

Abstract
In the striatum, medium spiny neurons (MSNs) are heavily involved in controlling movement and reward. MSNs form two distinct populations expressing either dopamine receptor 1 (D1-MSN) or dopamine receptor 2 (D2-MSN), which differ in their projection targets and neurochemical composition. The activity of both types of MSNs is shaped by multiple neuromodulatory inputs processed by GPCRs that fundamentally impact their synaptic properties biasing behavioral outcomes. How these GPCR signaling cascades are regulated and what downstream targets they recruit in D1-MSN and D2-MSN populations are incompletely understood. In this study, we examined the cellular and molecular mechanisms underlying the action of RGS9–2, a key GPCR regulator in MSNs implicated in both movement control and actions of addictive drugs. Imaging cultured striatal neurons, we found that ablation of RGS9–2 significantly reduced calcium influx through NMDARs. Electrophysiological recordings in slices confirmed inhibition of NMDAR function in MSNs, resulting in enhanced AMPAR/NMDAR ratio. Accordingly, male mice lacking RGS9–2 displayed behavioral hypersensitivity to NMDAR blockade by MK-801 or ketamine. Recordings from genetically identified populations of striatal neurons revealed that these changes were selective to D2-MSNs. Surprisingly, we found that these postsynaptic effects resulted in remodeling of presynaptic inputs to D2-MSNs increasing the frequency of mEPSC and inhibiting paired-pulse ratio. Pharmacological dissection revealed that these adaptations were mediated by the NMDAR-dependent inhibition of retrograde endocannabinoid signaling from D2-MSNs to CB1 receptor on presynaptic terminals. Together, these data demonstrate a novel mechanism for pathway selective regulation of synaptic plasticity in MSNs controlled by GPCR signaling.
SIGNIFICANCE STATEMENT This study identifies a role for a major G-protein regulator in controlling synaptic properties of striatal neurons in a pathway selective fashion.
Keywords: endocannabinoids, GPCR, medium spiny neurons, NMDAR, RGS, striatum
Introduction
The striatum integrates excitatory inputs from the cortex and thalamus to coordinate voluntary movement, reward valuation, motor learning, and decision-making (Corbit and Balleine, 2003; Balleine et al., 2007; Smith et al., 2011). These functions critically rely on medium spiny neurons (MSNs), which account for ∼95% of neurons in this region (Kemp and Powell, 1971). In the dorsal striatum, two types of MSNs project to different areas and exert opposite effects on behavior. MSNs forming the direct pathway (D1-MSNs) project primarily to substantia nigra pars reticulata and internal globus pallidus, whereas the indirect pathway (D2-MSNs) projects to the external segment of globus pallidus (Smith et al., 1998). In addition, these two types of MSNs differentially express distinct signaling molecules. The most notable example is the selective expression of D1-type dopamine receptors (D1R) in D1-MSNs and D2-type dopamine receptors (D2R) in D2-MSNs. While D1Rs activate Gαs/olf, D2Rs signal via Gαi/o to activate or inhibit adenylate cyclase activity and cAMP production, respectively (Gerfen et al., 1990; Taussig et al., 1994). Behaviorally, activation of D1-MSNs generally stimulates movements, whereas activation of D2-MSNs inhibits them (Gerfen et al., 1990; Smith et al., 1998; Kravitz et al., 2012). However, recent models propose a more intricate interplay that relies on synergy and synchronization of D1-MSN and D2-MSN in action selection (Jin et al., 2014; Tecuapetla et al., 2016; Nakamura et al., 2017). In addition, striatal MSNs play essential roles in processing reward signals; and their modulation by addictive drugs, such as cocaine, amphetamines, heroin, and cannabinoids, underlies dependence, craving, and relapse (Nestler, 2001; Kosten and George, 2002). In general, the prevalent models posit that the modulation of D1-MSNs and D2-MSNs activity by neuromodulatory inputs plays a key role in reward seeking, motivation, and motor control.
Most neuromodulators and addictive drugs acting on MSNs exert their effects through GPCRs, which are abundantly expressed in this region. In addition to segregated dopamine D1R and D2R receptors, others include opioids that act on μ-, κ-, and δ-opioid receptors (Winzer-Serhan et al., 2003; Le Merrer et al., 2009), and endocannabinoids (eCBs) that engage Type 1 or Type 2 cannabinoid receptors (CB1 or CB2) (Araque et al., 2017). How these various GPCR inputs are regulated to control synaptic properties of MSNs and striatal-mediated behaviors is poorly understood.
A key role in regulation of GPCR signaling belongs to regulators of G-protein signaling (RGS) proteins, which limit G-protein signaling by accelerating the rate of GTP hydrolysis on Gα subunits (for review, see Siderovski and Willard, 2005). The long splice isoform of the ninth member of RGS family (RGS9–2) is selectively enriched in striatal MSNs (Rahman et al., 1999; Zachariou et al., 2003). In the striatum, RGS9–2 exists in a macromolecular complex with the Type 5 G-protein β subunit and R7 binding-protein (Xie and Martemyanov, 2011). The RGS9–2 complex is well documented to have an essential role in motor control and reward responses (Traynor et al., 2009). Mice lacking RGS9–2 display augmented responses to the rewarding and motor-stimulatory effects of morphine and cocaine (Rahman et al., 2003; Psifogeorgou et al., 2007), as well as deficits in motor learning tasks (Blundell et al., 2008). At the molecular level, RGS9–2 has been shown to control signaling initiated by D2R and μ-opioid receptor (Rahman et al., 2003; Cabrera-Vera et al., 2004; Psifogeorgou et al., 2011). However, the downstream effector targets of RGS9–2 complex are not well defined. One of the prominent targets of μ-opioid receptor signaling is the NMDAR (Rusin and Randić, 1991; Xie and Lewis, 1997; Rodríguez-Muñoz et al., 2012). Accordingly, RGS9–2 has been implicated in the regulation of NMDAR function (Kovoor et al., 2005; Bouhamdan et al., 2006) and synaptic plasticity (Busse et al., 2014). However, the cellular mechanisms of RGS9–2 mediated effects on synaptic adaptations, and their relevance in behavior and circuit specificity remains unclear.
In this study, we examined the role of RGS9–2 in controlling synaptic functions of MSN at the molecular level and within the context of the intact striatal circuitry. We report a mechanism whereby RGS9–2 acts specifically to adjust synaptic strength of inputs to D2-MSN via a retrograde mechanism that involved the action of the postsynaptic NMDARs to control eCB release and modulation of the presynaptic function via CB1R.
Materials and Methods
Experimental models and subject details.
All studies were performed in accordance with the National Institutes of Health guidelines and were granted formal approval by the Institutional Animal Care and Use Committee. Generation and characterization of mouse knock-out strains with targeted disruption of RGS9 (Rgs9−/−) were described previously (Chen et al., 2000). For electrophysiological recording, Rgs9−/− mice were crossed with D2R-EGFP line so that GFP was selectively expressed in D2-MSN neurons. Mice were housed in groups on a 12 h light-dark cycle with food and water available ad libitum. Both male and female mice between 2 and 5 months of age were used.
Primary striatal neuron cultures.
Primary cultures of striatal neurons were prepared as developed by Ivkovic and Ehrlich (1999). Briefly, brains were first dissected from wild-type and RGS9–2 KO mice (all in C57BL/6J background) on postnatal day 1. Striatum were dissected in ice-cold dissection media [Ca2+/Mg2+ free Hank's buffered salt solution (CellGro) containing 3 mm HEPES, 1 mm kynurenic acid, 12 mm MgSO4, and 5.5 mm glucose, pH 7.4]. After dissection, tissues were treated by 250 μg/ml papain (Roche) in dissection media, triturated and plated onto 25-mm-diameter, 0.13-mm-thick glass coverslips (Thermo Fisher Scientific) coated with 5 μg/ml poly-L-lysine (Sigma-Aldrich) at a density of 2000 cells/mm2. Cultures were maintained in Neurobasal-A medium supplemented with 2% B27, 0.5 mm l-glutamine, and 100 U/ml penicillin/streptomycin (Invitrogen). The cultures were incubated at 37°C in a humidified 5% CO2 incubator. One-half of media was replaced with the fresh media every 72 h.
Ca2+ imaging experiments.
All Ca2+ imaging experiments were performed on striatal neurons at 10–12 DIV at room temperature (22°C–24°C). Neurons were loaded with the cell-permeant 2 μm Indo-1/AM (the acetoxymethyl derivative of Indo-1; Invitrogen) for 40 min in aCSF (137 mm NaCl, 5.4 mm KCl, 0.4 mm KH2PO4, 0.3 mm Na2HPO4, 3 mm NaHCO3, 0.5 mm MgCl2, 0.4 mm MgSO4, 1.3 mm CaCl2, 20 mm HEPES, 5.6 mm glucose, pH 7.4). The Indo-1/AM was then allowed to fully metabolize to Indo-1 by incubating in fresh aCSF for an additional 20–25 min before imaging acquisition. Coverslips were mounted in a perfusion chamber and positioned on a movable stage of a CTR6000 inverted microscope (Leica Microsystems) and perfused with aCSF by gravity at ∼2 ml/min. Indo-1 was excited using 380 nm UV light through a dichroic filter cube and a 63× objective (Leica Microsystems). Emitted light was then passed through a DV2 dual-view image splitter (MAG Biosystems) equipped with bandpass filters of 405 nm (30 nm cutoff) and 485 nm (25 nm cutoff). The 405 and 485 nm images were subsequently acquired using an Em CCD camera (Hamamatsu) at 1 s interval. Ratiometric images of Indo-1 fluorescence were analyzed by defining regions of interest (ROIs) outlining the cell soma using MetaFluor Software version 7.5.6.0 (MDS Analytical Technologies).
Fluorescence changes were converted to [Ca2+]i by using the formula [Ca2+]i = Kd β (R − Rmin)/(Rmax − R), where R is 405/490 nm fluorescence ratio (Grynkiewicz et al., 1985). The dissociation constant (Kd) for indo-1 was 250 nm and β was the ratio of fluorescence emitted at 490 nm and measured in the absence and presence of Ca2+. Rmin, Rmax, and β were determined by bathing intact cells in 2 μm ionomycin in Ca2+-free buffer (1 mm EGTA) and saturating Ca2+ (5 mm Ca2+). Values for Rmin, Rmax, and β were 0.25, 2.3, and 3.5, respectively. Background light levels were determined at each wavelength and subtracted before calculating ratios.
Patch-clamp recordings.
Coronal slices (300 μm) containing striatum (0.2–1.3 mm posterior to bregma) were cut in ice-cold aCSF (in mm as follows: 124 NaCl, 2.8 KCl, 1.25 NaH2PO4, 2 CaCl2, 1.25 MgSO4, 26 NaHCO3, 10 glucose, pH 7.5, bubbled with 95% O2/5% CO2) using a vibrating tissue slicer (VT1200, Leica Microsystems). The slice was divided into two hemispheres along the midline, and each hemisphere was placed into an individual well of a custom slice incubation chamber (Moyer and Brown, 2007) where they remained in oxygenated aCSF at 32°C–36°C until use. During recording, striatal slices were transferred to a submerged recording chamber where they were continuously perfused at 2 ml/min with oxygenated aCSF with picrotoxin (100 μm) and maintained at 32°C–36°C. Voltage-clamp whole-cell recordings were obtained with borosilicate glass pipettes (2–5 mΩ) filled with the following solution (in mm: 130 CsMeSO3, 20 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 TEA, 4 MgATP, 0.3 Na2GTP, with a pH of 7.3 and osmolarity of 290 mOsmol). Membrane capacitance (Cm) and series resistance (Rs) were determined by application of a depolarizing pulse of 5 mV for 5 ms. After the measurement of Cm and Rs, cells were briefly switched to current recording mode to obtain resting membrane potential. Genetic deletion of RGS9 did not affect resting membrane potential or Cm in either D2-MSNs or D1-MSNs (Table 1). Cells were recorded only if initial series resistance ≤20 mΩ and were excluded from analysis if the Rs changed >20% during recording period. For mEPSC recording, TTX (1 μm) was added to the perfusion solution and spontaneous activities were recorded 3 min after whole-cell mode was obtained for at least 3 min. Evoked EPSCs were recorded using a bipolar stimulating electrode located within the striatum and ∼200 μm away dorsal to the soma. AMPAR- and NMDAR-mediated components were identified according to their distinct activation mechanisms and deactivation kinetics (Myme et al., 2003; Chung et al., 2015). AMPAR-mediated EPSCs were recorded at −70 mV and measured as the peak response following the stimulus. NMDAR-mediated EPSCs were recorded at 40 mV and measured as the mean current over a 5 ms window, 50 ms after the stimulus. Mean EPSCs were an average of 10–15 sweeps obtained at 0.1 Hz. For paired-pulse ratio (PPR) recordings, a paired-pulse protocol of two stimuli at an interpulse interval of 50 ms was applied. PPR was defined as the second peak amplitude (P2) divided by the first peak amplitude (P1). Mean PPRs were an average of 10 sweeps acquired at 1/15 Hz. Fatty acid amide hydrolase (FAAH) activator PDP-EA (0.3 mm) was added to the internal solution to facilitate hydrolysis of anandamide (AEA). CB1 blocker AM251 (5 μm) and calcium chelator BAPTA-AM (10 μm) were added in the external solution. TTX (catalog #1069), PDP-EA (catalog #5210), AM251 (catalog #1117), and BAPTA-AM (catalog #2787) were obtained from Tocris Bioscience and made into appropriate stock solutions in DMSO before use. All other chemicals were purchased from Sigma-Aldrich. All data were collected with an EPC10 amplifier system (HEKA Instruments) and transferred to a personal computer using an ITC-16 digital-to-analog converter (HEKA Instruments). The signals were filtered at 2.9 kHz and digitized at 10 kHz using Patchmaster software (HEKA Instruments). Data were analyzed offline using Patchmaster. Voltages were not corrected for the liquid-liquid junction potential.
Table 1.
Basic membrane properties and evoked EPSCs of MSNs in dorsal striatuma
| RMP (mV) | Cm (pF) | IAMPA (pA) | INMDA (pA) | |
|---|---|---|---|---|
| iMSN Rgs9++ | −69.2 + 1.2 (21) | 124.2 + 5.4 (25) | −167.2 + 10.7 (19) | 43.6 + 3.4 (19) |
| iMSN Rgs9−/− | −66.8 + 1.3 (18) | 125.1 + 5.8 (22) | −153.7 + 8.4 (16) | 31.0 + 2.6 (16)* |
| dMSN Rgs9++ | −65.5 + 1.1 (16) | 136.4 + 5.8 (21) | −169.2 + 7.4 (17) | 49.8 + 5.6 (17) |
| dMSN Rgs9−/− | −66.9 + 1.1 (15) | 137.4 + 4.9 (25) | −167.2 + 12.4 (18) | 41.5 + 5.0 (17) |
aData are mean ± SE (no. of cells). IAMPA, AMPA receptor-mediated current; INMDA, NMDA receptor-mediated current; dMSN, direct pathway MSN; iMSN, indirect pathway MSN; RMP, resting membrane potential. iMSNs were identified by their presence of EGFP fluorescence under microscope. dMSNs were identified by their absence of EGFP fluorescence under microscope. RMPs were measured immediately after whole-cell mode was established by switching to current mode. EPSCs were evoked by a stimulating electrode located ∼200 μm away dorsal to the soma within the striatum. IAMPA was measured at the peak current when cells were held at −70 mV. INMDA was measured as the average current between 50 and 55 ms following the stimulation when the cells were held at 40 mV.
*t(33) = 2.8, p = 0.008, statistically different between iMSN Rgs9++ and iMSN Rgs9−/−.
Behavioral testing in the open field and rotarod.
Locomotor performance was evaluated using male mice in an open field arena (140 cm × 140 cm × 140 cm). All animals were allowed to habituate to the behavioral room and apparatus before the start of study. On the test day, mice were injected with MK-801 (0, 0.3, 1.0, 3.0, and 6.0 mg/kg, i.p.) and placed into the center of the arena; activity was recorded with ANY-Maze automated video tracking software (Stoelting) for 90 min. All animals were given several days of recovery between doses and monitored for normal behavior before each session.
Rotarod performance was tested using a five-station rotarod treadmill with a 1.25 inch rod diameter (IITC Life Sciences). Initially, male mice were trained on the accelerating rotarod (8–20 rpm) to reach a time of 60 s, with 5 min between intertrial intervals. The following day, the mice were retrained on the rotarod with a cutoff time of 60 s to ensure each mouse had the same initial motor performance. Mice were injected with saline or ketamine (3, 10, 20, or 30 mg/kg; i.p.), and their performance on the rotarod was assessed 15, 30, and 60 min following injection. Each trial ended when a mouse fell off the rod or completed one full revolution on the rod or reached 60 s and the time was scored as the latency to fall.
Statistical analysis.
Statistical analysis was performed using Prism 6.0 (GraphPad). Student's t test was used to compare means between two groups, and ANOVA followed by Bonferroni post hoc tests was used to determine significant differences among multiple groups. Repeated-measures ANOVA followed by Bonferroni post hoc tests was used to determine the effect of ketamine on rotarod test. All statistical tests were performed two-sided. Differences were considered significant if p < 0.05. Data are mean ± SEM. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally used in comparable studies.
Results
Knock-out of RGS9–2 suppresses calcium influx through NMDARs in MSNs
We began probing the role of RGS9–2 in controlling signaling in striatal neurons, by evaluating Ca2+ influx in response to glutamate application using ratiometric calcium imaging in neurons grown in primary culture (Fig. 1A,B). Bath application of glutamate (30 μm) induced a fast increase in the intracellular Ca2+ concentration in both Rgs9+/+ and Rgs9−/− neurons (Fig. 1C). However, the Ca2+ influx was significantly smaller in Rgs9−/− neurons (∼35% suppression) compared with Rgs9+/+ neurons (t(83) = 3.29, p = 0.002; two-tailed independent-samples t test), suggesting that RGS9–2 facilitates glutamate-activated Ca2+ entry (Fig. 1C,D). In response to glutamate application, Ca2+ may enter the cell through NMDARs or voltage-gated calcium channels (VGCCs). To dissect which route Ca2+ is affected by RGS9–2 loss, we sequentially blocked Ca2+ entry via NMDAR by bath application of MK-801 (10 μm) or via L-type VGCC by nimodipine (10 μm) (Fig. 1B). We found that MK-801 significantly suppressed the Ca2+ influx (74% in WT and 61% in Rgs9−/− neurons, respectively) in response to glutamate application (t(78) = 6.80, p < 0.001 and t(43) = 4.12, p < 0.001 for Rgs9+/+ and Rgs9−/− neurons, respectively) (Fig. 1C,D). Addition of nimodipine further suppressed glutamate-induced Ca2+ entry in both genotypes compared with MK-801 alone (t(41) = 4.27, p < 0.001; and t(24) = 2.82, p = 0.010 for Rgs9+/+ and Rgs9−/−, respectively). Notably, the Ca2+ influx in MSNs in response to glutamate was comparable between the genotypes in the presence of MK-801 alone or in the presence of the mixture of MK-801 and nimodipine (p > 0.6 for both experiments), suggesting that lower calcium influx in response to glutamate application seen in Rgs9−/− neurons is mediated through suppression of NMDAR activity, with negligible changes in entry via VGCC.
Figure 1.
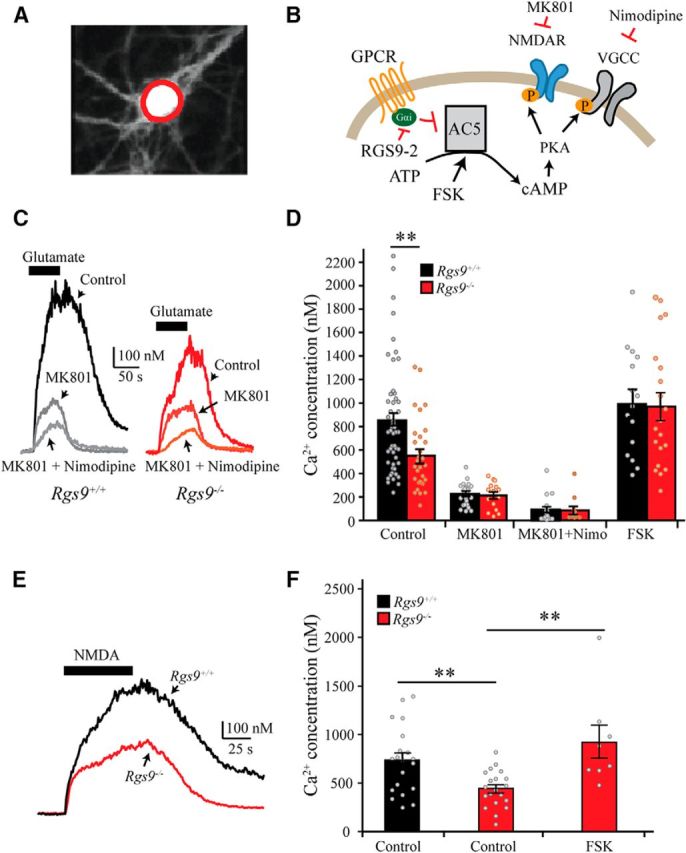
RGS9–2 deletion in primary striatal neurons inhibits calcium influx through NMDARs. A, Fluorescent image of a representative striatal neuron loaded with calcium indicator Indo-1 and excited with 380 nm UV light. Red circle represents the ROI (soma) used to measure intracellular calcium concentration. B, Schematic of the assay design to study the role RGS9–2 on calcium influx through NMDARs. MK-801 was used to block NMDARs, and nimodipine was used to block VGCCs. Forskolin (FSK) was used to activate adenylyl cyclase (AC) to increase cAMP. C, D, Representative traces and summarized bar graphs show that glutamate (30 μm) elicited a dramatic increase in intracellular calcium concentration, which was significantly suppressed in Rgs9−/− neurons (t(83) = 3.3, p = 0.002). MK-801 or a mixture of MK-801 + nimodipine significantly reduced the calcium influx evoked by glutamate, but no difference between genotypes was observed in the presence of the blockers. E, F, Representative traces and summarized bar graphs show NMDA (100 μm)-induced calcium influx in MSNs. One-way ANOVA (F(2,45) = 7.4, p = 0.007) followed by Bonferroni post hoc tests revealed that NMDA-evoked Ca2+ influx was significantly attenuated in Rgs9−/− neurons (p < 0.01), whereas FSK rescued the effect of Rgs9−/−. **p < 0.01, significantly different between groups.
Because RGS9–2 is involved in controlling cAMP levels by modulating adenylyl cyclase 5 (AC5) activity in MSN (Xie et al., 2012), we next investigated the role of cAMP in suppression of glutamate-induced Ca2+ influx seen in Rgs9−/− neurons. Application of AC activator forskolin did not significantly change glutamate-induced Ca2+ influx in wild-type (p = 0.38) but significantly enhanced Ca2+ influx in Rgs9−/− neurons (t(48) = 3.5, p < 0.01), abolishing the difference between the genotypes (Fig. 1D; p = 0.97). These data suggest that the inhibition of glutamate-induced Ca2+ influx in the absence of RGS9–2 involves the reduction in cAMP levels.
To confirm that the observed glutamate-induced Ca2+ influx was mediated by NMDARs, we measured intracellular Ca2+ concentration in response to application of selective NMDAR agonist NMDA (100 μm) in the absence of Mg2+. We found that, similarly to glutamate application, NMDA resulted in a robust increase in Ca2+ influx in MSNs (Fig. 1E). One-way ANOVA revealed a significant difference between the genotypes (Fig. 1F; F(2,45) = 7.44, p = 0.007). Bonferroni post hoc tests revealed that the Ca2+ influx was also significantly suppressed (40%) in Rgs9−/− neurons compared with Rgs9+/+ neurons (p < 0.01). Again, forskolin completely rescued reduction in Ca2+ influx deficits in Rgs9−/− neurons (p < 0.01). Collectively, these data suggest that RGS9–2 positively regulates mobilization of NMDAR-mediated Ca2+ influx in response to glutamate application in a cAMP-dependent manner.
Mice lacking RGS9–2 are hypersensitive to inhibition of NMDAR by MK-801 and ketamine
We next probed the behavioral relevance of the observed changes in NMDAR-mediated function associated with RGS9–2 loss. Given the role of the striatum in motor control, we first assessed the effect of NMDAR-selective blocker MK-801 on locomotor activity in an open field (Fig. 2A), which is commonly used to assess striatum-dependent motor control. Consistent with previous reports (Liljequist et al., 1991; Ouagazzal et al., 1993), systemic administration of MK-801 at low doses (0.3–1 mg/kg) caused dopaminergic psychomotor activation and facilitated locomotion in both genotypes (Fig. 2B) as evidenced by a significant main effect of dosage (F(2,28) = 116, p < 0.001). At these concentrations, no significant effect of genotype was noted (F(1,28) = 0.04, p = 0.85), neither was there an interaction between genotype and dose (F(2,28) = 1.6, p = 0.22). Interestingly, at higher doses that produce an anesthetic effect (3–6 mg/kg), MK-801 inhibited locomotion (Fig. 2C) as evidenced by a significant effect of dose (F(2,28) = 86.5, p < 0.001). Furthermore, mice lacking the RGS9 protein were more susceptible to MK-801 effects as demonstrated by a significant effect of genotype (F(1,28) = 41.2, p < 0.001), as well as a significant genotype and dose interaction (F(2,28) = 24.5, p < 0.001).
Figure 2.
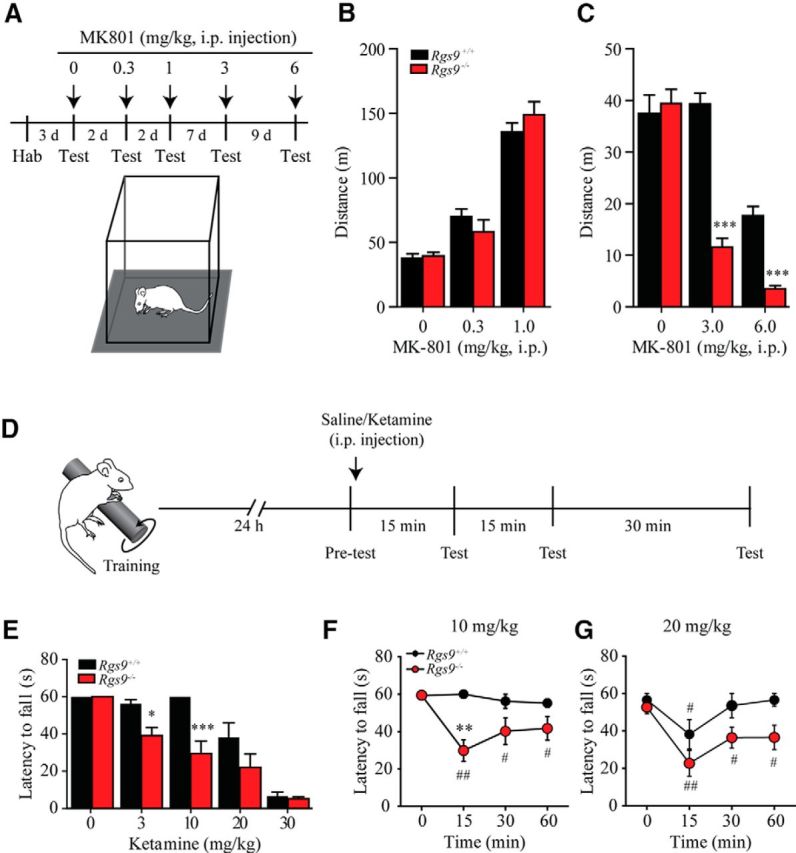
Rgs9 knock-out mice are supersensitive to ketamine administration. A, Schematic represents the experimental procedure for open field test. Mice first habituated (hab) to the open field arena before they were tested with saline or MK-801 injection. During the test, animals were placed into center of open field arena, and activities were recorded for 90 min. B, Systemic administration of MK-801 at low doses (0.3–1 mg/kg) significantly facilitated locomotion in both genotypes. Two-way ANOVA revealed a significant main effect of dosage (F(2,28) = 116, p < 0.001), but no significant effects of genotype (F(1,28) = 0.04, p = 0.85) or interaction (F(2,28) = 1.6, p = 0.22). C, Systemic administration of MK-801 at higher doses (3–6 mg/kg) significantly inhibited locomotion. Two-way ANOVA revealed significant main effects of dose (F(2,28) = 86.5, p < 0.001) and genotype (F(1,28) = 41.2, p < 0.001), as well as a significant genotype and dose interaction (F(2,28) = 24.5, p < 0.001). ***p < 0.001, significant difference between Rgs9+/+ and Rgs9−/−. D, Schematic represents the experimental procedure for rotarod test. Mice were trained on the accelerating rotarod for 60 s on day 1. The next day, the latency to fall within 60 s was tested immediately before and 15, 30, and 60 min following saline or ketamine injection. E, Summarized graph shows latency to fall recorded 15 min after saline or ketamine administration. RGS9–2-deficient mice were supersensitive to ketamine at 3–10 mg/kg tested 15 min following intraperitoneal injection. Two-way ANOVA revealed a main effect of ketamine (Figure 6A; F(4,40) = 46.2, p = 0.010), genotype (F(1,10) = 19.9, p = 0.001), and a significant interaction of dose and genotype(F(4,40) = 3.8, p = 0.010). *p < 0.05, significant difference between Rgs9+/+ and Rgs9−/−. ***p < 0.001, significant difference between Rgs9+/+ and Rgs9−/−. F, Time course of motor performance on the rotarod test following 10 mg/kg ketamine administration. Repeated-measures ANOVA revealed a significant main effect of time (F(3,30) = 5.7, p < 0.01), genotype (F(1,30) = 7.8, p < 0.05), and interaction (F(3,30) = 5.6, p < 0.01). G, Time course of motor performance on the rotarod test following 20 mg/kg ketamine administration. Repeated-measures ANOVA revealed significant main effect of time (F(3,30) = 10.9, p < 0.001), but neither significant main effect of genotype (F(1,30) = 4.8, p = 0.054) nor interaction (F(3,30) = 1.4, p = 0.28) was observed. Statistically different between genotypes: *p < 0.05; **p < 0.01; ***p < 0.001. Statistically different from time point 0: #p < 0.05; ##p < 0.01. ** Number of animals per group was 8 each for Rgs9+/+ and Rgs9−/− for experiments presented in B and C, and 5 and 7 for Rgs9+/+ and Rgs9−/−, respectively, in E–G.
To further evaluate the behavioral effect of NMDAR dysregulation, we assessed the effect of ketamine, another NMDAR antagonist on motor coordination using a standard rotarod assay. The Rgs9−/− mice have been previously shown to have mild impairments in motor learning (Blundell et al., 2008); therefore, we first trained the mice on the rotarod with a cutoff time of 60 s to ensure that each mouse had the same initial motor performance. Once the criteria had been reached, we investigated the effect of ketamine on their motor performance. One day following training, mice were subjected to a test on the rotarod before and 15, 30, and 60 min after either saline or ketamine injection (Fig. 2D). At the first time point following either saline or ketamine injection, we found that Rgs9−/− mice displayed a greater sensitivity to ketamine on the rotarod as indicated by the reduced latency time across several doses (Fig. 2E). Two-way ANOVA revealed a main effect of ketamine (F(4,40) = 46.2, p = 0.010), genotype (F(1,10) = 19.9, p = 0.001), and a significant interaction of dose and genotype (F(4,40) = 3.8, p = 0.010). Further post hoc analysis with Bonferroni correction revealed that the latency to fall from the rotarod was significantly reduced in Rgs9−/− mice at 3 mg/kg (p < 0.05) and 10 mg/kg (p < 0.001) but were ineffective in Rgs9+/+ mice. There were no significant differences in performance between genotypes in control experiments involving saline (0) or at higher ketamine concentrations (20 and 30 mg/kg) that induce anesthesia (Fig. 2E). At a subthreshold dose of 10 mg/kg, only Rgs9−/− mice displayed reduced motor performance at 15 min following ketamine injection, and this effect lasted for >60 min (Fig. 2F; main effect of genotype as revealed by repeated-measures ANOVA followed by Bonferroni post hoc test: F(1,30) = 7.8, p = 0.019). At a higher dose of 20 mg/kg, Rgs9+/+ mice showed a reduced motor performance 15 min after injection but were able to recover (30 min after injection), whereas Rgs9−/− mice were unable recover as evident from the reduced performance at 60 min (Fig. 2G; main effect of time as revealed by repeated-measures ANOVA followed by Bonferroni post hoc test; F(3,30) = 10.9, p < 0.001). These data indicate that elimination of RGS9–2 in mice substantially augments their behavioral sensitivity to NMDAR blockade, consistent with the reduction in the NMDAR function observed in cultured neurons and brain slices.
Deletion of RGS9–2 suppresses NMDAR current and enhances AMPA/NMDA ratio selectively in D2-MSNs
To test the relevance of the observed NMDAR deficits associated with RGS9–2 loss in an intact neuronal circuit, we further used whole-cell patch-clamp recordings of MSNs in brain slices directly measuring the NMDAR currents. We reasoned that high variability of Ca2+ responses in cultured neurons (Fig. 1D,F) could be related to differences in the behavior from the two distinct populations of MSNs, D1-MSNs and D2-MSNs, which have distinct electrophysiological properties and regulation (Planert et al., 2013). Therefore, in the rest of our studies, we distinguished between MSN populations by crossing Rgs9−/− mice with the D2R-EGFP line, which expresses GFP selectively in D2-MSN neurons and recording from both fluorescent (D2-MSN) and nonfluorescent (D1-MSNs) neurons containing or lacking RGS9–2 expression (Fig. 3A,B). NMDAR current in MSNs was elicited by direct bath application of NMDA (30 μm), and the cells were held at −70 mV in the presence of GABAergic blocker picrotoxin. This manipulation induced a large inward current in both Rgs9+/+ D2-MSNs and D1-MSNs. The amplitude of this current was significantly attenuated in Rgs9−/− D2-MSNs (Fig. 3C; t(23) = 2.17, p = 0.041). In contrast, we observed no significant differences in the amplitude of NMDA-elicited current between genotypes in D1-MSNs (Fig. 3D; t(19) = 0.30, p = 0.77). These data reinforce our observations that loss of RGS9–2 leads to inhibition of NMDAR activity in D2-MSNs.
Figure 3.
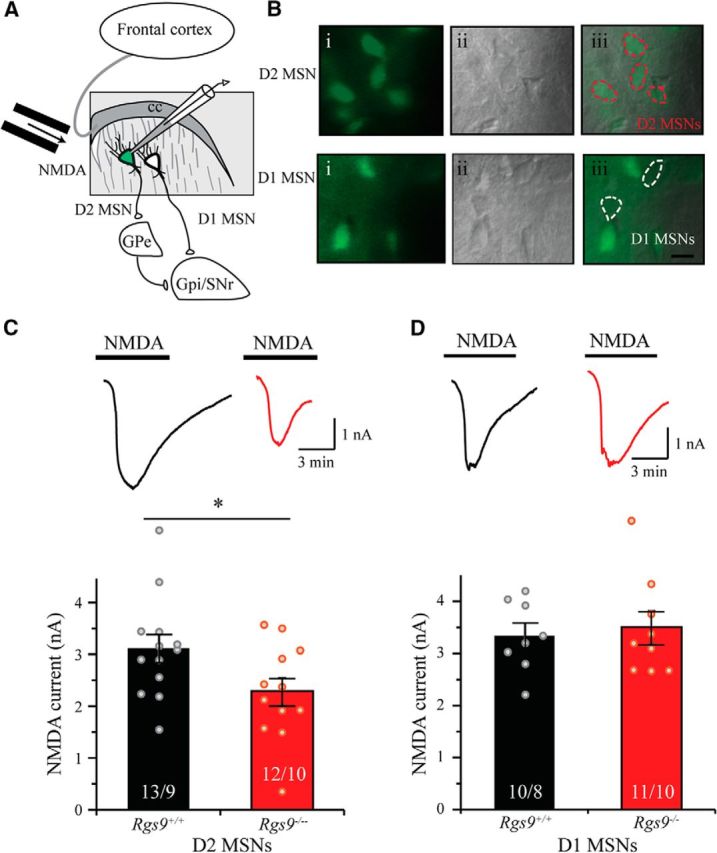
Selective suppression of NMDA current in D2-MSN lacking RGS9–2. A, Schematic shows whole-cell patch-clamp recording on dorsal striatal MSNs, where fluorescently (EGFP) labeled cells are D2-MSNs and nonfluorescent cells are considered as D1-MSNs. B, Representative fluorescent (left), IR-DIC (middle), and merged (right) images show D2 MSNs (top) and D1-MSNs (bottom) were recorded from dorsal striatum. C, Representative traces and summarized bar graphs show that NMDAR current was significantly smaller in Rgs9−/− D2-MSNs compared with Rgs9+/+ D2-MSNs (t(23) = 2.2, p = 0.041). D, Representative traces and summarized bar graph show that the NMDA current was comparable between Rgs9+/+ and Rgs9−/− D1-MSNs (t(19) = 0.3, p = 0.77). Bar represents number of animals and cells used in each group. *p < 0.05, significantly different between groups.
We next examined the contribution of RGS9–2 to synaptic events mediated by NMDAR. In these experiments, glutamatergic inputs from the cortex onto MSNs were electrically stimulated by concentric electrode placed ∼200 μm dorsal to the cell body evoking EPSCs while recording from genetically identified D2-MSN and D1-MSN neurons in the presence of picrotoxin (Fig. 4A). Contributions of AMPAR and NMDAR to EPSCs were isolated by holding cells at −70 mV and 40 mV, respectively (Fig. 4B,C). Consistent with reduction in chemically evoked NMDAR current in Rgs9−/− mice, we found that the NMDAR component was significantly reduced (t(33) = 2.8, p = 0.008) in Rgs9−/− neurons, whereas the AMPAR component was comparable (t(32) = 1.1, p = 0.278) between genotypes in D2-MSNs (Table 1). Furthermore, deletion of RGS9–2 significantly increased the AMPA/NMDA ratio in D2-MSNs (Fig. 4B; t(33) = 2.24, p = 0.032). In contrast, no significant differences were observed between genotypes in any of these measurements in D1-MSNs (Fig. 4C; Table 1). Together, these data indicate that the loss of RGS9–2 inhibits NMDAR function selectively in D2-MSN neurons, which results in greater contribution of AMPAR relative to NMDAR to synaptic responses.
Figure 4.

Ablation of RGS9–2 enhances AMPA/NMDA ratio in D2-MSNs. A, Schematic shows whole-cell patch-clamp recording on dorsal striatal MSNs. A concentric bipolar electrode was used to evoke EPSCs located ∼200 μm away dorsal from the soma. B, Representative traces and summarized bar graph show that genetic deletion of RGS9–2 significantly enhanced the AMPA/NMDA ratio in D2-MSNs (t(33) = 2.2, p = 0.033). C, Representative traces and summarized bar graph show that the AMPAR/NMDA ratio was comparable between Rgs9+/+ and Rgs9−/− neurons in D1-MSNs (t(32) = 1.0, p = 0.33). Bar represents number of animals and cells used in each group. *p < 0.05, significantly different between groups.
Loss of postsynaptic RGS9–2 selectively facilitates presynaptic glutamate release onto D2-MSNs
To further examine the mechanisms underlying RGS9–2-mediated modulation of synaptic properties, we recorded mEPSCs in dorsal striatal MSNs in the presence of TTX and picrotoxin. As shown in the representative traces in Figure 5A and summarized in Figure 5C, we found that deletion of RGS9–2 had no effect on the amplitude of mEPSC (t(18) = 0.64, p = 0.530) in D2-MSNs. Because mEPSCs are predominantly, if not exclusively, contributed by AMPAR currents, this further confirms no influence of RGS9–2 on the synaptic AMPAR function and demonstrates its exclusivity of NMDAR modulation. Surprisingly, we found that D2-MSN neurons from Rgs9−/− mice showed significantly increased frequency of mEPSC compared with wild-type neurons (Fig. 5D; t(20) = 3.78, p = 0.0012), suggesting additional presynaptic adaptations. This effect was again specific to the D2-MSN population as neither frequency nor amplitude of mEPSC was significantly different between genotypes in D1-MSNs (Fig. 5B,E,F; t(20) = 0.23, p = 0.823, and t(20) = 1.39, p = 0.180 for frequency and amplitude, respectively).
Figure 5.

Genetic deletion of Rgs9 increases the frequency of mEPSC in D2-MSNs. A, C, D, Representative traces, plots of cumulative probability, and summarized graphs show that genetic deletion of RGS9–2 significantly increased the frequency (t(20) = 3.78 p < 0.01), but not the amplitude (t(18) = 0.64, p = 0.53), of mEPSC in D2 MSNs. B, E, F, Representative traces and summarized graphs show that genetic deletion of RGS9–2 did not significantly change the frequency (t(20) = 0.23, p = 0.82) and amplitude (t(20) = 1.39, p = 0.18) of mEPSC in D1-MSNs. Bar represents number of animals and cells used in each group. **p < 0.01, significantly different between groups.
Enhanced inputs onto D2-MSNs lacking RGS9–2 occur via weakening of retrograde endo-cannabinoid feedback
To confirm the presynaptic modulation of glutamate release onto D2-MSNs triggered by the loss of postsynaptic RGS9–2, we recorded PPR in aCSF with picrotoxin while considering the molecular context of the key events involved in retrograde modulation of neurotransmitter release (Fig. 6A). Consistent with prior reports (Kim et al., 2011; Atwood et al., 2014), we found that glutamatergic inputs onto both D2-MSN and D1-MSN neurons to be of low probability, showing prominent facilitation upon second pulse delivery as evidenced by representative traces and summarized graphs in Figure 6B (aCSF). Remarkably, deletion of RGS9–2 substantially reduced PPR in D2-MSNs (t(28) = 3.03, p = 0.005), eliminating synaptic facilitation of these synapses (PPRs were 1.42 ± 0.14 and 0.97 ± 0.05 for Rgs9+/+ and Rgs9−/− neurons, respectively). This effect was specific to D2-MSN and not observed in D1-MSNs where loss of RGS9–2 still resulted in strong facilitation (Fig. 6C, aCSF; 1.29 ± 0.10 and 1.12 ± 0.051 for Rgs9+/+ and Rgs9−/− neurons, respectively; t(33) = 1.55, p = 0.131). These data suggest that postsynaptic RGS9–2 in D2-MSNs regulates synaptic feedback that selectively adjusts release probability in presynaptic terminals.
Figure 6.

Involvement of RGS9–2 in regulation of presynaptic glutamate release via modulation of eCB signaling. A, Schematic of the assay design to study possible signaling pathways involved in retrograde synaptic signaling that regulates presynaptic vesicle release in MSN. NAPE, N-arachidonoyl phosphatidylethanolamine. B, Representative traces and summarized bar graph show that genetic deletion of RGS9–2 significantly reduced PPR in D2-MSNs (aCSF; t(28) = 3.0, p = 0.005), whereas application of CB1 receptor blocker AM251, calcium chelator BAPTA, or FAAH activator PDP-EA mimicked the effect of RGS9–2 ablation. No significant differences between genotypes were observed in the presence of these drugs (t(21) = 0.92, p = 0.37; t(16) = 1.7, p = 0.1; and t(17) = 0.37, p = 0.72 for AM251, BAPTA, and PDP-EA, respectively). C, Representative traces and summarized bar graph show that genetic knock-out of RGS9–2 did not significantly change PPR in D1-MSNs (aCSF; t(33) = 1.6, p = 0.13). Application of AM251, BAPTA, or PDP-EA did not have a significant effect on PPR in D1-MSNs (t(17) = 0.35, p = 0.73; t(14) = 0.67, p = 0.51; and t(13) = 1.2, p = 0.24 for AM251, BAPTA, and PDP-EA, respectively, compared between genotypes). Bar represents number of animals and cells used in each group. **p < 0.01, significantly different between groups.
To determine the mechanisms by which the loss of RGS9–2 in postsynaptic D2-MSNs regulates presynaptic release probability, we tested a hypothesis about the involvement of retrograde eCB signaling, which plays a prominent role in retrograde feedback in striatal synapses (Gerdeman and Lovinger, 2001). Our strategy relied on blocking different steps in the retrograde eCB signaling in D2-MSN and D1-MSN neurons from wild-type and Rgs9−/− mice: blocking CB1R with AM251, chelating intracellular calcium with BAPTA-AM, and facilitating the breakdown of AEA with a FAAH activator PDP-EA (Fig. 6A).
We found that addition of CB1R blocker AM251 reduced the PPR of Rgs9+/+, but not Rgs9−/− D2-MSNs, eliminating the PPR differences between the genotypes (Fig. 6B; t(21) = 0.92, p = 0.37). No significant effect of AM251 on D1-MSNs in both genotypes was observed (Fig. 6C; t(17) = 0.35, p = 0.73, between Rgs9+/+ and Rgs9−/− neurons). Similarly, PDP-EA also removed the difference of PPR between wild-type and RGS9–2 knock-out neurons in D2-MSNs (t(17) = 0.37, p = 0.72). Again, no significant effect was observed when PDP-EA was applied to D1-MSNs (t(13) = 1.2, p = 0.24), compared between genotypes. Given the impact of RGS9–2 deletion on Ca2+ influx through the NMDAR, we further tested the effect of blocking Ca2+ by BAPTA-AM. Consistent with the expectations, it also selectively inhibited facilitation bringing PPR ratio in wild-type synapses to the level seen in Rgs9−/− neurons and eliminating the PPR differences between genotypes in D2-MSNs (t(16) = 1.7, p = 0.11). There was still no significant difference between genotypes in D1-MSNs in the presence of BAPTA-AM (t(14) = 0.67, p = 0.51).
These data suggest that the increase in release probability observed in D2-MSN following RGS9–2 deletion was mediated by reduced activation of the eCB signaling system, which might be associated with the reduction in NMDAR activation.
Discussion
The current study provides the first evidence that RGS9–2 specifically regulates NMDAR function and synaptic transmission in D2-MSNs in the dorsal striatum. Both calcium imaging from primary culture and electrophysiological recording from brain slices reveal that NMDAR function was significantly attenuated following RGS9–2 deletion. These observations are consistent with earlier reports that noticed similar dysregulation of NMDAR function (Kovoor et al., 2005). The experiments performed to dissect the consequences of this alteration led to propose a model where NMDAR hypofunction increased AMPAR/NMDAR ratio and suppressed the eCB signaling system, which led to increased presynaptic glutamate release (Fig. 7). In this model, RGS9–2 suppresses signaling via Gi/o-coupled GPCRs, which in turn inhibit adenylyl cyclase to control cAMP production. Consequently, deletion of RGS9–2 would increase inhibitory tone suppressing cAMP production and reducing the activity of its key effector PKA, well known for its stimulatory effects on NMDAR mediated by phosphorylation (Raman et al., 1996; Crump et al., 2001; Scott et al., 2001; Skeberdis et al., 2006). This would reduce Ca2+ permeability of NMDARs suppressing eCB release and engagement of presynaptic CB1R relieving their inhibitory effects on neurotransmitter release. At the behavioral level, suppression of NMDAR function made mice lacking RGS9–2 hypersensitive to the dissociative effects of ketamine and MK-801 administration. Collectively, these data clarify cellular and molecular mechanisms by which RGS9–2 provides pathway-selective control of psychomotor functions mediated by the dorsal striatum.
Figure 7.

Proposed model for RGS9–2 regulation of NMDAR function and synaptic signaling in D2-MSNs. Within MSNs, RGS9–2 inactivates Gi-coupled GPCR, which inhibits the production of cAMP, which in turn plays a key role in positive modulation of NMDAR function. The change in Ca2+ influx through NMDARs then affects the production of AEA, which retrogradely inhibits presynaptic neurotransmitter release.
RGS9–2 specifically modulates synaptic properties of D2R-expressing MSNs
Although highly restricted to striatum, RGS9–2 is reported to be equally expressed in D1-MSNs and D2-MSNs (Kovoor et al., 2005; Tecuapetla et al., 2016). Interestingly, in the current study, we observed that effects induced by RGS9–2 deletion were specific to D2-MSNs, including changes in NMDAR current, eCB signaling, AMPA/NMDA ratio, mEPSC frequency, and PPRs. We think that the pathway selectivity of RGS9–2 effects may be explained by specificity of RGS9–2 for the Gαi/o protein (Hooks et al., 2003; Xie et al., 2012; Masuho et al., 2013), which mediates the effects of D2R in the D2-MSNs. This explanation is supported by the observations that RGS9–2 when ectopically coexpressed in transfected cells interacts with D2Rs, but not D1Rs (Rahman et al., 2003; Kovoor et al., 2005), and that NMDARs interact differently with the two subtypes of dopamine receptors such that activation of D1Rs potentiates, whereas activation of D2Rs attenuates, NMDAR responses (Cepeda et al., 1993, 2008). Furthermore, genetic deletion of RGS9–2 causes locomotor deficits (e.g., autonomic involuntary movements) that are related to D2R, but not D1R, functions (Kovoor et al., 2005). Conversely, overexpression of RGS9–2 in rat nucleus accumbens reduced locomotor responses to cocaine and to D2R, but not to D1R, receptor agonists (Rahman et al., 2003). The function of RGS9–2 in D1-MSNs is presently unclear and will require further investigation. It is possible that it may be related to the action of the Gi/o-coupled opioid receptors, clearly regulated by the RGS9–2 at the behavioral level and present in both MSN populations (Gaspari et al., 2014; Oude Ophuis et al., 2014).
RGS9–2 regulates NMDAR function and synaptic plasticity
Our data obtained in calcium imaging experiments in cultured neurons and electrophysiological recordings of acute slices indicate prominent inhibition of NMDAR function upon RGS9–2 loss. This is consistent with a previous study showing that NMDAR, but not AMPAR, current was attenuated in MSNs in Rgs9 knock-out mice (Kovoor et al., 2005). These data are also consistent with the increase in AMPAR/NMDAR ratio in D2-MSNs lacking RGS9–2 and suggest that the behavioral deficits observed in Rgs9 knock-out animals might be at least in part related to the hypofunction of NMDAR and ensuing synaptic adaptations.
At the behavioral level, NMDAR function is critical for striatum-dependent motor control and learning. For example, striatum-specific ablation of NMDAR increases locomotor activity (Mohn et al., 1999; Ohtsuka et al., 2008) and significantly impairs behavioral performance in rotarod test (Dang et al., 2006). This is reminiscent of behavioral deficits in the same behavioral test following RGS9 deletion (Blundell et al., 2008). The observation that mice lacking RGS9 were supersensitive to ketamine reported in this study further reinforces the role of RGS9–2 in NMDAR modulation at the behavioral level.
At the molecular level, the hypofunction of NMDAR in RGS9–2 deficient mice is likely driven by alterations in NMDARs function. RGS9–2 deletion is expected to enhance the activity of Gi/o-linked GPCRs prominent in the region, such as D2R or μ-opioid receptor. The ensuing suppression of PKA activity would reduce its ability to phosphorylate NMDARs (Rusin and Randić, 1991; Martin et al., 1997; Xie and Lewis, 1997; Rodríguez-Muñoz et al., 2012). Phosphorylation of NMDARs by PKA has been shown to increase the surface expression, gating, and Ca2+ permeability of the receptors (Scott et al., 2001; Chen and Roche, 2007; Aman et al., 2014). Conversely, blocking PKA dramatically inhibits NMDAR function (Skeberdis et al., 2006). Thus, loss of RGS9–2 likely leads to changes in NMDAR gating and/or targeting to plasma membrane. Furthermore, RGS9–2 may also exert its facilitatory effects on NMDAR activity by suppressing its calcium-dependent inactivation through interacting with α-actinin-2 (Bouhamdan et al., 2006). The functional nature of the effects observed is also supported by no significant changes in the expression of NMDAR transcript in the striatum upon RGS9 ablation (Busse et al., 2014).
RGS9–2 regulates eCB system
In the course of this study, we made a surprising observation that postsynaptic RGS9–2 expressed in MSN modulates mEPSC frequency and PPR of excitatory inputs to D2-MSNs by a presynaptic mechanism. These results are consistent with a previous study where deletion of RGS9–2 was noted to increase spontaneous EPSC accompanied by impaired induction of LTD (Busse et al., 2014). Our pharmacological dissection indicates that changes in presynaptic release probability are likely due to an inhibition of eCB signaling caused by RGS9–2 deletion. Accordingly, blockade of presynaptic CB1R or inhibition of eCB production mimicked the effect of RGS9–2 ablation. Previous studies showed that eCBs are released from MSNs triggered by calcium influx upon membrane depolarization (Di Marzo et al., 1994; Alger, 2004), and this process is potentiated when cAMP levels rise (Cadas et al., 1996). After being released, eCBs act as retrograde messengers that regulate synaptic transmission through presynaptic CB1Rs and suppress neurotransmitter release (for review, see Heifets and Castillo, 2009). eCB signaling is required for synaptic plasticity in striatum, especially LTD (Gerdeman and Lovinger, 2001; Gerdeman et al., 2002). Genetic deletion of CB1R significantly impaired skill learning on rotarod in adult mice (Bilkei-Gorzo et al., 2005), indicating that the eCB signaling system is also involved in motor control. Together, these observations suggest that RGS9–2 may ultimately regulate motor learning through modulating GPCR-NMDAR-eCB signaling axis.
Footnotes
This work was supported by National Institutes of Health Grants DA036596 and DA026405 to K.A.M. We thank Natalia Martemyanova for help with breeding and maintenance of mouse lines used in these experiments; and members of K.A.M. laboratory for critical feedback on the results and their interpretations.
The authors declare no competing financial interests.
References
- Alger BE. (2004) Endocannabinoids: getting the message across. Proc Natl Acad Sci U S A 101:8512–8513. 10.1073/pnas.0402935101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman TK, Maki BA, Ruffino TJ, Kasperek EM, Popescu GK (2014) Separate intramolecular targets for protein kinase A control N-methyl-D-aspartate receptor gating and Ca2+ permeability. J Biol Chem 289:18805–18817. 10.1074/jbc.M113.537282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Castillo PE, Manzoni OJ, Tonini R (2017) Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 124:13–24. 10.1016/j.neuropharm.2017.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA, Lovinger DM (2014) Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci 17:540–548. 10.1038/nn.3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine BW, Delgado MR, Hikosaka O (2007) The role of the dorsal striatum in reward and decision-making. J Neurosci 27:8161–8165. 10.1523/JNEUROSCI.1554-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilkei-Gorzo A, Racz I, Valverde O, Otto M, Michel K, Sastre M, Sarstre M, Zimmer A (2005) Early age-related cognitive impairment in mice lacking cannabinoid CB1 receptors. Proc Natl Acad Sci U S A 102:15670–15675. 10.1073/pnas.0504640102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Hoang CV, Potts B, Gold SJ, Powell CM (2008) Motor coordination deficits in mice lacking RGS9. Brain Res 1190:78–85. 10.1016/j.brainres.2007.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhamdan M, Yan HD, Yan XH, Bannon MJ, Andrade R (2006) Brain-specific regulator of G-protein signaling 9–2 selectively interacts with alpha-actinin-2 to regulate calcium-dependent inactivation of NMDA receptors. J Neurosci 26:2522–2530. 10.1523/JNEUROSCI.4083-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse K, Strotmann R, Strecker K, Wegner F, Devanathan V, Gohla A, Schöneberg T, Schwarz J (2014) Adaptive gene regulation in the striatum of RGS9-deficient mice. PLoS One 9:e92605. 10.1371/journal.pone.0092605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Hernandez S, Earls LR, Medkova M, Sundgren-Andersson AK, Surmeier DJ, Hamm HE (2004) RGS9–2 modulates D2 dopamine receptor-mediated Ca2+ channel inhibition in rat striatal cholinergic interneurons. Proc Natl Acad Sci U S A 101:16339–16344. 10.1073/pnas.0407416101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D (1996) Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci 16:3934–3942. 10.1523/JNEUROSCI.16-12-03934.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS (1993) Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A 90:9576–9580. 10.1073/pnas.90.20.9576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, André VM, Yamazaki I, Wu N, Kleiman-Weiner M, Levine MS (2008) Differential electrophysiological properties of dopamine D1 and D2 receptor-containing striatal medium-sized spiny neurons. Eur J Neurosci 27:671–682. 10.1111/j.1460-9568.2008.06038.x [DOI] [PubMed] [Google Scholar]
- Chen BS, Roche KW (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53:362–368. 10.1016/j.neuropharm.2007.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Burns ME, He W, Wensel TG, Baylor DA, Simon MI (2000) Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9–1. Nature 403:557–560. 10.1038/35000601 [DOI] [PubMed] [Google Scholar]
- Chung W, Choi SY, Lee E, Park H, Kang J, Park H, Choi Y, Lee D, Park SG, Kim R, Cho YS, Choi J, Kim MH, Lee JW, Lee S, Rhim I, Jung MW, Kim D, Bae YC, Kim E (2015) Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat Neurosci 18:435–443. 10.1038/nn.3927 [DOI] [PubMed] [Google Scholar]
- Corbit LH, Balleine BW (2003) The role of prelimbic cortex in instrumental conditioning. Behav Brain Res 146:145–157. 10.1016/j.bbr.2003.09.023 [DOI] [PubMed] [Google Scholar]
- Crump FT, Dillman KS, Craig AM (2001) cAMP-dependent protein kinase mediates activity-regulated synaptic targeting of NMDA receptors. J Neurosci 21:5079–5088. 10.1523/JNEUROSCI.21-14-05079.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y (2006) Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci U S A 103:15254–15259. 10.1073/pnas.0601758103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372:686–691. 10.1038/372686a0 [DOI] [PubMed] [Google Scholar]
- Gaspari S, Papachatzaki MM, Koo JW, Carr FB, Tsimpanouli ME, Stergiou E, Bagot RC, Ferguson D, Mouzon E, Chakravarty S, Deisseroth K, Lobo MK, Zachariou V (2014) Nucleus accumbens-specific interventions in RGS9–2 activity modulate responses to morphine. Neuropsychopharmacology 39:1968–1977. 10.1038/npp.2014.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman G, Lovinger DM (2001) CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol 85:468–471. 10.1152/jn.2001.85.1.468 [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5:446–451. 10.1038/nn832 [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr, Sibley DR (1990) D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250:1429–1432. 10.1126/science.2147780 [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440–3450. [PubMed] [Google Scholar]
- Heifets BD, Castillo PE (2009) Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol 71:283–306. 10.1146/annurev.physiol.010908.163149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks SB, Waldo GL, Corbitt J, Bodor ET, Krumins AM, Harden TK (2003) RGS6, RGS7, RGS9, and RGS11 stimulate GTPase activity of Gi family G-proteins with differential selectivity and maximal activity. J Biol Chem 278:10087–10093. 10.1074/jbc.M211382200 [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Ehrlich ME (1999) Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J Neurosci 19:5409–5419. 10.1523/JNEUROSCI.19-13-05409.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Tecuapetla F, Costa RM (2014) Basal ganglia subcircuits distinctively encode the parsing and concatenation of action sequences. Nat Neurosci 17:423–430. 10.1038/nn.3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JM, Powell TP (1971) The structure of the caudate nucleus of the cat: light and electron microscopy. Philos Trans R Soc Lond B Biol Sci 262:383–401. 10.1098/rstb.1971.0102 [DOI] [PubMed] [Google Scholar]
- Kim J, Park BH, Lee JH, Park SK, Kim JH (2011) Cell type-specific alterations in the nucleus accumbens by repeated exposures to cocaine. Biol Psychiatry 69:1026–1034. 10.1016/j.biopsych.2011.01.013 [DOI] [PubMed] [Google Scholar]
- Kosten TR, George TP (2002) The neurobiology of opioid dependence: implications for treatment. Sci Pract Perspect 1:13–20. 10.1151/spp021113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovoor A, Seyffarth P, Ebert J, Barghshoon S, Chen CK, Schwarz S, Axelrod JD, Cheyette BN, Simon MI, Lester HA, Schwarz J (2005) D2 dopamine receptors colocalize regulator of G-protein signaling 9–2 (RGS9–2) via the RGS9 DEP domain, and RGS9 knock-out mice develop dyskinesias associated with dopamine pathways. J Neurosci 25:2157–2165. 10.1523/JNEUROSCI.2840-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, Kreitzer AC (2012) Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci 15:816–818. 10.1038/nn.3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Merrer J, Becker JA, Befort K, Kieffer BL (2009) Reward processing by the opioid system in the brain. Physiol Rev 89:1379–1412. 10.1152/physrev.00005.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljequist S, Ossowska K, Grabowska-Andén M, Andén NE (1991) Effect of the NMDA receptor antagonist, MK-801, on locomotor activity and on the metabolism of dopamine in various brain areas of mice. Eur J Pharmacol 195:55–61. 10.1016/0014-2999(91)90381-Y [DOI] [PubMed] [Google Scholar]
- Martin G, Nie Z, Siggins GR (1997) mu-Opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J Neurosci 17:11–22. 10.1523/JNEUROSCI.17-01-00011.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Xie K, Martemyanov KA (2013) Macromolecular composition dictates receptor and G protein selectivity of regulator of G protein signaling (RGS) 7 and 9–2 protein complexes in living cells. J Biol Chem 288:25129–25142. 10.1074/jbc.M113.462283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH (1999) Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98:427–436. 10.1016/S0092-8674(00)81972-8 [DOI] [PubMed] [Google Scholar]
- Moyer JR Jr, Brown TH (2007) Visually-guided patch-clamp recordings in brain slices. In: Advanced techniques for patch-clamp analysis, Ed 2 (Walz W, ed), pp 169–227. Totowa, NJ: Humana. [Google Scholar]
- Myme CI, Sugino K, Turrigiano GG, Nelson SB (2003) The NMDA-to-AMPA ratio at synapses onto layer 2/3 pyramidal neurons is conserved across prefrontal and visual cortices. J Neurophysiol 90:771–779. 10.1152/jn.00070.2003 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nagata M, Yagi T, Graybiel AM, Yamamori T, Kitsukawa T (2017) Learning new sequential stepping patterns requires striatal plasticity during the earliest phase of acquisition. Eur J Neurosci 45:901–911. 10.1111/ejn.13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. (2001) Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2:119–128. 10.1038/35053570 [DOI] [PubMed] [Google Scholar]
- Ohtsuka N, Tansky MF, Kuang H, Kourrich S, Thomas MJ, Rubenstein JL, Ekker M, Leeman SE, Tsien JZ (2008) Functional disturbances in the striatum by region-specific ablation of NMDA receptors. Proc Natl Acad Sci U S A 105:12961–12966. 10.1073/pnas.0806180105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouagazzal A, Nieoullon A, Amalric M (1993) Effects of dopamine D1 and D2 receptor blockade on MK-801-induced hyperlocomotion in rats. Psychopharmacology (Berl) 111:427–434. 10.1007/BF02253532 [DOI] [PubMed] [Google Scholar]
- Oude Ophuis RJ, Boender AJ, van Rozen AJ, Adan RA (2014) Cannabinoid, melanocortin and opioid receptor expression on DRD1 and DRD2 subpopulations in rat striatum. Front Neuroanat 8:14. 10.3389/fnana.2014.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planert H, Berger TK, Silberberg G (2013) Membrane properties of striatal direct and indirect pathway neurons in mouse and rat slices and their modulation by dopamine. PLoS One 8:e57054. 10.1371/journal.pone.0057054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psifogeorgou K, Papakosta P, Russo SJ, Neve RL, Kardassis D, Gold SJ, Zachariou V (2007) RGS9–2 is a negative modulator of mu-opioid receptor function. J Neurochem 103:617–625. 10.1111/j.1471-4159.2007.04812.x [DOI] [PubMed] [Google Scholar]
- Psifogeorgou K, Psigfogeorgou K, Terzi D, Papachatzaki MM, Varidaki A, Ferguson D, Gold SJ, Zachariou V (2011) A unique role of RGS9–2 in the striatum as a positive or negative regulator of opiate analgesia. J Neurosci 31:5617–5624. 10.1523/JNEUROSCI.4146-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman Z, Gold SJ, Potenza MN, Cowan CW, Ni YG, He W, Wensel TG, Nestler EJ (1999) Cloning and characterization of RGS9–2: a striatal-enriched alternatively spliced product of the RGS9 gene. J Neurosci 19:2016–2026. 10.1523/JNEUROSCI.19-06-02016.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman Z, Schwarz J, Gold SJ, Zachariou V, Wein MN, Choi KH, Kovoor A, Chen CK, DiLeone RJ, Schwarz SC, Selley DE, Sim-Selley LJ, Barrot M, Luedtke RR, Self D, Neve RL, Lester HA, Simon MI, Nestler EJ (2003) RGS9 modulates dopamine signaling in the basal ganglia. Neuron 38:941–952. 10.1016/S0896-6273(03)00321-0 [DOI] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE (1996) Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron 16:415–421. 10.1016/S0896-6273(00)80059-8 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M, Sánchez-Blázquez P, Vicente-Sánchez A, Berrocoso E, Garzón J (2012) The mu-opioid receptor and the NMDA receptor associate in PAG neurons: implications in pain control. Neuropsychopharmacology 37:338–349. 10.1038/npp.2011.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusin KI, Randić M (1991) Modulation of NMDA-induced currents by mu-opioid receptor agonist DAGO in acutely isolated rat spinal dorsal horn neurons. Neurosci Lett 124:208–212. 10.1016/0304-3940(91)90095-B [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD (2001) An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci 21:3063–3072. 10.1523/JNEUROSCI.21-09-03063.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siderovski DP, Willard FS (2005) The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci 1:51–66. 10.7150/ijbs.1.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS (2006) Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci 9:501–510. 10.1038/nn1664 [DOI] [PubMed] [Google Scholar]
- Smith Y, Bevan MD, Shink E, Bolam JP (1998) Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 86:353–387. 10.1016/S0306-4522(98)00004-9 [DOI] [PubMed] [Google Scholar]
- Smith Y, Surmeier DJ, Redgrave P, Kimura M (2011) Thalamic contributions to basal ganglia-related behavioral switching and reinforcement. J Neurosci 31:16102–16106. 10.1523/JNEUROSCI.4634-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussig R, Tang WJ, Hepler JR, Gilman AG (1994) Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J Biol Chem 269:6093–6100. [PubMed] [Google Scholar]
- Tecuapetla F, Jin X, Lima SQ, Costa RM (2016) Complementary contributions of striatal projection pathways to action initiation and execution. Cell 166:703–715. 10.1016/j.cell.2016.06.032 [DOI] [PubMed] [Google Scholar]
- Traynor JR, Terzi D, Caldarone BJ, Zachariou V (2009) RGS9–2: probing an intracellular modulator of behavior as a drug target. Trends Pharmacol Sci 30:105–111. 10.1016/j.tips.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Chen Y, Leslie FM (2003) Expression of opioid peptides and receptors in striatum and substantia nigra during rat brain development. J Chem Neuroanat 26:17–36. 10.1016/S0891-0618(03)00031-0 [DOI] [PubMed] [Google Scholar]
- Xie CW, Lewis DV (1997) Involvement of cAMP-dependent protein kinase in mu-opioid modulation of NMDA-mediated synaptic currents. J Neurophysiol 78:759–766. 10.1152/jn.1997.78.2.759 [DOI] [PubMed] [Google Scholar]
- Xie K, Martemyanov KA (2011) Control of striatal signaling by G protein regulators. Front Neuroanat 5:49. 10.3389/fnana.2011.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Masuho I, Brand C, Dessauer CW, Martemyanov KA (2012) The complex of G protein regulator RGS9–2 and Gβ(5) controls sensitization and signaling kinetics of type 5 adenylyl cyclase in the striatum. Sci Signal 5:ra63. 10.1126/scisignal.2002922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou V, Georgescu D, Sanchez N, Rahman Z, DiLeone R, Berton O, Neve RL, Sim-Selley LJ, Selley DE, Gold SJ, Nestler EJ (2003) Essential role for RGS9 in opiate action. Proc Natl Acad Sci U S A 100:13656–13661. 10.1073/pnas.2232594100 [DOI] [PMC free article] [PubMed] [Google Scholar]