

Abstract
Background
Pseudogymnoascus destructans (Pd), the causative fungal agent of white-nose syndrome (WNS), has led to the deaths of millions of hibernating bats in the United States of America (USA) and Canada. Efficient strategies are needed to decontaminate Pd from the bat hibernacula to interrupt the disease transmission cycle without affecting the native microbes. Previously, we discovered a novel Trichoderma polysporum (Tp) strain (WPM 39143), which inhibited the growth of Pd in autoclaved soil samples. In the present investigation, we used culture-based approaches to determine Tp-induced killing of native and enriched Pd in the natural soil of two bat hibernacula. We also assessed the impact of Tp treatment on native microbial communities by metagenomics.
Results
Our results demonstrated that Tp at the concentration of 105 conidia/g soil caused 100% killing of native Pd in culture within 5 weeks of incubation. A 10-fold higher concentration of Tp (106 conidia/g soil) killed an enriched Pd population (105 conidia/g soil). The 12,507 fungal operational taxonomic units (OTUs, dominated by Ascomycota and Basidiomycota) and 27,427 bacterial OTUs (dominated by Acidobacteria and Proteobacteria) comprised the native soil microbes of the two bat hibernacula. No significant differences in fungal and bacterial relative abundances were observed between untreated and Tp-treated soil (105 Tp conidia/g soil, p ≤ 0.05).
Conclusions
Our results suggest that Tp-induced killing of Pd is highly specific, with minimal to no impact on the indigenous microbes present in the soil samples. These findings provide the scientific rationale for the field trials of Tp in the WNS-affected hibernacula for the effective decontamination of Pd and the control of WNS.
Electronic supplementary material
The online version of this article (10.1186/s40168-018-0512-6) contains supplementary material, which is available to authorized users.
Keywords: White-nose syndrome, Pseudogymnoascus destructans, Trichoderma polysporum, Biological decontamination, Biocontrol agent, Selective inhibition, Bat hibernacula, Native soil microbiota, Metagenomics
Background
Pseudogymnoascus destructans (Pd), the etiological agent of white-nose syndrome (WNS), has caused significant reductions in hibernating bat populations across the USA and Canada [1–6]. Mortality was first observed in hibernating bats in Howes Cave near Albany, New York (NY), in 2006, and has since spread extensively to 32 US states and 7 Canadian provinces [7]. Recently Pd was detected in a little brown bat (Myotis lucifugus) as far away as Washington State [8]. Several species of bats are already threatened with extinction, including Myotis lucifugus (little brown bat), Myotis sodalis (Indiana bat), and Myotis septentrionalis (northern long-eared bat) [3, 9]. Pseudogymnoascus destructans, a psychrophilic fungus, is well adapted to grow in the cold conditions prevailing in caves and mines [2, 10]. It has been shown to secrete proteolytic enzymes similar to the fungi that cause skin infections (dermatophytes); [2] and has a clonal population in the USA [11–13]. Low body temperature, along with reduced immune system in hibernating bats, provides the optimal growth environment for Pd [14–17]. Alarmingly, Pd has been found to survive in the affected hibernacula, even in the absence of bats [18–20]. Thus, an infected hibernaculum could remain contaminated with Pd for prolonged periods of time and serve as the foci for new infections [21]. Mathematical models have predicted that reducing Pd in caves and mines may prevent WNS-associated bat mortality [20, 22]. Currently, efforts are being devoted to the development and testing of chemical and biological agents for the effective eradication of Pd from bat hibernacula and hibernating bats. Although these control strategies appear to be promising, they are not being used for the large-scale decontamination of hibernacula, because of the likely off-target effects on the native microbial communities [23–27]. Considering the mass mortality of bats caused by WNS and the economic loss of 22.9 billion dollars to agricultural pest control in the USA annually [28], imminent steps are needed to decontaminate Pd from bat hibernacula and break its transmission cycle.
Previously, we characterized a novel, psychrotolerant Trichoderma polysporum (Tp) strain (WPM 39143) from the William Preserve Mine, Ulster County, NY, one of the mines at the epicenter of the WNS zoonotic [29]. Tp grew well at low temperatures of 6–15 °C and inhibited Pd in laboratory media and autoclaved soil samples [29]. The present study aimed at further evaluation of Tp as an effective biocontrol agent against Pd. Specifically, we examined (a) if Tp could act as an effective biocontrol agent against Pd in the natural soil from bat hibernacula and (b) whether Tp treatment impacted the native microbial communities in the natural soil from bat hibernacula.
Methods
Fungal strains and media
Pseudogymnoascus destructans (Pd) strain M1379 and Tp strain WPM 39143 were used as described previously [29, 30]. All fungal isolates were maintained on Sabouraud dextrose agar (SDA) slants at 4 °C and stored in 20% glycerol at − 70 °C in sterile cryogenic vials. Sabouraud dextrose agar fortified with an enhanced panel of antibacterials (SDA-A; Additional file 1), SDA-A with cycloheximide (0.2 g/L), and rose bengal agar with chloramphenicol (RBC; 100 μg/ml) were used for the isolation of Pd, Tp, and other fungi from bat hibernacula as described previously [30]. Potato dextrose agar (PDA) and water agar (WA) were used to induce spore formation in Pd and Tp. Millet seeds extract was used to fortify nutrients in the soil samples from bat hibernacula for Tp and Pd interaction studies. In brief, 50 g of millet seeds was added in 250 ml water, was autoclaved at 121 °C for 20 min, and was mixed with an additional 250 ml sterilized water. The resulting extract was passed through muslin cloth, autoclaved as above, allowed to cool, and stored at 4 °C until used. All the experiments were performed in the biosafety cabinet in a biosafety level 2 laboratory.
Biocontrol application of Tp in natural soil
To determine Tp-induced killing of Pd in a natural soil, one soil sample from Aeolus cave (AC), Bennington County, VT (dark black in color; collection date of 11/13/2015), and one soil sample from Barton Hill Mine (BHM), Essex County, NY (course sediments; collection date of 12/28/2015), were used. Soil samples, approximately 100 g, from each site were weighed, transferred to an autoclaved mortar and pestle, and mixed gently to obtain a homogeneous mixture. The mixture was aliquoted into eight vials for each site, with each vial containing 5 g of soil. Four vials were inoculated with Pd (105 conidia/g soil), and the remaining four vials received sterilized water. Following 1-week post-incubation, two of the four Pd-containing vials received Tp (105 conidia/g soil) to obtain a 1:1 ratio of Pd to Tp. The other two Pd containing vials received sterilized water (Pd only). Of the remaining four vials in which Pd was not inoculated, two received Tp (105 conidia/g soil), which served as Tp only controls, while the remaining two vials received sterilized water and served as soil only controls (Additional file 2). All the vials were incubated at 10 °C for 5 weeks.
To determine the number of Tp required for killing Pd in subsequent experiments, 10-fold (106 conidia/g soil) and 100-fold (107 conidia/g soil) higher concentrations of Tp conidia than Pd conidia (105 conidia/g soil) were added and these soil samples were incubated at 10 °C and then processed at 1, 3, and 5 weeks post-incubation (Additional file 3).
Culture recovery of Pd, Tp, and other fungi
Following incubation of soil samples at 10 °C for 1–5 weeks, 100 mg of soil sample was removed from each vial in duplicate and transferred into 2-ml screw cap vials. The soil sample was suspended in 500 μl of sterilized water and vortexed vigorously. Ten-fold dilutions of the supernatant were prepared, and 50 μl of each dilution was plated onto various media plates (150 mm diameter) in duplicate. The plates were incubated at 10 °C and checked periodically for the recovery of Pd, Tp, and other fungi as described previously [30]. In brief, SDA-A medium was used to determine the Tp colony-forming units (CFUs), SDA-A with cycloheximide was used to determine Pd CFU, and the RBC medium was used for the determination of other fungi present in the cave and mine soil samples. The percent killing of Pd by Tp was calculated using a formula 1 − (Pd CFU experiment/Pd CFU control) × 100.
Dual culture challenge studies
For dual culture challenge studies, approximately 1 × 1 mm of freshly grown Tp fungal hyphal mat was inoculated on one side of the SDA plate. Similarly, freshly grown fungal hyphal mat from various fungi obtained from both AC and BHM were inoculated on the opposite side of the plate. The interactions between Tp and other fungal isolates were assessed by measuring colony diameter following 18 days post-incubation at 10 °C. The Tp and different fungal isolate cultures alone served as a control.
Statistical analysis for culture-based assays (CFU enumeration and colony diameter) was performed using GraphPad Prism software (GraphPad, San Diego, CA, USA). The comparison of two groups was performed using a two-tailed unpaired t-test with a p value of ≤ 0.05 accepted as significant.
Metagenomics of soil samples
DNA libraries
For the microbial community analysis, DNA from 100 mg of untreated and Tp-treated (105 and 106 conidia/g soil for AC and 105 conidia/g soil for BHM) soil samples (in duplicate) were extracted with the Powersoil DNA isolation kit (MO BIO Laboratories, Carlsbad, CA, USA) (Additional file 4). DNA concentrations were measured using a Nanodrop spectrophotometer ND 2000 (Nano-Drop Technologies, Wilmington, DE, USA). DNA libraries were prepared using a two-step PCR. In the first PCR, the primer sets targeted the ITS2 region of the ribosomal RNA (rRNA) gene of fungi [31] and the hypervariable region V4 of the 16S rRNA gene of bacteria and archaea [32]. The same Illumina adaptor sequences were added to both fungal and bacterial/archaeal primers. Thus, the primer set used for the first PCR for fungi had the following sequence: Forward primer 5′- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAACTTTYRRCAAYGGATCWCT-3′ (locus specific sequence in bold) and Reverse primer 5′- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGAGCCTCCGCTTATTGATATGCTTAART -3′ (locus-specific sequence in bold). The primer set used for the first PCR for bacteria/archaea had the following sequence: Forward primer 5′- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGYCAGCMGCCGCGGTAA-3′ (locus-specific sequence in bold) and Reverse primer 5′- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGACTACNVGGGTWTCTAAT-3′ (locus-specific sequence in bold). PCR reactions were carried out in a total volume of 25 μl. Three microliters of extracted DNA (5 ng/μl) was added to the PCR reaction containing 15.1 μl of sterile water, 0.2 μl of bovine serum albumin, 2.5 μl of Accutaq LA buffer, 2 μl of dNTPs, 0.2 μl of Accutaq™ LA DNA polymerase (Sigma-Aldrich, St. Louis, MO, USA), and 1 μl each of fungal and bacterial/archaeal forward and reverse primers. All the PCR reactions were carried out in triplicate on a C1000 Touch Thermal Cycler (BioRad, Hercules, CA, USA). The thermocycling conditions for fungal-specific PCR were initial denaturation at 95 °C for 1 min, followed by 27 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 1 min, extension at 68 °C for 1 min, followed by final extension at 68 °C for 5 min. For bacterial/archaeal-specific PCR, thermocycling conditions were 95 °C for 1 min, followed by 27 cycles of denaturation at 94 °C for 30 s, annealing at 63 °C for 1 min, extension at 68 °C for 1 min, and a final extension at 68 °C for 5 min. The first-stage PCR products were run on the agarose gel to confirm the length of the respective bands. Triplicate samples were pooled to limit possible PCR artifacts. An Agencourt AMPure XP PCR purification kit (Beckman Coulter, Inc., Indianapolis, IN, USA) was used for the purification of the PCR products. The second PCR step added dual indices (barcode) along with Illumina sequencing adaptors using the Nextera XT index kit (Illumina-16S Metagenomics protocol). PCR reactions were carried out in a total volume of 50 μl, which contained 5 μl of PCR product from the first PCR and the 2× KAPA Hifi HotStart Ready mix. The reaction conditions were initial denaturation at 95 °C for 3 min, followed by eight cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 30 s, and a final extension at 72 °C for 5 min. All PCR reactions were performed in triplicate. The PCR products were cleaned using an Agencourt AMPure XP PCR purification kit. DNA concentration was measured using a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and the size was measured with an Agilent Tapestation using the D1000 High Sensitivity kit. The resulting DNA libraries were pooled, denatured, and sequenced on MiSeq using MiSeq Reagent kit v3 (600 cycles) (Illumina Inc., San Diego, CA, USA) by the Wadsworth Center Advanced Sequencing Core Facility.
Sequence processing
The Wadsworth Sequencing Core demultiplexed reads and removed primer and linker sequences before analysis. The subroutines of BBTools v36.38 [33], BBmerge and BBduk, merged all pairs of forward and reverse reads, quality trimmed merged pairs (with the parameters trimq = 20, minq = 20, minlength = 150, minavgquality = 20, efilter = 3, mininsert = 250, mininsert0 = 250), and removed any remaining forward and reverse primers for fungal and bacterial samples. The ITS2 region for taxonomic identification was extracted from fungal sequences using ITSx v.1.0.11 [34], which removed 5.8S and 28S regions from merged sequences. Chimeric sequences were removed using USearch61 with a modified Unite “ITS2-only” reference dataset [35] (version 7.2 release 28.06.2017) which was designed to serve as a chimera-free reference database. Chimeric 16S sequences were removed from bacterial samples in QIIME v.1.9.0 [36] via Usearch61 [37, 38] and the most recent version of the Greengenes database (gg_13_8 minor release of gg_13_5 from 15.08.2013). Using default parameters, QIIME further processed quality-filtered sequences, which were then clustered into operational taxonomic units (OTUs) at 97% similarity by the UCLUST algorithm [37] and assigned taxonomy by mothur v.1.25 [39, 40] with the pick open reference OTU option and a default confidence level of 0.5. The latest releases of the Greengenes 16s (gg_13_8) and UNITE datasets (version 7 release 28.06.2017) [41], both clustered at 97% similarity, served as the reference databases for clustering and taxonomic assignment of bacterial and fungal sequences, respectively.
Sample statistics
For bacterial and fungal datasets, Chao1, observed species, and Simpson’s index estimates of alpha diversity were calculated in QIIME with rarefied OTU tables. Significant differences across treatments and locations were assessed via a non-parametric t-test with 999 permutations of the p value. QIIME also calculated beta diversity with Bray Curtis, and Euclidean metrics and significant differences in community composition were identified with an analysis of similarities (ANOSIM). Principal component analysis plots were generated in R v.3.3.0 [42] with Euclidean distance matrices to confirm consistency among replicates and verify that most of the variation in datasets was derived from differences in treatment and location (data not shown).
Bacterial and fungal samples were rarefied to depths of 190,000 and 92,226, respectively, and relative abundances were summarized to a genus level (if possible). Abundance count at the phylum and genus levels were exported from QIIME and analyzed in DESeq2 v.3.5 [43] in R to identify significant changes in taxonomic composition. Only taxa with four or more counts across samples were included in DESeq2 analyses to remove sparse OTUs.
Fungal identification
All fungi recovered from the soil samples were identified by morphological and molecular methods [44]. For molecular testing, DNA from pure fungal colonies was extracted using MasterPureTM Complete DNA and RNA purification kits (Epicenter, Madison, WI, USA) as per manufacturer’s instructions. The extracted DNA was used for the amplification of internal transcribed spacer (ITS) regions 1 and 2 (ITS1, 5.8S, and ITS2) of the ribosomal gene as described previously [30]. In some instances, where the ITS region failed to provide fungal identification, the D1/D2 region of the large subunit (LSU) of the 28S rDNA gene was amplified [30]. PCR was carried out as described in White et al. [45]. PCR products were cleaned with ExoSAP-IT (USB Corp., Cleveland, OH, USA) and sequenced at the Wadsworth Center Advanced Genomics Core. The sequences were assembled and edited for accuracy using Sequencher software 4.8 (Gen Codes Corp., Ann Arbor, MI, USA). All unknown sequences were compared to the NCBI GenBank database with blast [46] and the Westerdijk Fungal Biodiversity Institute database [47] for fungal identifications; % identity of ≥ 97 was used for species confirmation. In case of discrepant results between the two databases, the Westerdijk Fungal Biodiversity Institute database was preferred as it has curated sequences.
Results
Biocontrol of Pd in natural soil – A culture-based approach
Pd was recovered from native AC soil at ~ 104 CFU/g soil (Fig. 1). Pd recovery was very low from BHM (~ 8 CFU/g soil), consistent with our previous observations [30]. Treatment of AC soil with Tp (105 conidia/g soil) resulted in 100% inhibition of the native Pd population within 5 weeks post-incubation at 10 °C, confirming high biocontrol potential of Tp (Fig. 1). However, when AC and BHM soil samples were enriched with Pd (105 conidia/g soil), the Tp-induced killing of Pd was markedly reduced to approximately 40 to 43% (data not shown). Maximum killing of Pd in enriched soil was observed, when Tp inoculum was increased from 1-fold (105conidia/g soil) to 10-fold (106 conidia/g soil) to that of Pd (105 conidia/g soil). Although Tp killed Pd as early as 1-week post-incubation, 95% killing was achievable at 5-week post-incubation (Fig. 2a). Further increase in Tp to 100-fold of Pd was not effective as the killing of Pd was observed to be only 84% at 5-week post-incubation (Fig. 2a). Enriching soil nutrients by adding millet seeds extract did not impact Tp-induced killing of Pd as only 87 and 72% Pd was killed with the additions of 10-fold and 100-fold more Tp, respectively (Fig. 2b). These results indicated that the ratio of Tp to Pd of 10:1 appeared to optimally render the maximum inhibitory effect. The soil microbial communities did not hamper Tp growth in any of the soil samples tested. The Tp growth was not only sustained but also increased with prolonged incubation (data not shown).
Fig. 1.
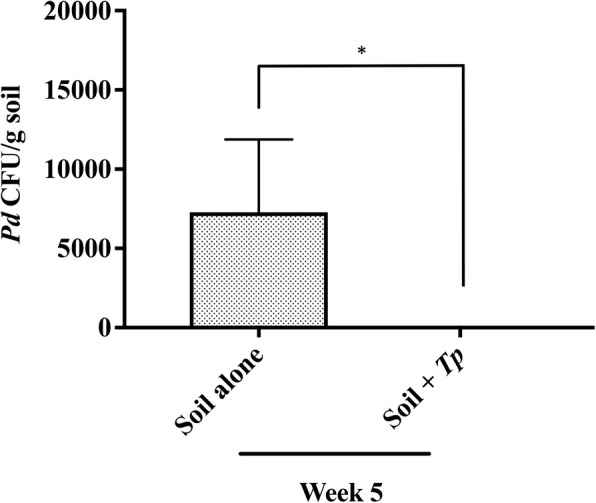
Inhibition of native Pd by Tp in AC soil samples. Untreated soil (soil alone) and soil treated with Tp (105 conidia/g soil) were incubated at 10 °C. Five weeks post-incubation, two aliquots from each sample were processed for the recovery of Pd. Tp induced 100% killing of native Pd in the AC soil samples (p < 0.05)
Fig. 2.

Inhibition of enriched Pd by Tp in AC soil samples. a Soil samples from AC were inoculated with Pd (105 conidia/g soil) in duplicate. Following 7 days post-incubation at 10 °C, Tp at 10-fold (106 conidia/g soil) or 100-fold (107 conidia/g soil) to that of Pd (105 conidia/g soil) was added. Untreated soil (soil alone) and soil containing Tp served as controls. Two aliquots from each sample were processed for the recovery of Pd in culture at week 1, 3, and 5 post-incubation. Approximately, 50 and 67% killing of Pd was observed with 10-fold and 100-fold higher Tp at week 1 post-incubation, which gradually increased to 57 and 72% at week 3 post-incubation and 95 and 84% at week 5 post-incubation. b Soil samples were inoculated with Pd and treated with Tp as explained in a, except that these samples also received 1% millet seeds extract. Addition of millet seeds extract did not enhance Tp killing of Pd.
Effect of Tp treatment on microbial communities of cave and mine soil samples—a metagenomics approach
Sequence analysis
A total of 2,003,427 fungal sequences passed initial quality filters. Of these, 96% (1,921,639) passed QIIME’s quality standards as well, while 0.07% (1347) were flagged as chimeric and removed (Table 1). This resulted in an average of 192,163 sequences per sample (N = 10), which were clustered into 12,507 OTUs. For all bacterial samples, a total of 3,295,648 sequences passed the initial quality filters of BBtools with 97% of these sequences (3,177,499) accepted by QIIME and 0.6% (21,503) identified as chimeric and removed (Table 1). On average, there were 317,749 sequences per sample (N = 10), which clustered into 27,427 OTUs.
Table 1.
Fungal and bacterial community analyses by high throughput sequencing
| Sample | Fungi | Bacteria | ||
|---|---|---|---|---|
| BBmerge and bbduk | QIIME and Usearch61 | BBmerge and bbduk | QIIME and Usearch61 | |
| AC Soil alone 1 | 96,993 | 92,225 | 247,465 | 237,561 |
| AC Soil alone 2 | 161,501 | 153,529 | 321,874 | 308,610 |
| AC Soil + Tp A1 | 214,333 | 204,336 | 393,209 | 377,754 |
| AC Soil + Tp A2 | 166,355 | 158,564 | 344,568 | 330,694 |
| AC Soil + Tp B1 | 323,672 | 312,063 | 444,201 | 427,840 |
| AC Soil + Tp B2 | 116,711 | 105,622 | 207,965 | 199,979 |
| BH Soil alone 1 | 156,003 | 150,631 | 275,597 | 267,273 |
| BH Soil alone 2 | 233,462 | 225,964 | 306,924 | 298,202 |
| BH Soil + Tp A1 | 205,092 | 200,594 | 304,892 | 295,134 |
| BH Soil + Tp A2 | 329,305 | 318,111 | 448,953 | 434,452 |
| Total | 2,003,427 | 1,921,639 | 3,295,648 | 3,177,499 |
Soil samples from Aeolus Cave (AC) and Barton Hill Mine (BHM) were treated with Tp at the concentration of 105 conidia/g soil (A1, A2) or 106 conidia/g soil (B1, B2) followed by gDNA extraction, PCR, and high throughput sequencing. Untreated soil samples (soil alone) were included for comparison. Note, due to small number of Pd recovered from BHM soil, the Tp treatment was limited to 105 conidia/g soil
Tp treatment and location effects on alpha and beta diversity of soil samples
No significant differences in the alpha diversity were observed across treatments or geographical locations for either bacterial or fungal communities (Additional file 5). Fungal community composition changes, as assessed by the beta diversity estimates, also did not differ significantly due to Tp treatment of either AC or BHM soils. However, there was a significant difference in community composition between the two locations (ANOSIM test statistic = 1.0, p value 0.003). Similarly, bacterial beta diversity was not significantly altered by Tp treatment within AC or BHM but varied significantly between sites.
Treatment effect on the abundance of fungal and bacterial communities at the phylum and genus levels
For fungi, 97% of all sequences could be assigned at the phylum level, with seven phyla identified from AC and five phyla identified from BHM. Both AC and BHM were dominated by OTUs from Ascomycota and Basidiomycota, which comprised over 80% of the fungal communities (Fig. 3a). Soil subjected to Tp treatment (105 conidia/g soil) showed no significant differences in the phylum abundances for fungi at either AC or BHM (Additional file 6). However, there was a significant increase in Ascomycota for AC soil subjected to a higher Tp treatment (106 conidia/g soil, Additional file 6), which reflected the dramatic increase in Trichoderma in these samples (Fig. 3b). Similarly, analysis at the genus level revealed highly significant increases in Trichoderma for both AC and BHM for both treatments, with few other taxa being affected (Fig. 3b, Additional file 7). Although no significant changes in Pseudogymnoascus abundance were observed (Additional file 7), the genus encompasses several species, which might not be influenced by Tp treatment.
Fig. 3.

Relative abundance of fungal and bacterial communities in soil samples from bat hibernacula with or without Tp treatment. The soil samples from Aeolus Cave (AC) and Barton Hill Mine (BHM) were treated with Tp at the concentration of 105 conidia/g soil (A1 & A2) or 106 conidia/g soil (B1 & B2). Untreated soil samples (soil alone) were included for comparison. gDNA was extracted followed by PCR and high throughput sequencing. a The relative abundance of fungal phyla is shown. Ascomycota dominated, followed by Basidiomycota and early diverging fungal lineages (Chytridiomycota, Entomophthoromycota, Mortierellomycota, Mucormycota, and Rozellomycota) from both AC and BHM. b The relative distribution of 25 most abundant fungal genera is shown. The increases in Trichoderma as represented by “T” in both AC and BHM were due to the exogenous addition of Tp. c The relative abundance of bacterial phyla is shown. Acidobacteria dominated AC soil and Protobacteria dominated BHM soil
For bacteria/archaea, a total of 50 phyla were identified from AC, whereas only 37 were identified from BHM with 0.27% of the sequences designated as unclassified (Bacteria; Other). The five most abundant phyla in AC were Acidobacteria (28%), Proteobacteria (18%), Planctomycetes (13%), Chloroflexi (10%), and Actinobacteria (9.5%) (Fig. 3c). The most abundant phyla identified from BHM included Planctomycetes (11%), Acidobacteria (10%), Firmicutes (10%), and Actinobacteria (8.8%), with Proteobacteria (44%) comprising almost half of the BHM bacterial community (Fig. 3c). Despite apparent differences in the community richness and evenness between the AC and BHM, there were no significant differences in alpha diversity (Additional file 5). There were also no significant differences in bacterial relative abundances between untreated and Tp-treated soil (105 Tp conidia/g soil) for AC at the phylum or genus level (Additional files 8 and 9). For AC soil samples treated with 106 Tp conidia/g soil, 12 phyla and 40 genera were significantly affected compared to untreated soil samples (Additional files 8 and 9). For BHM soil samples treated with 105 Tp conidia/g soil, no phyla and only 12 genera showed significant changes (Additional files 8 and 9).
Tp interaction with other cave/mine fungi—a culture-based approach
A total of 39 fungal species were recovered from AC, and of these, 85% belonged to Ascomycota, 13% belonged to early diverging fungal lineage (EDFL), and 2% belonged to Basidiomycota (Fig. 4a, Additional file 10). Of the 31 fungal species recovered from BHM, 87% belonged to Ascomycota, 10% belonged to EDFL, and 3% belonged to Basidiomycota (Fig. 4a, Additional file 11). Thus, Ascomycota dominated the fungal species followed by EDFL and Basidiomycota from both AC and BHM.
Fig. 4.

Fungal recovery from bat hibernacula and their interaction with Tp. a Soil samples from Aeolus Cave (AC) and Barton Hill Mine (BHM) were suspended in sterile water and then spread on culture media plates. Colonies recovered were identified by sequencing of the ITS and D1/D2 regions of the ribosomal gene followed by BLAST search. Pie charts represent the relative distributions of fungal phyla. Ascomycota dominated followed by Early Diverging Fungal Lineage, and Basidiomycota in both AC and BHM. b Interaction of Tp with other fungal species isolated from AC and BHM was carried out on SDA plate and the results of these interactions were assessed 18 days post-incubation at 10 °C. Upper panel denotes Tp-induced inhibition of Pd and Microascus species (Ms). Lower panel denotes fungal species not inhibited by Tp. Two such examples are Mortierella clonocystis (Mc) and Penicillium sopii (Ps)
Dual challenge study
The dual interaction studies indicated that Tp is highly specific in inhibiting Pd. In addition to Pd, only one fungal isolate belonging to genus Microascus was inhibited. The rest of the 67 fungal species collectively identified from both AC and BHM were not inhibited, including the closely related species Pseudogymnoascus pannorum (Fig. 4b, Additional files 10 and 11).
Discussion
We demonstrated that the biocontrol agent Tp inhibited Pd in the presence of microbes that are native to the soil from the affected hibernacula. This finding further expanded our earlier observations of the efficacy of Tp in killing of Pd in autoclaved soil samples [29]. Another important finding of this study was the specificity of Tp for killing Pd with minimal to no impact on the microbial diversity and community structures of soil samples tested from both AC and BHM. Even though the microbial community compositions of AC and BHM were significantly different, they were not affected by Tp treatment, suggesting that the soil communities are relatively robust and indifferent to Tp treatment. The combined culture-based and metagenomics approaches allowed us to follow the fate of the biocontrol agent and its target in the treated soil. Culture-based monitoring of Pd and Tp was important to estimate loss of viable organisms while DNA-based approaches provided better census of microbial communities.
Several DNA and culture-based studies have revealed wide distribution and persistence of Pd in bat hibernacula [18, 21, 48, 49]. Other published cave fungal surveys indicate that Pd could survive and persist in bat hibernacula for prolonged periods and can have impact on both WNS disease management and epidemiology [18]. To eradicate Pd from bat hibernacula, we need highly competent biocontrol agent, which can grow, sustain, and selectively kill Pd without impacting the hibernacula ecosystem. The Tp strain used in this investigation fulfills all these criteria, thereby strengthening the argument for application of Tp as a potential biocontrol agent against Pd in caves and mines.
In concordance with metagenomics analysis, the dual-culture challenge studies of Tp with several fungi recovered from AC and BHM revealed that except for one isolate of Microascus species, the growth of other fungi was not affected by Tp. Microascus is a soil saprophyte and a common agent of bio-deterioration [50]. Since other species in the genus Microascus were not inhibited by Tp, we do not anticipate deleterious effects on cave/mine ecosystem. Conversely, Tp grew well in the presence of both fast- and slow-growing fungi, as well as in the presence of other microbial communities indigenous to the soil samples tested. The high survival potential of Tp in hibernacula soil suggests its ability to survive under unfavorable conditions and high reproductive capacity.
Microbial communities play several critical roles in the soil, including organic matter decomposition and control of its cycle, regulation of mineral nutrient availability, and nitrogen fixation [51]. Thousands of bacterial, archaeal, and eukaryotic organisms are present in natural soil and collectively contribute to maintaining the myriad of functions of soil. Microbial inoculation of a biocontrol agent can cause tremendous changes in the number and composition of taxonomic groups. These changes can increase the diversity of the soil samples while also having toxic effects on the indigenous microbes [52]. Thus, the practical use of any microbial inoculation should be rigorously tested in a laboratory setting to avoid any deleterious effects to microbial diversity in soils. To this end, we have rigorously tested the use of Tp as a biocontrol agent for the eradication of Pd in cave and mine soil samples and Tp treatment in large had no impact on the native microbial communities other than Pd.
Conclusions
The present study demonstrates the remarkable specificity and high potency of Tp for killing Pd in the presence of indigenous microbial communities with minimal to no impact on the microbial community structure or diversity present in AC and BHM soil samples. The study rigorously tested the application of Tp in natural soil samples in a lab setting, with results that strengthens the argument for Tp’s application as a biocontrol agent under field conditions.
Additional files
Concentrations of extended panel of antibiotics in Sabouraud dextrose agar. (DOCX 13 kb)
Flow chart of Pd and Tp (1:1 ratio) interaction in AC and BHM soil samples. (TIF 294 kb)
Flow chart of Pd and Tp (1:10 and 1:100 ratio) interaction in AC soil samples. (TIF 324 kb)
Flow chart of Pd and Tp interaction (1:1 and 1:10 ratio) in AC soil and Pd and Tp interaction (1:1 ratio) in BHM soil followed by DNA extraction and metagenomics analysis. (TIF 184 kb)
Comparison of alpha-diversity among Tp treatments and locations using a non-parametric t-test. (XLSX 55 kb)
DESeq2 analysis of changes in the abundance of fungal phyla in soil from bat hibernacula with or without Tp treatment. (XLSX 55 kb)
DESeq2 analysis of changes in the abundance of fungal genera in soil from bat hibernacula with or without Tp treatment. (XLSX 97 kb)
DESeq2 analysis of changes in the abundance of bacterial phyla in soil from bat hibernacula with or without Tp treatment. (XLSX 19 kb)
DESeq2 analysis of changes in abundance of bacterial genera in soil from bat hibernacula with or without Tp treatment. (XLSX 193 kb)
Fungi recovered from AC soil and assessed for growth inhibition by Tp. (DOCX 20 kb)
Fungi recovered from BHM soil and assessed for growth inhibition by Tp. (DOCX 17 kb)
Acknowledgements
We are thankful to the Wadsworth Center Media & Tissue Culture Core for the preparation of various media and reagents and Wadsworth Center Advanced Genomics Core for sequencing of DNA samples. We also thank Mr. Al Hicks for providing us soil samples for both AC and BHM. The members of Chaturvedi Lab are acknowledged for their support and help.
Funding
This study was supported in part with funds from Fish and Wildlife Service (F15AS00188) to SC and VC and in part with funds from the National Science Foundation (1203528) to VC and SC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The datasets generated and analyzed during the current study have been deposited in the NCBI BioProject database (accession number PRJNA433166) (https://www.ncbi.nlm.nih.gov/bioproject/). Raw sequences and metadata are available in the SRA database with the following accession numbers: SRR6676481, Aeolus Cave Soil Alone, fungal data, replicate 1; SRR6676487, Aeolus Cave Soil Alone, fungal data, replicate 2; SRR6676483, Aeolus Cave Soil + Tp (105 conidia/g soil), fungal data, replicate 1; SRR6676489, Aeolus Cave Soil + Tp (105 conidia/g soil), fungal data, replicate 2; SRR6676477, Aeolus Cave Soil + Tp (106 conidia/g soil), fungal data, replicate 1; SRR6676491, Aeolus Cave Soil + Tp (106 conidia/g soil), fungal data, replicate 2; SRR6676479, Barton Hill Mine Soil Alone, fungal data, replicate 1; SRR6676493, Barton Hill Mine Soil Alone, fungal data, replicate 2; SRR6676475, Barton Hill Mine Soil + Tp (105 conidia/g soil), fungal data, replicate 1; SRR6676485, Barton Hill Mine Soil + Tp (106 conidia/g soil), fungal data, replicate 2; SRR6676480, Aeolus Cave Soil Alone, bacterial data, replicate 1; SRR6676486, Aeolus Cave Soil Alone, bacterial data, replicate 2; SRR6676482, Aeolus Cave Soil + Tp (105 conidia/g soil), bacterial data, replicate 1; SRR6676488, Aeolus Cave Soil + Tp (105 conidia/g soil) bacterial data, replicate 2; SRR6676476, Aeolus Cave Soil + Tp (106 conidia/g soil) bacterial data, replicate 1; SRR6676490, Aeolus Cave Soil + Tp (106 conidia/g soil), bacterial data, replicate 2; SRR6676478, Barton Hill Mine Soil Alone, bacterial data, replicate 1; SRR6676492, Barton Hill Mine Soil Alone, bacterial data, replicate 2; SRR6676474, Barton Hill Mine Soil + Tp (105 conidia/g soil), bacterial data, replicate 1; SRR6676484, Barton Hill Mine Soil + Tp (105 conidia/g soil) bacterial data, replicate 2. A permanent link to the data can be found at https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP132276.
Abbreviations
- AC
Aeolus cave
- ANOSIM
Analysis of similarities
- BHM
Barton Hill Mine
- CFU
Colony-forming unit
- EDFL
Early diverging fungal lineage
- ITS
Internal transcribed spacer
- LSU
Large subunit
- OTUs
Operational taxonomic units
- Pd
Pseudogymnoascus destructans
- PDA
Potato dextrose agar
- RBC
Rose bengal agar with chloramphenicol
- rRNA
Ribosomal RNA
- SDA
Sabouraud dextrose agar
- Tp
Trichoderma polysporum
- WA
Water agar
- WNS
White-nose syndrome
Authors’ contributions
APS and SC conceived and designed the experiments. APS performed the experiments. APS, ELN, and SC analyzed the data. APS, ELN, SC, and VC wrote the paper. All authors read and approved the final manuscript.
The collection of soil samples from Barton Hill Mine in New York State (NYS) was done with the permission and cooperation of DEC, as supplemental activities during standard winter hibernacula surveys conducted by the NYS Department of Environmental Conservation (NYSDEC). Aeolus Cave soil samples were collected with the permission and cooperation of the Vermont Department of Fish and Wildlife (VDFW) as a supplemental activity during a scheduled visit to the site. The samples were collected as members of the agency teams were counting the bats, with no additional disturbance events. No visitation and sampling permits were required in either state.
Not applicable.
The authors declare that they have no competing interests.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Blehert DS, Hicks AC, Behr M, Meteyer CU, Berlowski-Zier BM, Buckles EL, et al. Bat white-nose syndrome: an emerging fungal pathogen? Science. 2009;323(5911):227. doi: 10.1126/science.1163874. [DOI] [PubMed] [Google Scholar]
- 2.Chaturvedi V, Springer DJ, Behr MJ, Ramani R, Li X, Peck MK, et al. Morphological and molecular characterizations of psychrophilic fungus Geomyces destructans from New York bats with white-nose syndrome (WNS) PLoS One. 2010;5(5):e10783. doi: 10.1371/journal.pone.0010783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frick WF, Pollock JF, Hicks AC, Langwig KE, Reynolds DS, Turner GG, et al. An emerging disease causes regional population collapse of a common North American bat species. Science. 2010;329(5992):679–682. doi: 10.1126/science.1188594. [DOI] [PubMed] [Google Scholar]
- 4.Blehert DS. Fungal disease and the developing story of bat white-nose syndrome. PLoS Pathog. 2012;8(7):e1002779. doi: 10.1371/journal.ppat.1002779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Reagan SM, Magori K, Pulliam JT, Zokan MA, Kaul RB, Barton HD, et al. Multi-scale model of epidemic fade-out: will local extirpation events inhibit the spread of white-nose syndrome? Ecol Appl. 2015;25(3):621–633. doi: 10.1890/14-0417.1. [DOI] [PubMed] [Google Scholar]
- 6.White-nosesyndrome.org. http://www.whitenosesyndrome.org/about/where-is-it-now. Accessed 10 June 2018.
- 7.White-nosesyndrome.org. http://www.whitenosesyndrome.org/resources/map. Accessed 10 June 2018.
- 8.Lorch JM, Palmer JM, Lindner DL, Ballmann AE, George KG, Griffin K, et al. First detection of bat white-nose syndrome in western North America. mSphere. 2016;1(4):e00148–e00116. doi: 10.1128/mSphere.00148-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langwig KE, Frick WF, Bried JT, Hicks AC, Kunz TH, Kilpatrick AM. Sociality, density-dependence and microclimates determine the persistence of populations suffering from a novel fungal disease, white-nose syndrome. Ecol Lett. 2012;15(9):1050–1057. doi: 10.1111/j.1461-0248.2012.01829.x. [DOI] [PubMed] [Google Scholar]
- 10.Gargas A, Trest M, Christensen M, Volk TJ, Blehert DS. Geomyces destructans sp. nov. associated with bat white-nose syndrome. Mycotaxon. 2009;108(1):147–154. doi: 10.5248/108.147. [DOI] [Google Scholar]
- 11.Ren P, Haman KH, Last LA, Rajkumar SS, Keel MK, Chaturvedi V. Clonal spread of Geomyces destructans among bats, midwestern and southern United States. Emerg Infect Dis. 2012;18(5):883–885. doi: 10.3201/eid1805.111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajkumar SS, Li X, Rudd RJ, Okoniewski JC, Xu J, Chaturvedi S, et al. Clonal genotype of Geomyces destructans among bats with white nose syndrome, New York, USA. Emerg Infect Dis. 2011;17(7):1273–1276. doi: 10.3201/eid1707.102056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drees KP, Lorch JM, Puechmaille SJ, Parise KL, Wibbelt G, Hoyt JR, et al. Phylogenetics of a fungal invasion: origins and widespread dispersal of white-nose syndrome. MBio. 2017;8(6):e01941–e01917. doi: 10.1128/mBio.01941-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyles JG, Willis CKR. Could localized warm areas inside cold caves reduce mortality of hibernating bats affected by white-nose syndrome? Frontiers Ecol Environment. 2010;8(2):92–98. doi: 10.1890/080187. [DOI] [Google Scholar]
- 15.Webb PI, Speakman JR, Racey PA. How hot is a hibernaculum? A review of the temperatures at which bats hibernate. Canadian J Zoology. 1996;74(4):761–765. doi: 10.1139/z96-087. [DOI] [Google Scholar]
- 16.Bouma HR, Carey HV, Kroese FG. Hibernation: the immune system at rest? J Leukoc Biol. 2010;88(4):619–624. doi: 10.1189/jlb.0310174. [DOI] [PubMed] [Google Scholar]
- 17.Verant ML, Boyles JG, Waldrep W, Jr, Wibbelt G, Blehert DS. Temperature-dependent growth of Geomyces destructans, the fungus that causes bat white-nose syndrome. PloS one. 2012;7(9):e46280. doi: 10.1371/journal.pone.0046280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lorch JM, Muller LK, Russell RE, O'Connor M, Lindner DL, Blehert DS. Distribution and environmental persistence of the causative agent of white-nose syndrome, Geomyces destructans, in bat hibernacula of the eastern United States. Appl Environment Microbiol. 2013;79(4):1293–1301. doi: 10.1128/AEM.02939-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raudabaugh DB, Miller AN. Nutritional capability of and substrate suitability for Pseudogymnoascus destructans, the causal agent of bat white-nose syndrome. PLoS One. 2013;8(10):e78300. doi: 10.1371/journal.pone.0078300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reynolds HT, Ingersoll T, Barton HA. Modeling the environmental growth of Pseudogymnoascus destructans and its impact on the white-nose syndrome epidemic. J Wildl Dis. 2015;51(2):318–331. doi: 10.7589/2014-06-157. [DOI] [PubMed] [Google Scholar]
- 21.Lindner DL, Gargas A, Lorch JM, Banik MT, Glaeser J, Kunz TH, et al. DNA-based detection of the fungal pathogen Geomyces destructans in soils from bat hibernacula. Mycologia. 2011;103(2):241–246. doi: 10.3852/10-262. [DOI] [PubMed] [Google Scholar]
- 22.Meyer AD, Stevens DF, Blackwood JC. Predicting bat colony survival under controls targeting multiple transmission routes of white-nose syndrome. J Theor Biol. 2016;409:60–69. doi: 10.1016/j.jtbi.2016.08.033. [DOI] [PubMed] [Google Scholar]
- 23.Hoyt JR, Cheng TL, Langwig KE, Hee MM, Frick WF, Kilpatrick AM. Bacteria isolated from bats inhibit the growth of Pseudogymnoascus destructans, the causative agent of white-nose syndrome. PLoS One. 2015;10(4):e0121329. doi: 10.1371/journal.pone.0121329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cornelison CT, Keel MK, Gabriel KT, Barlament CK, Tucker TA, Pierce GE, et al. A preliminary report on the contact-independent antagonism of Pseudogymnoascus destructans by Rhodococcus rhodochrous strain DAP96253. BMC Microbiol. 2014;14:246. doi: 10.1186/s12866-014-0246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng TL, Mayberry H, McGuire LP, Hoyt JR, Langwig KE, Nguyen H, et al. Efficacy of a probiotic bacterium to treat bats affected by the disease white-nose syndrome. J Appl Ecol. 2017;54(3):701–708. doi: 10.1111/1365-2664.12757. [DOI] [Google Scholar]
- 26.Cornelison CT, Gabriel KT, Barlament C, Crow SA., Jr Inhibition of Pseudogymnoascus destructans growth from conidia and mycelial extension by bacterially produced volatile organic compounds. Mycopathologia. 2014;177(1–2):1–10. doi: 10.1007/s11046-013-9716-2. [DOI] [PubMed] [Google Scholar]
- 27.Wilcox A, Willis CKR. Energetic benefits of enhanced summer roosting habitat for little brown bats (Myotis lucifugus) recovering from white-nose syndrome. Conservation Physiol. 2016;4(1):cov070–covcov. doi: 10.1093/conphys/cov070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyles JG, Cryan PM, McCracken GF, Conservation KTH. Economic importance of bats in agriculture. Science. 2011;332(6025):41–42. doi: 10.1126/science.1201366. [DOI] [PubMed] [Google Scholar]
- 29.Zhang T, Chaturvedi V, Chaturvedi S. Novel Trichoderma polysporum strain for the biocontrol of Pseudogymnoascus destructans, the fungal etiologic agent of bat white-nose syndrome. PLoS One. 2015;10(10):e0141316. doi: 10.1371/journal.pone.0141316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang T, Victor TR, Rajkumar SS, Li X, Okoniewski JC, Hicks AC, et al. Mycobiome of the bat white nose syndrome affected caves and mines reveals diversity of fungi and local adaptation by the fungal pathogen Pseudogymnoascus (Geomyces) destructans. PLoS One. 2014;9(9):e108714. doi: 10.1371/journal.pone.0108714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor DL, Walters WA, Lennon NJ, Bochicchio J, Krohn A, Caporaso JG, et al. Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for Illumina amplicon sequencing. Appl Environment Microbiol. 2016;82(24):7217–7226. doi: 10.1128/AEM.02576-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Earth Microbiome Project. http://www.earthmicrobiome.org/emp-standard-protocols/16S/. Accessed Jun 2017.
- 33.BBMap. https://sourceforge.net/projects/bbmap/.
- 34.Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol. 2013;4(10):914–919. [Google Scholar]
- 35.Nilsson RH, Tedersoo L, Ryberg M, Kristiansson E, Hartmann M, Unterseher M, et al. A comprehensive, automatically updated fungal ITS sequence dataset for reference-based chimera control in environmental sequencing efforts. Microbes Environments. 2015;30(2):145–150. doi: 10.1264/jsme2.ME14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics (Oxford, England) 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 38.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (Oxford, England) 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environment Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environment Microbiol. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Unite community. https://unite.ut.ee.
- 42.The R Project for Statistical Computing. https://www.r-project.org/.
- 43.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Hoog GSGJ, Gene J, Figueras MJ. Atlas of clinical fungi. Utrecht: Centraalbureau voor Schimmelcultures/Universitat Rovira i Virgili; 2000. [Google Scholar]
- 45.White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogeneticcs. In: Innis DHG MA, Snisky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. New York: NY Academic Press Inc; 1990. p. 315–24.
- 46.Basic Local Alignment Search Tool (BLAST)-NCBI-NIH. https://blast.ncbi.nlm.nih.gov/Blast.cgi. [DOI] [PubMed]
- 47.WESTERDIJK Fungal Biodiveristy Institute. http://www.westerdijkinstitute.nl.
- 48.Puechmaille SJ, Wibbelt G, Korn V, Fuller H, Forget F, Muhldorfer K, et al. Pan-European distribution of white-nose syndrome fungus (Geomyces destructans) not associated with mass mortality. PLoS One. 2011;6(4):e19167. doi: 10.1371/journal.pone.0019167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shuey MM, Drees KP, Lindner DL, Keim P, Foster JT. Highly sensitive quantitative PCR for the detection and differentiation of Pseudogymnoascus destructans and other Pseudogymnoascus species. Appl Environmental Microbiol. 2014;80(5):1726–1731. doi: 10.1128/AEM.02897-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Onions AHS, Allsopp D, Eggins HOW. UK: Cambridge University Press ISBN 978-0521427821. 1991. Smith's introduction to industrial mycology (7th ed.). Cambridge. [Google Scholar]
- 51.Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL, et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl Acad Sci. 2012;109(52):21390–21395. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trabelsi D, Mhamdi R. Microbial inoculants and their impact on soil microbial communities: a review. BioMed Res Internat. 2013;2013:11. doi: 10.1155/2013/863240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Concentrations of extended panel of antibiotics in Sabouraud dextrose agar. (DOCX 13 kb)
Flow chart of Pd and Tp (1:1 ratio) interaction in AC and BHM soil samples. (TIF 294 kb)
Flow chart of Pd and Tp (1:10 and 1:100 ratio) interaction in AC soil samples. (TIF 324 kb)
Flow chart of Pd and Tp interaction (1:1 and 1:10 ratio) in AC soil and Pd and Tp interaction (1:1 ratio) in BHM soil followed by DNA extraction and metagenomics analysis. (TIF 184 kb)
Comparison of alpha-diversity among Tp treatments and locations using a non-parametric t-test. (XLSX 55 kb)
DESeq2 analysis of changes in the abundance of fungal phyla in soil from bat hibernacula with or without Tp treatment. (XLSX 55 kb)
DESeq2 analysis of changes in the abundance of fungal genera in soil from bat hibernacula with or without Tp treatment. (XLSX 97 kb)
DESeq2 analysis of changes in the abundance of bacterial phyla in soil from bat hibernacula with or without Tp treatment. (XLSX 19 kb)
DESeq2 analysis of changes in abundance of bacterial genera in soil from bat hibernacula with or without Tp treatment. (XLSX 193 kb)
Fungi recovered from AC soil and assessed for growth inhibition by Tp. (DOCX 20 kb)
Fungi recovered from BHM soil and assessed for growth inhibition by Tp. (DOCX 17 kb)
Data Availability Statement
The datasets generated and analyzed during the current study have been deposited in the NCBI BioProject database (accession number PRJNA433166) (https://www.ncbi.nlm.nih.gov/bioproject/). Raw sequences and metadata are available in the SRA database with the following accession numbers: SRR6676481, Aeolus Cave Soil Alone, fungal data, replicate 1; SRR6676487, Aeolus Cave Soil Alone, fungal data, replicate 2; SRR6676483, Aeolus Cave Soil + Tp (105 conidia/g soil), fungal data, replicate 1; SRR6676489, Aeolus Cave Soil + Tp (105 conidia/g soil), fungal data, replicate 2; SRR6676477, Aeolus Cave Soil + Tp (106 conidia/g soil), fungal data, replicate 1; SRR6676491, Aeolus Cave Soil + Tp (106 conidia/g soil), fungal data, replicate 2; SRR6676479, Barton Hill Mine Soil Alone, fungal data, replicate 1; SRR6676493, Barton Hill Mine Soil Alone, fungal data, replicate 2; SRR6676475, Barton Hill Mine Soil + Tp (105 conidia/g soil), fungal data, replicate 1; SRR6676485, Barton Hill Mine Soil + Tp (106 conidia/g soil), fungal data, replicate 2; SRR6676480, Aeolus Cave Soil Alone, bacterial data, replicate 1; SRR6676486, Aeolus Cave Soil Alone, bacterial data, replicate 2; SRR6676482, Aeolus Cave Soil + Tp (105 conidia/g soil), bacterial data, replicate 1; SRR6676488, Aeolus Cave Soil + Tp (105 conidia/g soil) bacterial data, replicate 2; SRR6676476, Aeolus Cave Soil + Tp (106 conidia/g soil) bacterial data, replicate 1; SRR6676490, Aeolus Cave Soil + Tp (106 conidia/g soil), bacterial data, replicate 2; SRR6676478, Barton Hill Mine Soil Alone, bacterial data, replicate 1; SRR6676492, Barton Hill Mine Soil Alone, bacterial data, replicate 2; SRR6676474, Barton Hill Mine Soil + Tp (105 conidia/g soil), bacterial data, replicate 1; SRR6676484, Barton Hill Mine Soil + Tp (105 conidia/g soil) bacterial data, replicate 2. A permanent link to the data can be found at https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP132276.