

 A novel compound binding with inactive IKKβ and allosterically blocking its activation.
A novel compound binding with inactive IKKβ and allosterically blocking its activation.
Abstract
I kappa B kinase β (IKKβ) is one of the primary targets to regulate canonical NF-κB activity. The misregulation of NF-κB is associated with various diseases, including chronic inflammation and cancers. Most of the known IKKβ inhibitors target its active form and suffer from poor selectivity. In the present study, we aim to design inhibitors that can bind to the IKKβ inactive form and block its activation. We identified a potential allosteric site between the kinase domain (KD) and ubiquitin-like domain (ULD) of human IKKβ and used it to virtually screen a chemical library for allosteric inhibitors. Among the 133 compounds tested, 16 inhibited NF-κB activity by over 50% at 50 μM in a reporter gene assay. Further quantitative measurements and cytotoxicity study gave one compound 124 (3,4-dichloro-2-ethoxy-N-(2,2,6,6-tetramethylpiperidin-4-yl)benzenesulfonamide) which specifically targets the IKKβ inactive form. In cells, 124 inhibited IκBα phosphorylation and NF-κB transcriptional activity for the reporter gene with an IC50 of 35 μM by decreasing the phosphorylation level of Ser177/181 on IKKβ and blocking its activation upon TNFα stimulation. Molecular dynamics simulations demonstrated that 124 binds to the pocket between KD and ULD in the inactive conformation of IKKβ rather than the active conformation. As the first allosteric inhibitor that prevents IKKβ activation, 124 provides a good starting point for further inhibitor discovery and a probe for IKKβ enzyme cycle and regulatory mechanism study.
Introduction
Nuclear factor-kappa B (NF-κB) is an important transcription factor controlling the expression of hundreds of genes associated with inflammation, immune response, cell survival, etc. NF-κB can be activated by various stimuli such as tumor necrosis factor (TNF), lipopolysaccharide (LPS), interleukin 1 (IL-1), radiation, pressure and pathogens.1 The dysfunction of NF-κB is implicated in chronic inflammation, autoimmune disorders and cancer.2–4 In resting cells, NF-κB is arrested in the cytoplasm by binding to its inhibitor protein IκBα. Upon stimulation, IκBα is phosphorylated by the IKK kinase complex, and then ubiquitinated and degraded by proteasome, liberating free NF-κB into the nucleus to initiate target gene transcription.5 The IκB kinase (IKK) complex is composed of three major components, two kinases IKKα and IKKβ and a regulatory subunit NEMO. It is generally agreed that IKKβ plays a key role in activating the NF-κB pathway.6 IKKβ was considered as a promising pharmaceutical target for drug discovery. Currently known IKKβ inhibitors, including ATP-competitive inhibitors (such as TPCA-1),7 substrate-competitive inhibitors (BMS-345541)8 and some natural products (such as ainsliadimer A),9 all directly interact with the active conformation of IKKβ to block its kinase activity. Three IKKβ inhibitors IMD-1041, CHS-828 and its prodrug EB-1627 have been reported to progress into clinical studies, but none of them have entered into phase III due to various problems. Thus, new strategies for IKKβ inhibitor discovery need to be explored.
IKK activity is controlled by a kinase cycle comprising a poised, an active and an inactive state.10 Upon the upstream TNFα signal, the poised state IKKβ is activated by the phosphorylation of Ser177 and Ser188 residues on its activation loop. Immediately, self-inhibitory trans-autophosphorylation in the C-terminal of IKKβ significantly reduces its activity. The hyper-phosphorylated inactive kinase then becomes available for a new round of activation through dephosphorylation by phosphatase. And this poised state can be activated again in response to upstream effectors. IKK activity should be tunable by regulating any single steps of this control cycle. Given that the activation of IKK is the precondition for initiating the kinase cycle and critical for the NF-κB activity, inhibiting this step should be able to block signal transmission to NF-κB most effectively.
The precise mechanisms by which IKKβ becomes phosphorylated and active remain unclear. Two general ideas about this process prevail: one is that there are upstream IKKβ kinases (IKKK) responsible for activating IKKβ, while the other point of view supports that it is possible for IKKβ kinases to autophosphorylate each other. As all currently identified IKKK candidates appear to be dispensable and some of them are cell type specific,11 IKKβ autophosphorylation seems to be the key step in IKKβ activation. Inhibition of this activation process provides a new opportunity for developing specific IKKβ inhibitors.
In the present study, we aim to discover novel inhibitors that selectively target the activation process of IKKβ using structure-based drug design methods. Fortunately, the inactive conformation of human IKKβ was captured in the X-ray crystal structure of an asymmetric dimer of human IKKβ, which contains one protomer in the active conformation with phosphorylated Ser177 and Ser181, and the other protomer is in the inactive conformation with unphosphorylated Ser177 and Ser181 in the activation loop.12 Considering the high similarity of active sites in the active and inactive conformations, compounds that bind to the ATP or substrate sites are unlikely to selectively capture the inactive conformation. A unique allosteric site in the inactive conformation might provide a solution to this problem. We set out to search for a potential allosteric site that only exists in the inactive protomer and used it to virtually screen for allosteric inhibitors for IKKβ activation. Compared with conventional orthosteric drugs, allosteric drugs often have higher selectivity and avoid competing with endogenous high concentration substrates and ligands, such as ATP for kinases.13
Results and discussion
The crystal structure of human IKKβ (PDB ID: 4KIK) was used for virtual screening. Each chain in the dimer is composed of one kinase domain (KD), one ubiquitin-like domain (ULD) and one scaffold dimerization domain (SDD). We first optimized the crystal structure by completing and refining the missing residues and loops, and then used the Cavity V1.1 program to predict potential ligand binding sites.14,15 We found one druggable binding site between KD and ULD of the inactive chain with a volume of 551 Å3 and a predicted maximal pKd of 10.9 (Fig. 1). The pocket is surrounded by three α helixes (R118-S127, L265-L273 and L303-H313) of KD and a loop (T368-L386) between two β sheets of ULD. Similar to TAK1 (transforming growth factor beta-activated kinase 1) activation by TAB (TAK1 binding protein 1),16 KD–ULD interaction is likely to enhance kinase activity, suggesting that small molecule binding in the pocket between KD and ULD is probable to interfere with the kinase function by disrupting the interaction between these two domains. Furthermore, the extensive interactions between domains in IKKβ have been illustrated important for the activation of IKKβ.17 So, we performed virtual screening against this pocket (Fig. 2). The programs Dock6 & AutoDock4 were used to virtually screen about 200 000 compounds in the SPECS library.18,19 Compounds with a score less than –6.16 kcal mol–1 were selected and then subjected to manual selection following the criteria reported previously.20 A total of 133 compounds were purchased for the cell assay.
Fig. 1. Predicted allosteric binding site in the inactive IKKβ.

Fig. 2. Overview of the virtual screening workflow.

The dual-luciferase reporter system was used to evaluate the inhibitory effect of these compounds on NF-κB transcriptional activity upon TNFα stimulation. In the first round of screening, 16 compounds inhibited the NF-κB activity by over 50% at 50 μM. Among them, 7 compounds showed dose-dependent inhibition (Fig. S1†). We measured the IC50s of the 5 compounds that passed the PAINS (pan-assay interference compounds) screening,21 and all of them inhibited luciferase expression at micromolar concentrations (Table S1†).
To eliminate the possible interference from cytotoxicity, the viability of cells treated with the compounds and TNFα with the same concentrations used in the NF-κB luciferase assay was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) method (Table S1†). Compound 124 (3,4-dichloro-2-ethoxy-N-(2,2,6,6-tetramethylpiperidin-4-yl)benzenesulfonamide) showed NF-κB inhibition with an IC50 of 35 μM in HEK293T and a similar activity in HeLa and SK-N-AS cells with negligible cytotoxicity (Fig. 3). HEK293T and HeLa cells were received from Professor Jincai Luo and Xing Chen (Peking University, China), respectively. SK-N-AS cells were purchased from ATCC. We further demonstrated that 124 blocked IκBα phosphorylation in TNFα-treated HeLa cells with an IC50 of 32.7 ± 3.7 μM using the phospho-IκBα (Ser32/36) ELISA Kit (Invitrogen) (the dose–response curve is provided in Fig. S2†). We then tested whether 124 can abrogate the TNFα-induced nuclear translocation of NF-κB/p65. Before TNFα stimulation, compound 124 or fresh medium was pre-incubated with SK-N-AS cells for at least 30 min at 37 °C (5% CO2). The time lapse images of NF-κB nuclear translocation were captured as described before.22 At 50 μM, 124 markedly abrogated the TNFα-induced nuclear translocation of NF-κB/p65 (Fig. 4). The bioactivity of 124 was not reported before according to SciFinder. Its purity has been confirmed by 1H-NMR (Fig. S8†) and mass spectra (Fig. S9†).
Fig. 3. Compound 124 blocks NF-κB transcription activity towards the luciferase reporter gene in various cells.

Fig. 4. Compound 124 inhibited TNFα-induced NF-κB nuclear translocation in SK-N-AS cells expressing the NF-κB/p65 subunit fused to the red fluorescent protein. Scale bar, 20 μm.

We further explored possible mechanisms of IKKβ inhibition by 124. The most significant difference between the inactive and active protomer of IKKβ lies in the activation loop, whereas the rest of their structures assume similar conformations. We performed molecular dynamics (MD) simulations for the apo structure of the asymmetrical IKKβ dimer (for details, see the ESI†). Three trajectories of 100 ns were obtained. Residues in the allosteric site, especially those from the T368-L386 loop, exhibited distinct dynamic characteristics. This loop showed higher flexibility in the active chain than in the inactive chain (the root mean square fluctuations of the two chains are shown in Fig. S4a†). We performed cluster analysis of the conformations from the MD simulations. The T368-L386 loop in the active chain exhibits more diversified conformations than that in the inactive chain (Fig. S4b†). We hypothesize that the allosteric pocket in the active chain might be too flexible to stably accommodate 124. We then docked 124 into both the active and inactive chains in the asymmetrical dimer and performed MD simulations (for details, see the ESI†). Three trajectories of 100 ns were obtained. In the inactive chain, 124 stably binds to the pocket between KD and ULD in all simulations. In contrast, 124 rapidly flies out of the pocket in the active chain in two trajectories after 15 ns simulation. In the third trajectory, 124 dissociates from the pocket and wanders around for most of the time (Fig. S6†). In the trajectories of the inactive chain complex, 124 forms a hydrogen bond with the nitrogen atom of the imidazole ring of His380 using the amide hydrogen atom of the pyridine ring, and a hydrogen bond with the main-chain amide hydrogen atom of Leu311 using its sulfonyl oxygen atom for most of the time (Fig. 5 and Table S3†). Our simulations support that 124 inhibits IKKβ activation through stabilizing the inactive conformation and preventing its conversion to the active state.
Fig. 5. Predicted binding mode of compound 124 in the potential allosteric site of inactive IKKβ. IKKβ is shown as a cyan cartoon. Compound 124 is illustrated in rose red sticks. The key residues from IKKβ are shown in yellow sticks and the predicted hydrogen bonds are displayed in green dashed lines.

To confirm that 124 hampers IKKβ activation in cells, we detected the level of active IKKβ in HeLa cells pre-treated with 124 before TNFα induction using western blot analysis (for details, see the ESI†). We found that 124 remarkably decreased the phosphorylation of Ser177/181 on IKKβ, thus blocking IKKβ activation under TNFα stimulation (Fig. 6). Our modeling and experimental studies all indicate that 124 inhibits IKKβ activation.
Fig. 6. Compound 124 inhibited IKKβ activation upon TNFα stimulation.

We further verified this using an in vitro kinase assay with activated IKKβ. At 100 μM, 124 shows very weak inhibition of IKKβ kinase activity and no detectable inhibition activity for other 17 kinases (Fig. S7†). This again demonstrates that 124 selectively inhibits the activation of IKKβ, making it highly selective toward other kinases.
We further searched for analogues of 124 in the SPECS library using the Shape Screening program (Schrödinger 2014-2) with the default settings and compared their inhibition effect on NF-κB transcription activity as described above. Five 2D analogues of 124 and six 3D derivatives were purchased for the cell assay. Among the 11 tested compounds, 5 showed dose-responsive inhibition with micromolar IC50s. As shown in Table 1, substituents on both sides of the sulfonamide group significantly affected the compounds' inhibitory activity. Compared to 124, the inhibitory effect of compounds with piperidinyl substituted by a rigid phenyl ring on the sulfonamide amino group slightly decreased (A38, A39 and A4), while compounds with piperidinyl substituted by flexible groups were completely inactive (A60 and A62). Two 3D derivatives A49 and A51 displayed similar activity to 124, and the size of the aromatic group was critical for the inhibition. Compounds with a too small (A50 and A52) or too large (A85 and A88) aryl sulfonamide group almost lost inhibition activity. Based on these analyses, further optimization of 124 can be performed.
Table 1. Analogue screening and activity comparison.
| ID | Structure | IC50 a (μM) | ID | Structure | IC50 a (μM) |
| 124 |
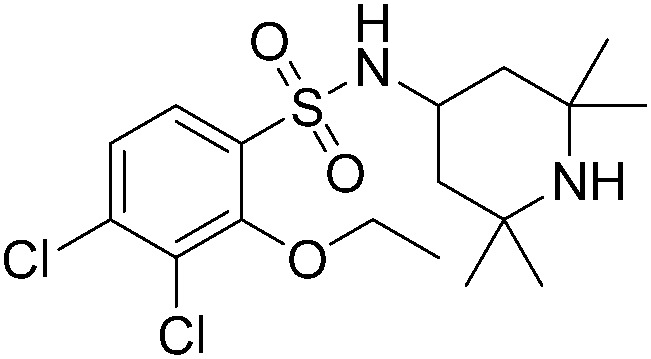
|
35.4 ± 5.3 | A49 |
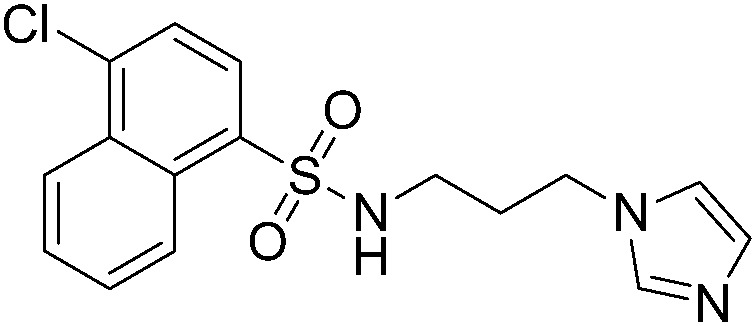
|
64.3 ± 2.5 |
| A60 |
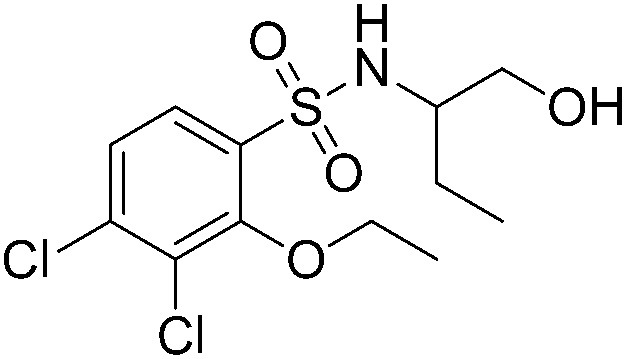
|
>200 | A51 |
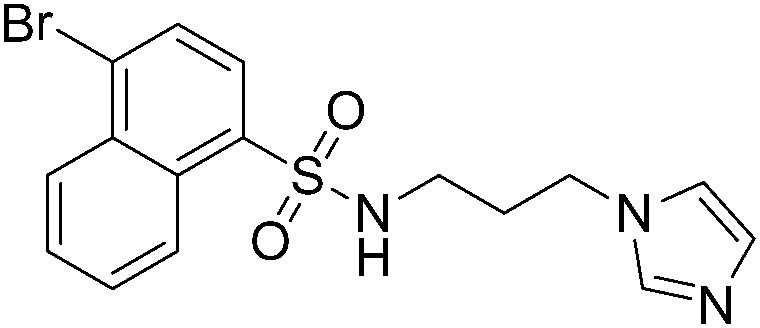
|
54.7 ± 3.6 |
| A62 |
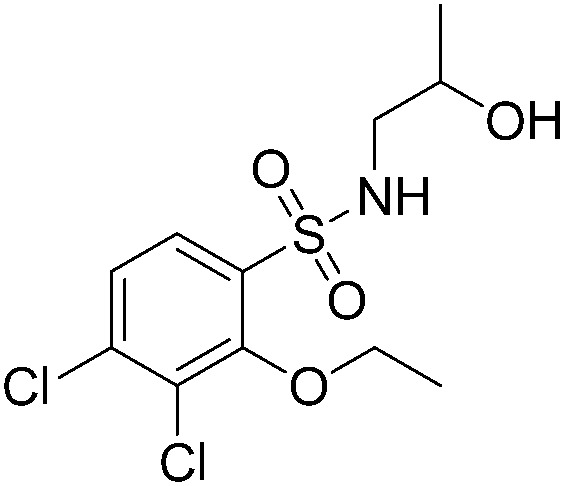
|
>200 | A50 |
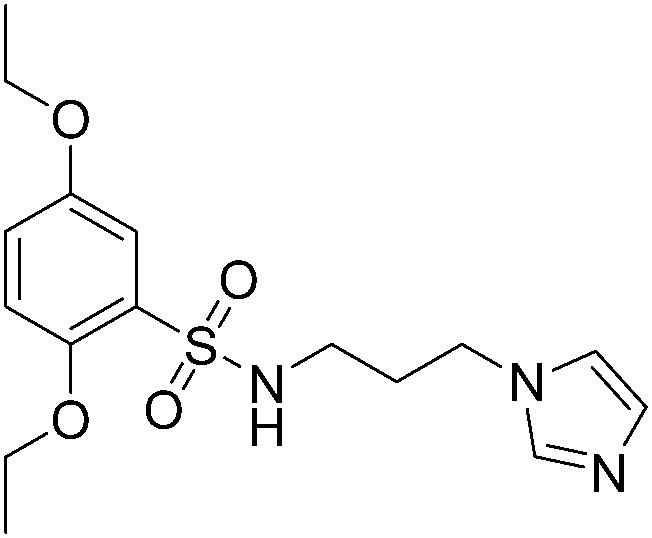
|
>200 |
| A38 |
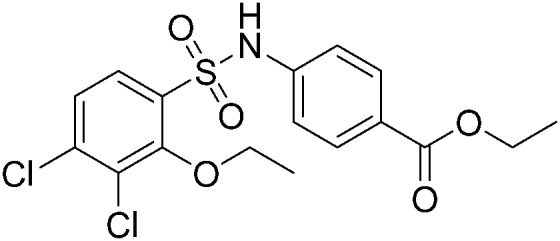
|
71.3 ± 2.0 | A52 |
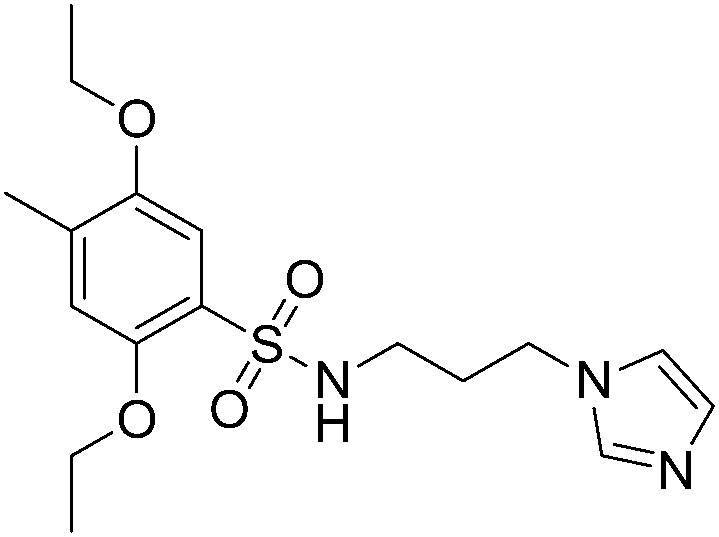
|
>200 |
| A39 |
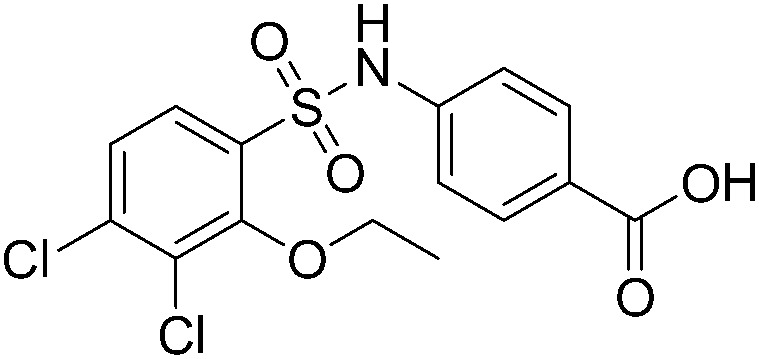
|
68.0 ± 1.6 | A85 |
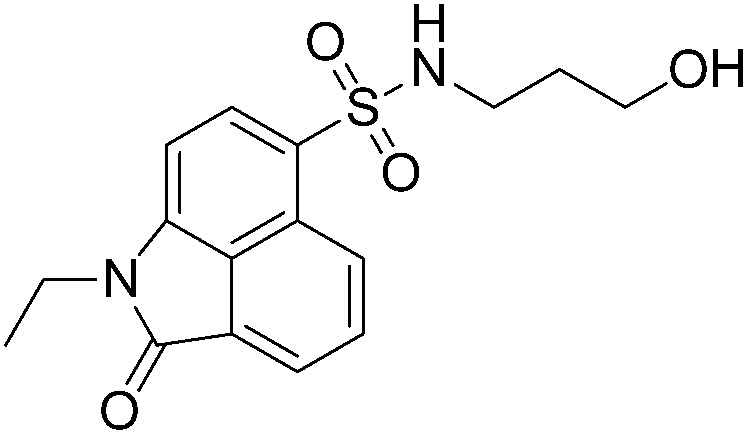
|
>200 |
| A4 |
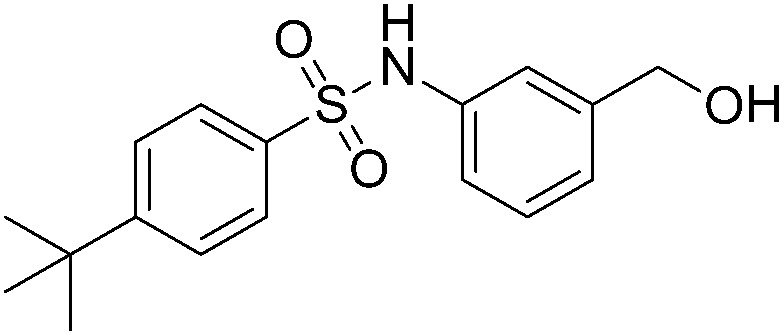
|
98.6 ± 3.5 | A88 |
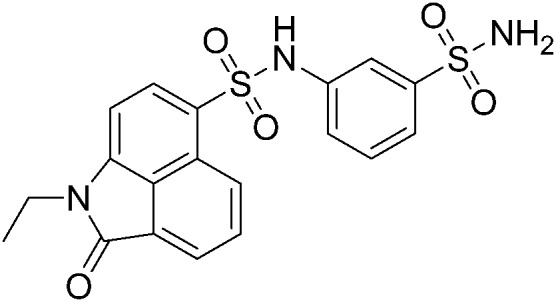
|
>200 |
aThe data represent the average and standard deviation (mean ± SD) of triplicate measurements.
Conclusions
In brief, we identified a compound that inhibits the activation of IKKβ for the first time. This compound was discovered by allosteric site prediction, structure-based virtual screen and experimental study. We demonstrate that blocking the activation of IKKβ can effectively prevent NF-κB nuclear translocation. Inhibitors blocking the activation of IKKβ activity provide a novel opportunity for developing selective inhibitors and chemical tools to investigate the molecular mechanisms of IKKβ activation, as well as to develop new drugs against cancer and inflammation.
Conflicts of interest
The authors declare no competing interests.
Supplementary Material
Acknowledgments
This work was supported in part by the Ministry of Science and Technology of China (2015CB910300 and 2016YFA0502303) and the National Natural Science Foundation of China (21633001). The simulations and analysis were performed on the High Performance Computing Platform of the Center for Life Sciences.
Footnotes
†Electronic supplementary information (ESI) available: Supplementary data and experimental methods. See DOI: 10.1039/c7md00599g
References
- Pahl H. L. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Ben-Neriah Y., Karin M. Nat. Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S., Karin M. Annu. Rev. Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Karin M. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Li Q. T., Verma I. M. Nat. Rev. Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Scheidereit C. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- Podolin P. L., Callahan J. F., Bolognese B. J., Li Y. H., Carlson K., Davis T. G., Mellor G. W., Evans C., Roshak A. K. J. Pharmacol. Exp. Ther. 2005;312:373–381. doi: 10.1124/jpet.104.074484. [DOI] [PubMed] [Google Scholar]
- Burke J. R., Pattoli M. A., Gregor K. R., Brassil P. J., MacMaster J. F., McIntyre K. W., Yang X. X., Iotzova V. S., Clarke W., Strnad J., Qiu Y. P., Zusi F. C. J. Biol. Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- Dong T., Li C., Wang X., Dian L., Zhang X., Li L., Chen S., Cao R., Li L., Huang N., He S., Lei X. Nat. Commun. 2015;6:6522. doi: 10.1038/ncomms7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar M., Hoffmann A. Biophys. J. 2013;105:231–241. doi: 10.1016/j.bpj.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Xia Y., Parker A. S., Verma I. M. Immunol. Rev. 2012;246:239–253. doi: 10.1111/j.1600-065X.2012.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Misquitta Y. R., Olland A., Johnson M. A., Kelleher K. S., Kriz R., Lin L. L., Stahl M., Mosyak L. J. Biol. Chem. 2013;288:22758–22767. doi: 10.1074/jbc.M113.482596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeDecker B. S. Chem. Biol. 2000;7:R103–R107. doi: 10.1016/s1074-5521(00)00115-0. [DOI] [PubMed] [Google Scholar]
- Yuan Y., Pei J., Lai L. J. Chem. Inf. Model. 2011;51:1083–1091. doi: 10.1021/ci100350u. [DOI] [PubMed] [Google Scholar]
- Yuan Y., Pei J., Lai L. Curr. Pharm. Des. 2013;19:2326–2333. doi: 10.2174/1381612811319120019. [DOI] [PubMed] [Google Scholar]
- Xu G., Lo Y., Li Q., Napolitano G., Wu X., Jiang X., Dreano M., Karin M., Wu H. Nature. 2011;472:325–330. doi: 10.1038/nature09853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polley S., Huang D., Hauenstein A. V., Fusco A. J., Zhong X., Vu D., Schroefelbauer B., Kim Y., Hoffmann A., Verma I. M., Ghosh G., Huxford T. PLoS Biol. 2013;11:e1001581. doi: 10.1371/journal.pbio.1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang P. T., Brozell S. R., Mukherjee S., Pettersen E. F., Meng E. C., Thomas V., Rizzo R. C., Case D. A., James T. L., Kuntz I. D. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Qi Y., Yin N., Lai L. PLoS One. 2014;9:e94829. doi: 10.1371/journal.pone.0094829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B., Holloway G. A. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Zhang X., Yin N., Guo A., Zhang Q., Zhang Y., Xu Y., Liu H., Tang B., Lai L. Biochem. Biophys. Res. Commun. 2017;489:287–292. doi: 10.1016/j.bbrc.2017.05.149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.