vitamin C in the treatment of pain: new insights. J Transl Med 15:77. 10.1186/s12967-017-1179-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288:306–313. 10.1126/science.288.5464.306 [DOI] [PubMed] [Google Scholar]
- Chakrabarty A, Liao Z, Smith PG (2013) Angiotensin II receptor type 2 activation is required for cutaneous sensory hyperinnervation and hypersensitivity in a rat hind paw model of inflammatory pain. J Pain 14:1053–1065. 10.1016/j.jpain.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty A, Liao Z, Mu Y, Smith PG (2018) Inflammatory renin-angiotensin system disruption attenuates sensory hyperinnervation and mechanical hypersensitivity in a rat model of provoked vestibulodynia. J Pain 19:264–277. 10.1016/j.jpain.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Chen TC, Lee CT, Yang CY, Wang HW, Wang CC, Hsieh SL (2008) Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood 111:5054–5063. 10.1182/blood-2007-12-130609 [DOI] [PubMed] [Google Scholar]
- Chen J, Joshi SK, DiDomenico S, Perner RJ, Mikusa JP, Gauvin DM, Segreti JA, Han P, Zhang XF, Niforatos W, Bianchi BR, Baker SJ, Zhong C, Simler GH, McDonald HA, Schmidt RG, McGaraughty SP, Chu KL, Faltynek CR et al. (2011) Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 152:1165–1172. 10.1016/j.pain.2011.01.049 [DOI] [PubMed] [Google Scholar]
- Cridland RA, Henry JL (1988) Effects of intrathecal administration of neuropeptides on a spinal nociceptive reflex in the rat: VIP, galanin, CGRP, TRH, somatostatin and angiotensin II. Neuropeptides 11:23–32. 10.1016/0143-4179(88)90024-8 [DOI] [PubMed] [Google Scholar]
- Danser AH, Anand P (2014) The angiotensin II type 2 receptor for pain control. Cell 157:1504–1506. 10.1016/j.cell.2014.05.030 [DOI] [PubMed] [Google Scholar]
- Davidson S, Golden JP, Copits BA, Ray PR, Vogt SK, Valtcheva MV, Schmidt RE, Ghetti A, Price TJ, Gereau RW 4th (2016) Group II mGluRs suppress hyperexcitability in mouse and human nociceptors. Pain 157:2081–2088. 10.1097/j.pain.0000000000000621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deen M, Correnti E, Kamm K, Kelderman T, Papetti L, Rubio-Beltrán E, Vigneri S, Edvinsson L, Maassen Van Den Brink A, Maassen Van Den Brink A (2017) Blocking CGRP in migraine patients: a review of pros and cons. J Headache Pain 18:96. 10.1186/s10194-017-0807-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T (2000) International union of pharmacology. XXIII. the angiotensin II receptors. Pharmacol Rev 52:415–472. [PubMed] [Google Scholar]
- de Kloet AD, Wang L, Ludin JA, Smith JA, Pioquinto DJ, Hiller H, Steckelings UM, Scheuer DA, Sumners C, Krause EG (2016) Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct Funct 221:891–912. 10.1007/s00429-014-0943-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhande I, Ma W, Hussain T (2015) Angiotensin AT2 receptor stimulation is anti-inflammatory in lipopolysaccharide-activated THP-1 macrophages via increased interleukin-10 production. Hypertens Res 38:21–29. 10.1038/hr.2014.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes AM, Bouwense SAW, Campbell CM, Ceyhan GO, Delhaye M, Demir IE, Garg PK, van Goor H, Halloran C, Isaji S, Neoptolemos JP, Olesen SS, Palermo T, Pasricha PJ, Sheel A, Shimosegawa T, Szigethy E, Whitcomb DC, Yadav D; Working group for the International (IAP – APA – JPS – EPC) Consensus Guidelines for Chronic Pancreatitis (2017) Guidelines for the understanding and management of pain in chronic pancreatitis. Pancreatology 17:720–731. 10.1016/j.pan.2017.07.006 [DOI] [PubMed] [Google Scholar]
- Eid SR, Crown ED, Moore EL, Liang HA, Choong KC, Dima S, Henze DA, Kane SA, Urban MO (2008) HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain 4:48. 10.1186/1744-8069-4-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewert S, Laesser M, Johansson B, Holm M, Aneman A, Fandriks L (2003) The angiotensin II receptor type 2 agonist CGP 42112A stimulates NO production in the porcine jejunal mucosa. BMC Pharmacol 3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte BL, Slosky LM, Zhang H, Arnold MR, Staatz WD, Hay M, Largent-Milnes TM, Vanderah TW (2016) Angiotensin-(1–7)/Mas receptor as an antinociceptive agent in cancer-induced bone pain. Pain 157:2709–2721. 10.1097/j.pain.0000000000000690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron L, Laflamme L, Rivard N, Asselin C, Payet MD, Gallo-Payet N (1999) Signals from the AT2 (angiotensin type 2) receptor of angiotensin II inhibit p21ras and activate MAPK (mitogen-activated protein kinase) to induce morphological neuronal differentiation in NG108–15 cells. Mol Endocrinol 13:1615–1626. 10.1210/mend.13.9.0344 [DOI] [PubMed] [Google Scholar]
- Ghasemlou N, Chiu IM, Julien JP, Woolf CJ (2015) CD11b+Ly6G- myeloid cells mediate mechanical inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A 112:E6808–E6817. 10.1073/pnas.1501372112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleissner CA, Shaked I, Little KM, Ley K (2010) CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol 184:4810–4818. 10.4049/jimmunol.0901368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami SC, Mishra SK, Maric D, Kaszas K, Gonnella GL, Clokie SJ, Kominsky HD, Gross JR, Keller JM, Mannes AJ, Hoon MA, Iadarola MJ (2014) Molecular signatures of mouse TRPV1-lineage neurons revealed by RNA-seq transcriptome analysis. J Pain 15:1338–1359. 10.1016/j.jpain.2014.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte RP, Kadunganattil S, Shepherd AJ, Merrill R, Planer W, Bruchas MR, Strack S, Mohapatra DP (2016) Convergent phosphomodulation of the major neuronal dendritic potassium channel Kv4.2 by pituitary adenylate cyclase-activating polypeptide. Neuropharmacology 101:291–308. 10.1016/j.neuropharm.2015.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafko R, Villapol S, Nostramo R, Symes A, Sabban EL, Inagami T, Saavedra JM (2013) Commercially available angiotensin II At(2) receptor antibodies are nonspecific. PLoS One 8:e69234. 10.1371/journal.pone.0069234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein L, Barsh GS, Pratt RE, Dzau VJ, Kobilka BK (1995) Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor in mice. Nature 377:744–747. 10.1038/377744a0 [DOI] [PubMed] [Google Scholar]
- Ho TC, Horn NA, Huynh T, Kelava L, Lansman JB (2012) Evidence TRPV4 contributes to mechanosensitive ion channels in mouse skeletal muscle fibers. Channels 6:246–254. 10.4161/chan.20719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallenborn-Gerhardt W, Hohmann SW, Syhr KM, Schröder K, Sisignano M, Weigert A, Lorenz JE, Lu R, Brune B, Brandes RP, Geisslinger G, Schmidtko A (2014) Nox2-dependent signaling between macrophages and sensory neurons contributes to neuropathic pain hypersensitivity. Pain 155:2161–2170. 10.1016/j.pain.2014.08.013 [DOI] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW 4th (2001) Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci 21:3771–3779. 10.1523/JNEUROSCI.21-11-03771.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson P, Nyengaard JR, Polydefkis M, Jensen TS (2015) Structural and functional assessment of skin nerve fibres in small-fibre pathology. Eur J Pain 19:1059–1070. 10.1002/ejp.645 [DOI] [PubMed] [Google Scholar]
- Khoury-Hanold W, Iwasaki A (2016) Transcriptome analysis of HSV-1 infected dorsal root ganglia and large intestinal musculara. Available from: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74215.
- Kielland A, Blom T, Nandakumar KS, Holmdahl R, Blomhoff R, Carlsen H (2009) In vivo imaging of reactive oxygen and nitrogen species in inflammation using the luminescent probe L-012. Free Radic Biol Med 47:760–766. 10.1016/j.freeradbiomed.2009.06.013 [DOI] [PubMed] [Google Scholar]
- Kim D, You B, Jo EK, Han SK, Simon MI, Lee SJ (2010) NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc Natl Acad Sci U S A 107:14851–14856. 10.1073/pnas.1009926107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner R. (2010) Central mechanisms of pathological pain. Nat Med 16:1258–1266. 10.1038/nm.2231 [DOI] [PubMed] [Google Scholar]
- Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP (2006) TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron 50:277–289. 10.1016/j.neuron.2006.03.042 [DOI] [PubMed] [Google Scholar]
- Lambeth JD. (2004) NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4:181–189. 10.1038/nri1312 [DOI] [PubMed] [Google Scholar]
- Liao Z, Chakrabarty A, Mu Y, Bhattacherjee A, Goestch M, Leclair CM, Smith PG (2017) A local inflammatory renin-angiotensin system drives sensory axon sprouting in provoked vestibulodynia. J Pain 18:511–525. 10.1016/j.jpain.2016.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Friedman JM (2003) Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci U S A 100:13698–13703. 10.1073/pnas.1735416100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo L, Shepherd AJ, Mickle AD, Lorca RA, Shutov LP, Usachev YM, Mohapatra DP (2012) The C-type natriuretic peptide induces thermal hyperalgesia through a noncanonical gbetagamma-dependent modulation of TRPV1 channel. J Neurosci 32:11942–11955. 10.1523/JNEUROSCI.1330-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A (2007a) Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445:541–545. 10.1038/nature05544 [DOI] [PubMed] [Google Scholar]
- Macpherson LJ, Xiao B, Kwan KY, Petrus MJ, Dubin AE, Hwang S, Cravatt B, Corey DP, Patapoutian A (2007b) An ion channel essential for sensing chemical damage. J Neurosci 27:11412–11415. 10.1523/JNEUROSCI.3600-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion E, Song OR, Christophe T, Babonneau J, Fenistein D, Eyer J, Letournel F, Henrion D, Clere N, Paille V, Guérineau NC, Saint André JP, Gersbach P, Altmann KH, Stinear TP, Comoglio Y, Sandoz G, Preisser L, Delneste Y, Yeramian E, et al. (2014) Mycobacterial toxin induces analgesia in buruli ulcer by targeting the angiotensin pathways. Cell 157:1565–1576. 10.1016/j.cell.2014.04.040 [DOI] [PubMed] [Google Scholar]
- Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Theurich S, Hausen AC, Schmitz J, Brönneke HS, Estevez E, Allen TL, Mesaros A, Partridge L, Febbraio MA, Chawla A, Wunderlich FT, Bruning JC (2014) Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol 15:423–430. 10.1038/ni.2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickle AD, Shepherd AJ, Mohapatra DP (2015a) Sensory TRP channels: the key transducers of nociception and pain. Prog Mol Biol Transl Sci 131:73–118. 10.1016/bs.pmbts.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickle AD, Shepherd AJ, Loo L, Mohapatra DP (2015b) Induction of thermal and mechanical hypersensitivity by parathyroid hormone-related peptide through upregulation of TRPV1 function and trafficking. Pain 156:1620–1636. 10.1097/j.pain.0000000000000224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickle AD, Shepherd AJ, Mohapatra DP (2016) Nociceptive TRP channels: sensory detectors and transducers in multiple pain pathologies. Pharmaceuticals (Basel) 9: pii: E72. 10.3390/ph9040072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RE, Malfait AM, Block JA (2017) Current status of nerve growth factor antibodies for the treatment of osteoarthritis pain. Clin Exp Rheumatol 35:85–87. [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C (2003) Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J Biol Chem 278:50080–50090. 10.1074/jbc.M306619200 [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C (2005) Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem 280:13424–13432. 10.1074/jbc.M410917200 [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Wang SY, Wang GK, Nau C (2003) A tyrosine residue in TM6 of the vanilloid receptor TRPV1 involved in desensitization and calcium permeability of capsaicin-activated currents. Mol Cell Neurosci 23:314–324. 10.1016/S1044-7431(03)00054-X [DOI] [PubMed] [Google Scholar]
- Moore RA, Wiffen PJ, Derry S, Toelle T, Rice AS (2014) Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev 4:CD007938. 10.1002/14651858.CD007938.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moparthi L, Kichko TI, Eberhardt M, Högestätt ED, Kjellbom P, Johanson U, Reeh PW, Leffler A, Filipovic MR, Zygmunt PM (2016) Human TRPA1 is a heat sensor displaying intrinsic U-shaped thermosensitivity. Sci Rep 6:28763. 10.1038/srep28763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan A, Wyse BD, Smith MT (2014) Analgesic efficacy and mode of action of a selective small molecule angiotensin II type 2 receptor antagonist in a rat model of prostate cancer-induced bone pain. Pain Med 15:93–110. 10.1111/pme.12258 [DOI] [PubMed] [Google Scholar]
- Okamura A, Rakugi H, Ohishi M, Yanagitani Y, Takiuchi S, Moriguchi K, Fennessy PA, Higaki J, Ogihara T (1999) Upregulation of renin-angiotensin system during differentiation of monocytes to macrophages. J Hypertens 17:537–545. 10.1097/00004872-199917040-00012 [DOI] [PubMed] [Google Scholar]
- Old EA, Nadkarni S, Grist J, Gentry C, Bevan S, Kim KW, Mogg AJ, Perretti M, Malcangio M (2014) Monocytes expressing CX3CR1 orchestrate the development of vincristine-induced pain. J Clin Invest 124:2023–2036. 10.1172/JCI71389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onysko M, Legerski P, Potthoff J, Erlandson M (2015) Targeting neuropathic pain: consider these alternatives. J Fam Pract 64:470–475. [PubMed] [Google Scholar]
- Park MH, Kim HN, Lim JS, Ahn JS, Koh JY (2013) Angiotensin II potentiates zinc-induced cortical neuronal death by acting on angiotensin II type 2 receptor. Mol Brain 6:50. 10.1186/1756-6606-6-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A, Tate S, Woolf CJ (2009) Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Discov 8:55–68. 10.1038/nrd2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert N, Rice AS (2014) Improving the translation of analgesic drugs to the clinic: animal models of neuropathic pain. Br J Pharmacol 171:2951–2963. 10.1111/bph.12645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrus M, Peier AM, Bandell M, Hwang SW, Huynh T, Olney N, Jegla T, Patapoutian A (2007) A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol Pain 3:40. 10.1186/1744-8069-3-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P, Torck A, Quigley L, Wangzhou A, Neiman M, Rao C, Lam T, Kim JY, Kim TH, Zhang MQ, Dussor G, Price TJ (2018) Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain 159:1325–1345. 10.1097/j.pain.0000000000001217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ASC, Dworkin RH, McCarthy TD, Anand P, Bountra C, McCloud PI, Hill J, Cutter G, Kitson G, Desem N, Raff M (2014) EMA401, an orally administered highly selective angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: a randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet 383:1637–1647. 10.1016/S0140-6736(13)62337-5 [DOI] [PubMed] [Google Scholar]
- Ristoiu V. (2013) Contribution of macrophages to peripheral neuropathic pain pathogenesis. Life Sci 93:870–881. 10.1016/j.lfs.2013.10.005 [DOI] [PubMed] [Google Scholar]
- Sapio MR, Goswami SC, Gross JR, Mannes AJ, Iadarola MJ (2016) Transcriptomic analyses of genes and tissues in inherited sensory neuropathies. Exp Neurol 283:375–395. 10.1016/j.expneurol.2016.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Mohapatra DP (2012) Tissue preparation and immunostaining of mouse sensory nerve fibers innervating skin and limb bones. J Vis Exp 59:e3485. 10.3791/3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Mohapatra DP (2018) Pharmacological validation of voluntary gait and mechanical sensitivity assays associated with inflammatory and neuropathic pain in mice. Neuropharmacology 130:18–29. 10.1016/j.neuropharm.2017.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Loo L, Gupte RP, Mickle AD, Mohapatra DP (2012) Distinct modifications in Kv2.1 channel via chemokine receptor CXCR4 regulate neuronal survival-death dynamics. J Neurosci 32:17725–17739. 10.1523/JNEUROSCI.3029-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Loo L, Mohapatra DP (2013) Chemokine co-receptor CCR5/CXCR4-dependent modulation of Kv2.1 channel confers acute neuroprotection to HIV-1 glycoprotein gp120 exposure. PLoS One 8:e76698. 10.1371/journal.pone.0076698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Mickle AD, Kadunganattil S, Hu H, Mohapatra DP (2018) Parathyroid hormone-related peptide elicits peripheral TRPV1-dependent mechanical hypersensitivity. Front Cell Neurosci 12:38. 10.3389/fncel.2018.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutov LP, Warwick CA, Shi X, Gnanasekaran A, Shepherd AJ, Mohapatra DP, Woodruff TM, Clark JD, Usachev YM (2016) The complement system component C5a produces thermal hyperalgesia via macrophage-to-nociceptor signaling that requires NGF and TRPV1. J Neurosci 36:5055–5070. 10.1523/JNEUROSCI.3249-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT, Wyse BD, Edwards SR (2013a) Small molecule angiotensin II type 2 receptor (AT(2)R) antagonists as novel analgesics for neuropathic pain: comparative pharmacokinetics, radioligand binding, and efficacy in rats. Pain Med 14:692–705. 10.1111/pme.12063 [DOI] [PubMed] [Google Scholar]
- Smith MT, Woodruff TM, Wyse BD, Muralidharan A, Walther T (2013b) A small molecule angiotensin II type 2 receptor (AT(2)R) antagonist produces analgesia in a rat model of neuropathic pain by inhibition of p38 mitogen-activated protein kinase (MAPK) and p44/p42 MAPK activation in the dorsal root ganglia. Pain Med 14:1557–1568. 10.1111/pme.12157 [DOI] [PubMed] [Google Scholar]
- Smith MT, Anand P, Rice AS (2016) Selective small molecule angiotensin II type 2 receptor antagonists for neuropathic pain: preclinical and clinical studies. Pain 157:S33–S341. 10.1097/j.pain.0000000000000369 [DOI] [PubMed] [Google Scholar]
- Swamydas M, Luo Y, Dorf ME, Lionakis MS (2015) Isolation of mouse neutrophils. Curr Protoc Immunol 110:3.20.1–3.20.15. 10.1002/0471142735.im0320s110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Mizuno Y, Kozai D, Yamamoto S, Kiyonaka S, Shibata T, Uchida K, Mori Y (2008) Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels 2:287–298. 10.4161/chan.2.4.6745 [DOI] [PubMed] [Google Scholar]
- Trevisan G, Benemei S, Materazzi S, De Logu F, De Siena G, Fusi C, Fortes Rossato M, Coppi E, Marone IM, Ferreira J, Geppetti P, Nassini R (2016) TRPA1 mediates trigeminal neuropathic pain in mice downstream of monocytes/macrophages and oxidative stress. Brain 139:1361–1377. 10.1093/brain/aww038 [DOI] [PubMed] [Google Scholar]
- Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andrè E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P (2007) 4-hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 104:13519–13524. 10.1073/pnas.0705923104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtcheva MV, Copits BA, Davidson S, Sheahan TD, Pullen MY, McCall JG, Dikranian K, Gereau RW 4th (2016) Surgical extraction of dorsal root ganglia and preparation of primary cultures for functional studies of sensory neurons. Nat Protoc 11:1877–1888. 10.1038/nprot.2016.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Callaghan BC, Smith AL, Feldman EL (2011) Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol 7:573–583. 10.1038/nrneurol.2011.137 [DOI] [PubMed] [Google Scholar]
- Weaver JL, Arandjelovic S, Brown G, K Mendu S, S Schappe M, Buckley MW, Chiu YH, Shu S, Kim JK, Chung J, Krupa J, Jevtovic-Todorovic V, Desai BN, Ravichandran KS, Bayliss DA (2017) Hematopoietic pannexin 1 function is critical for neuropathic pain. Sci Rep 7:42550. 10.1038/srep42550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ (1999) Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353:1959–1964. 10.1016/S0140-6736(99)01307-0 [DOI] [PubMed] [Google Scholar]
- Yekkirala AS, Roberson DP, Bean BP, Woolf CJ (2017) Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov 16:545–564. 10.1038/nrd.2017.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin K, Baillie GJ, Vetter I (2016) Neuronal cell lines as model dorsal root ganglion neurons: a transcriptomic comparison. Mol Pain 12: pii: 1744806916646111. 10.1177/1744806916646111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All of the data presented herein are available without any restriction upon request. The RNAseq data are available through the public database, as mentioned above.