

Abstract
Methamphetamine and mephedrone are designer drugs with high abuse liability and they share extensive similarities in their chemical structures and neuropharmacological effects. However, these drugs differ in one significant regard: methamphetamine elicits dopamine neurotoxicity and mephedrone does not. From a structural perspective, mephedrone has a β-keto group and a 4-methyl ring addition, both of which are lacking in methamphetamine. Our previous studies found that methcathinone, which contains only the β-keto substituent, is neurotoxic, while 4-methylmethamphetamine, which contains only the 4-methyl ring substituent, elicits minimal neurotoxicity. In the present study, it was hypothesized that the varying neurotoxic potential associated with these compounds is mediated by the drug-releasable pool of dopamine, which may be accessed by methamphetamine more readily than mephedrone, methcathinone, and 4-methylmethamphetamine. To test this hypothesis, L-DOPA and pargyline, compounds known to increase both the releasable pool of dopamine and methamphetamine neurotoxicity, were combined with mephedrone, 4-methylmethamphetamine and methcathinone. Methamphetamine was also tested because of its ability to increase releasable dopamine. All three regimens significantly enhanced striatal neurotoxicity and glial reactivity for 4-methylmethamphetamine. Methcathinone neurotoxicity and glial reactivity were enhanced only by L-DOPA. Mephedrone remained non-neurotoxic when combined with either L-DOPA or pargyline. Body temperature effects of each designer drug were not altered by the combined treatments. These results support the conclusion that the neurotoxicity of 4-methylmethamphetamine, methcathinone and methamphetamine may be differentially regulated by the drug-releasable pool of dopamine due to β-keto and 4-methyl substituents, but that mephedrone remains non-neurotoxic despite large increases in this pool of dopamine.
Keywords: Methamphetamine, Mephedrone, 4-Methylmethamphetamine, Methcathinone, Dopamine, Neurotoxicity
1. Introduction
The β-ketoamphetamine mephedrone (MEPH)1 is a common constituent of bath salts drug cocktails, and despite being a controlled substance of abuse, is still being used frequently (Hockenhull et al., 2016; Papaseit et al., 2016). Users of the drug report subjective effects including feelings of euphoria, well-being, and altered sensory perceptions, but acute toxicity and occasional deaths have also been reported (Papaseit et al., 2016). MEPH has a markedly similar neurotransmitter releasing effect on dopamine (DA) and serotonin (5-HT), mediated by their respective reuptake transporters, in comparison with methamphetamine (METH), its non-β-keto analog (Baumann et al., 2012; Cameron et al., 2013; Eshleman et al., 2013; Golembiowska et al., 2016; Lopez-Arnau et al., 2012; Simmler et al., 2013; Suyama et al., 2016). Additionally, MEPH shares similarities with METH in its acute effects on thermoregulation (Baumann et al., 2012; Martinez-Clemente et al., 2014; Shortall et al., 2016), locomotor stimulation (Baumann et al., 2012; Lopez-Arnau et al., 2012; Marusich et al., 2012; Motbey et al., 2012; Nguyen et al., 2016; Wright et al., 2012), and indicators of addictive liability (Creehan et al., 2015; Hadlock et al., 2011; Karlsson et al., 2014; Lisek et al., 2012). Despite these similarities, these two compounds differ in their ability to evoke long-term toxicity to DA nerve endings.
It has been well established in rodent models that the classic amphetamines, including METH, elicit long-lasting damage to DA nerve terminals. For METH, this is typically manifested as reductions in markers of presynaptic dopaminergic integrity such as DA, the dopamine transporter (DAT), and the synthetic enzyme tyrosine hydroxylase (TH) (Moratalla et al., 2015). This toxicity is thought to be mediated via pathways involving neuroinflammation and oxidative stress (Halpin et al., 2014a; Yamamoto and Raudensky, 2008). It has been proposed that the excessive DA release evoked by METH is a primary factor, as the metabolism and auto-oxidation of DA is known to generate reactive species that can contribute to neuronal damage (Halpin et al., 2014a). MEPH has generally not been found to evoke this long-lasting dopaminergic toxicity. Most rodent studies under standard conditions known to evoke neurotoxicity with METH have not reported similar neuro-chemical or inflammatory changes in MEPH-treated animals (Angoa-Perez et al., 2012, 2013; Anneken et al., 2015; Anneken et al., 2017; Baumann et al., 2012; den Hollander et al., 2013; Motbey et al., 2013), except under harsher environmental conditions (den Hollander et al., 2014; Martinez-Clemente et al., 2014).
MEPH differs from METH by 2 substituents: a β-keto group, and a 4-methyl group on the phenyl ring. A recent study in this lab investigated the toxicity of two intermediate compounds, methcathinone (β-keto; MeCa) and 4-methylmethamphetamine (4-methyl; 4 MM) (Anneken et al., 2017) (see Fig. 1). MeCa, although less potent, elicited dopaminergic toxicity resembling METH, while 4 MM resembled MEPH in that it had greatly diminished dopaminergic toxicity compared to METH. The mechanism of this differential toxicity remains to be elucidated. Multiple studies have shown that increases in the releasable pool of DA augment METH toxicity (Guillot et al., 2008; Kita et al., 1995; Kuhn et al., 2008; Thomas et al., 2008, 2009). MEPH, which is non-toxic, enhances METH toxicity as well and may do so via interactions with the releasable pool of DA (Angoa-Perez et al., 2013), as it has been reported that METH and MEPH both release DA via reverse transport through the DAT in vitro (Simmler et al., 2013). However, Eshleman et al. (2013) observed that MEPH is much less effective at releasing vesicular norepinephrine via the vesicular monoamine transporter (VMAT2) in vitro than METH, and also less effective in the amount of release it evokes via DAT reverse transport, releasing half the amount of DA observed with METH. The inability of MEPH alone to increase the cytosolic, drug-releasable pool of DA in a VMAT2-dependent manner could explain its low neurotoxic potential by comparison to METH, which releases DA from vesicles into the cytosol, and then through the DAT into the synapse. MeCa, which is neurotoxic, also released a greater amount of DA via the DAT when compared to non-toxic MEPH (Eshleman et al., 2013). In the same study, while MeCa had a lower binding affinity and release profile at VMAT2 compared to METH, it was found to release norepinephrine from VMAT2 in slightly higher amounts than MEPH (42% compared with 33%).
Fig. 1.
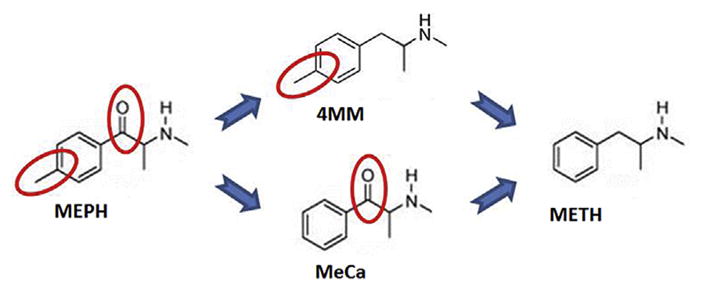
Comparative structures of MEPH, METH, and intermediate structures. Diagram depicts the structures of the related compounds methamphetamine (no structural substituents), 4-methlymethamphetamine (4-methyl), methcathinone (β-keto), and mephedrone (4-methyl and β-keto). Reprinted from Anneken et al. (2017) with permission from the American Society for Pharmacology and Experimental Therapeutics.
To test whether these variations in dopamine release could account for the differential toxic potential among these structural analogs, we hypothesized that increasing the drug-releasable pool of DA, by administration of either the DA precursor L-DOPA, the monoamine-oxidase (MAO) B inhibitor pargyline, or a mild dose of METH, which can release vesicular DA to the cytosol, would impart toxicity to MEPH, as well as enhance the dopaminergic toxicity of the two closely related compounds, 4 MM and MeCa.
2. Materials and methods
2.1. Drugs and reagents
(R,S)-N-Methcathinone HCl and (R,S)-mephedrone HCl were provided by the NIDA Research Resources Drug Supply Program. Racemic 4-methlymethamphetamine HCl was synthesized as described by Davis et al. (2012) from methylamine HCl and 4-methylphenylacetone purchased from Alfa Aesar (Ward Hill, MA, USA). (+)- Methamphetamine HCl, pargyline HCl, L-3,4-dihydroxyphenylalanine (L-DOPA), S-(−)-carbidopa, DA, polyclonal antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and all buffers and HPLC reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Bicinchoninic acid protein assay kits for Western blot analysis were obtained from Pierce (Rockford, IL, USA). Polyclonal antibodies against rat TH were produced as previously described (Kuhn and Billingsley, 1987). Monoclonal antibodies against rat DAT were generously provided by Dr. Roxanne Vaughan (University of North Dakota, Grand Forks, ND, USA). IRDye secondary antibodies for Odyssey Imaging Systems were purchased from LiCor Biosciences (Lincoln, NE, USA).
2.2. Animals
Female C57BL/6 mice (Harlan, Indianapolis, IN, USA) weighing 18–25 g at the time of experimentation were housed 5–7 per cage in large shoe-box cages in a light- (12 h light/dark) and temperature-controlled room. Female mice were used as they have been shown to be impacted by the neurotoxicity induced by amphetamines and to maintain consistency with our previous studies of METH and β-ketoamphetamine interactions (Angoa-Perez et al., 2012, 2013; Anneken et al., 2015; Anneken et al., 2017). Mice had free access to food and water. The mice used were randomly divided into treatment groups. The Institutional Care and Use Committee of Wayne State University approved the animal care and experimental procedures. All procedures were also in compliance with the NIH Guide for the Care and Use of Laboratory Animals and were conducted in compliance with ARRIVE guidelines and under IACUC-approved protocols.
2.3. Drug treatments
Mice were treated i.p. with saline (controls), 4 MM (40 mg/kg), MeCa (80 mg/kg), or MEPH (40 mg/kg) in a binge-like regimen, which involves 4 injections (0.2 mL) at 2 h intervals. This binge treatment regimen has been established by multiple prior studies in this laboratory and others to elicit significant neurotoxicity for amphetamine compounds. Doses of β-keto amphetamines and 4 MM eliciting mild to moderate DA depletion were selected based on prior studies (Angoa-Perez et al., 2013; Anneken et al., 2015, 2017; Gygi et al., 1997; Gygi et al., 1996; Sparago et al., 1996).
In order to increase the pool of releasable DA, 4 MM, MeCa, and MEPH were given in combination with either METH, which releases vesicular DA into the cytosol, or with L-DOPA or pargyline, which increase the amount of cytosolic DA through increased synthesis or inhibition of MAO-B metabolism, respectively. METH (4 × 2.5 mg/ kg) was given simultaneously with saline, 4 MM, MeCa, or MEPH, as this regimen is known to show enhanced toxicity when combined with β-ketoamphetamines (Angoa-Perez et al., 2013; Anneken et al., 2015). The dosing regimen for animals receiving pargyline or L-DOPA with the above drugs was modified from prior studies in this lab that have shown elevated DA at the time of each drug injection (Kuhn et al., 2008; Thomas et al., 2008, 2009). In those studies, HPLC analysis revealed that L-DOPA elevated striatal dopamine by 50% at the time of METH administration, while the MAO inhibitor clorgyline elevated striatal dopamine by 12% at the time of METH treatment. A study by Harun et al. (2015) using fast-scan cyclic voltammetry suggests that increases in DA via pharmacological methods such as L-DOPA do promote greater DA release in the striatum. At the time of analysis for METH toxicity 48 h following treatment, animals treated with either of these enhancing regimens exhibited no significant differences from controls, indicating a transient increase in striatal DA. Pargyline (25 mg/kg) was administered i.p. as a single injection (in 0.2 mL saline) 1 h prior to the first injection of saline, 4 MM, MeCa, or MEPH. 50 mg/kg L-DOPA (50 mg/ kg) and carbidopa (25 mg/kg; to inhibit peripheral conversion to DA) was administered 1 h prior to each injection of saline, 4 MM, MeCa, or MEPH in 0.4 mL of warm distilled water. Mice were sacrificed 48 h after the last drug treatment when METH-associated decreases in dopaminergic toxicity markers and increases in neuroinflammation are known to reach maximal levels (Thomas et al., 2004b, 2008). The range of ambient temperature for all drug treatment studies was maintained between 22 and 24 °C.
2.4. Determination of striatal DA content
Striatal tissue was dissected from the brain after treatment and stored at −80 °C. Frozen tissues were weighed and sonicated in 10 vol of 0.16 N perchloric acid at 4 °C. Insoluble protein was removed by centrifugation and DA was determined by HPLC with electrochemical detection as previously described (Angoa-Perez et al., 2012, 2013). Briefly, supernatant diluted 1:8 in 0.16 N PCA was injected via autosampler onto a C-18 reverse phase column in buffer (100 mM citric acid, 75 mM NaH2PO4, 176 mg/L octane-sulfonic acid, 50 mg/L EDTA, 16.5% methanol, pH 4.5). Peaks were quantified on the basis of known standards, and values were normalized to controls.
2.5. Determination of DAT and TH protein levels by immunoblotting
The effects of drug treatments on striatal DAT and TH levels, highly specific markers for striatal DA nerve endings, were determined by immunoblotting as an index of toxicity. Striatal tissue was dissected from the brain after treatment and stored at −80 °C. Frozen tissue was disrupted by sonication in 1% SDS at 95 °C and insoluble material was removed by centrifugation. The concentration of soluble protein was determined by the bicinchoninic acid method and equal amounts of protein (70 μg/lane) were resolved by SDS-polyacrylamide gel electrophoresis and then electroblotted to nitrocellulose. Blots were blocked in Odyssey blocking buffer (PBS) for 1 h at room temperature. Primary antibodies against DAT (1:1000), TH (1:1000), or GAPDH (1:10,000) were added to blots and allowed to incubate overnight at 4 °C. Blots were washed 3x in Tris-buffered saline to remove unreacted antibodies and then incubated with IRDye secondary antibodies (1:4000) for 1 h at room temperature. Immunoreactive bands were visualized by enhanced fluorescence and the relative densities of TH-, DAT-, and GAPDH-reactive bands were determined by imaging with an Odyssey CLx Infrared Image System (LiCor Biosciences, Lincoln, NE) and quantified using ImageJ software (NIH). DAT and TH relative densities were normalized to the GAPDH level for each lane to control for loading error.
2.6. Assessment of glial status in striatum
Whole brains were removed after the final drug treatment and post-fixed in 4% paraformaldehyde. Coronal sections (40 μm) containing striatum within the coordinates of 4.66 mm interaural and 0.18 mm bregma, and 3.94 mm interaural and 0.14 mm bregma were selected for fluorescence immunohistochemical analyses. Microglial activation was assessed as before (Thomas et al., 2004a) using anti-ionized calcium-binding adapter molecule 1 (Iba-1; 1:1000; Wako, Richmond, VA), and astrocyte activation was assessed as described (Angoa-Perez et al., 2013) using anti–glial fibrillary acidic protein antibody (GFAP; 1:1000; LabVision, Fremont, CA). Brain sections were incubated with primary antibodies at 4 °C overnight. Secondary antibodies conjugated to biotin (Vector Labs, Burlingame, CA) were incubated for 1 h at room temperature, followed by an incubation under similar conditions with streptavidin-conjugated Alexa Fluor-555 for Iba-1 and Alexa Fluor-488 for GFAP (Molecular Probes, Waltham, MA). Prolong Gold anti-fade mounting medium with DAPI was applied for nuclear counterstaining (Invitrogen, Carlsbad, CA). Fluorescence was viewed using the appropriate filters and photographed using an Olympus BX51 microscope with a DP71 camera. Images acquired at 20× magnification from 3 non-adjacent regions of interest per slice from 3 to 5 mice were used for analysis. ImageJ software version 1.48v (NIH, Bethesda, MD) was used to quantify immunoreactivity. The results are expressed as mean of fluorescence intensity (arbitrary units). In order to reduce the number of mice used, some groups (control, L-DOPA, pargyline, METH) were utilized in all analyses as these animals were run concurrently with every other treatment.
2.7. Body temperature
Core body temperatures were monitored in treated animals by telemetry using IPTT-300 implantable temperature transponders from Bio Medic Data Systems, Inc. (Seaford, DE, USA) inserted subcutaneously at least 24 h prior to the experiment. Temperatures were recorded non-invasively every 20 min starting 60 min before the first stimulant drug injection and continuing for 9 h thereafter using the DAS-7006/7s console system from Bio Medic. Data were then pooled and averaged for analysis.
2.8. Data analysis
The effects of drug treatments on core body temperature over time were analyzed using two-way ANOVAs and post hoc comparisons were carried out using Bonferroni's Test. One-way ANOVAs were performed to analyze the effects of drug treatments on striatal levels of DA, DAT, and TH, with post hoc comparisons carried out using Bonferroni's Test. A one-way ANOVA was also utilized for the effect of drug treatments on glial status, with post hoc comparisons carried out using Tukey's Test. Differences were considered significant if p < 0.05. Group sizes were: n = 5–16 for depletion experiments; n = 3–5 (3 image fields/animal) for glial status experiments; and n = 4–9 for body temperature experiments. All statistical analyses were carried out using GraphPad Prism version 6.01 for Windows (GraphPad Software, San Diego, CA, USA, www.graphpad.com).
3. Results
3.1. Effects of increased DA on drug-induced depletions of markers of striatal DA integrity
Binge treatment with 4 MM alone or in combination with L-DOPA, pargyline, or METH evoked significant depletions in striatal DA, as revealed by one-way ANOVA (Fig. 2A; L-DOPA: F(3,35) = 22.08, p < 0.0001; pargyline: F(3,36) = 29.74, p < 0.0001; METH: F(3,38) = 37.38, p < 0.0001). Following post-hoc analysis, it was found that L-DOPA, pargyline, or METH all significantly enhanced DA depletion compared to animals treated with 4 MM alone, which itself induced a 20% reduction in DA, in agreement with our prior study (Anneken et al., 2017). METH alone also evoked depletion of DA, but 4 MM + METH treated animals were significantly more depleted than either drug alone. The same pattern of significant enhancements was also evident for DAT (Fig. 2B; L-DOPA: F(3,32) = 20.02, p < 0.0001; pargyline: F(3,33) = 14.28, p < 0.0001; METH: F(3,32) = 34.46, p < 0.0001) and TH (Fig. 2C; L-DOPA: F(3,33) = 12.51, p < 0.0001; pargyline: F(3,33) = 9.642, p < 0.001; METH: F(3,32) = 13.73, p < 0.0001). Neither L-DOPA nor pargyline alone significantly altered striatal DA, DAT, or TH.
Fig. 2.
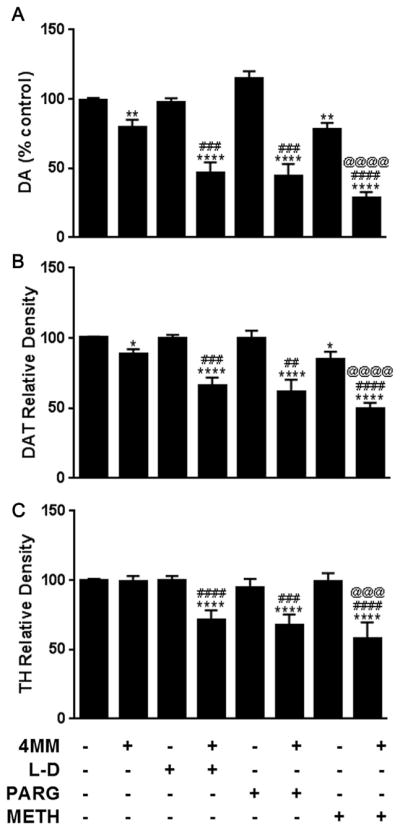
Effects of 4 MM ± L-DOPA, PARG or METH on markers of striatal dopaminergic toxicity. Levels of (A) DA, (B) DAT, and (C) TH were determined in the striatum following treatment with 4 MM, +/−L-DOPA (L–D), pargyline (PARG), or METH. *p < 0.05 **p < 0.01 ****p < 0.0001 compared to controls. ##p < 0.01 ###p < 0.001 ####p < 0.0001 compared to 4MM. @@@ p < 0.001 @@@@ p < 0.0001 compared to METH. Data are presented as mean + SEM. N = 5–16 mice per group.
MeCa caused significant depletion of striatal DA (Fig. 3A; L-DOPA: F(3,39) = 17.24, p < 0.0001; pargyline: F(3,39) = 7.41, p < 0.001; METH: F(3,51) = 8.037, p < 0.001). Post-hoc analysis revealed mice treated with L-DOPA + MeCa had significantly enhanced DA depletion when compared to mice treated with MeCa alone. METH alone elicited significant DA depletion, but the combination of METH + MeCa did not exhibit enhanced toxicity over either drug alone. Likewise, pargyline + MeCa evoked DA depletion that was not enhanced compared to MeCa alone. There was a significant treatment effect on DAT levels in the striatum (Fig. 3B; L-DOPA: F(3,37) = 13.69, p < 0.0001; pargyline: F(3,39) = 9.443, p < 0.0001; METH: F(3,50) = 7.595, p < 0.001), but post-hoc analysis revealed no significant enhancement of DAT toxicity by any treatment combination. However, there was both a significant MeCa depletion of striatal TH (Fig. 3C; L-DOPA: F(3,39) = 20.67, p < 0.0001; pargyline: F(3,38) = 8.755, p < 0.001; METH: F(3,48) = 7.956, p < 0.001), as well as a significant enhancement of MeCa-induced depletion by L-DOPA treatment. No other treatment combination significantly altered the effect seen in animals treated with MeCa alone.
Fig. 3.
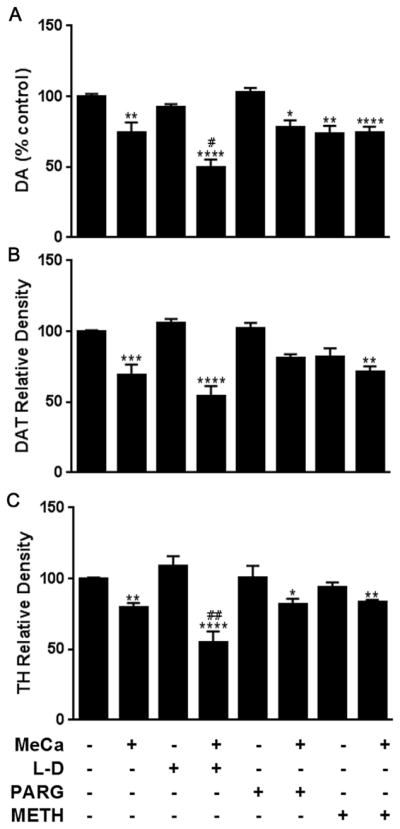
Effects of MeCa ± L-DOPA, PARG or METH on markers of striatal dopaminergic toxicity. Levels of (A) DA, (B) DAT, and (C) TH were determined in the striatum following treatment with MeCa, +/−L-DOPA (L–D), pargyline (PARG), or METH. *p < 0.05 **p < 0.01 ***p < 0.001 ****p < 0.0001 compared to controls. #p < 0.05 ##p < 0.01 compared to MeCa. Data are presented as mean + SEM. N = 5–16 mice per group.
There was a slight but significant elevation in striatal DA elicited by pargyline treatment (F(3,37) = 6.606, p < 0.01), but no other significant changes in any dopaminergic marker were elicited by binge treatment with MEPH ± L-DOPA or pargyline (Fig. 4A–C). Data from a prior published study in this lab showing MEPH-induced enhancement of METH neurotoxicity are included in Fig. 4 for comparison. (Angoa-Perez et al., 2013).
Fig. 4.
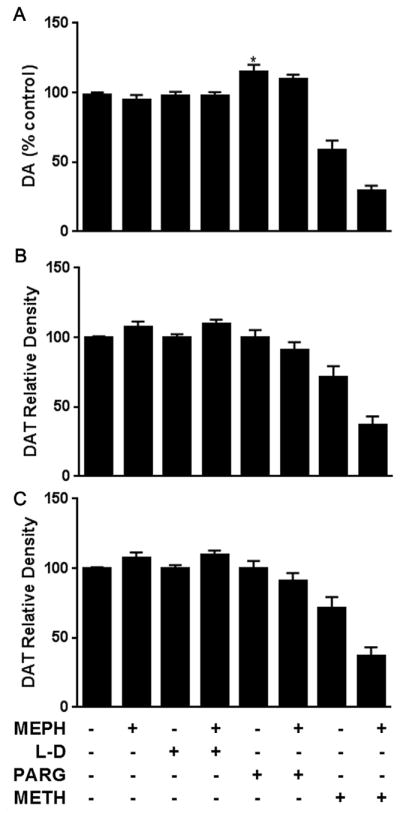
Effects of MEPH ± L-DOPA and PARG on markers of striatal dopaminergic toxicity. Levels of (A) DA, (B) DAT, and (C) TH were determined in the striatum following treatment with MEPH, +/−L-DOPA (L–D) or pargyline (PARG) are shown above. METH and MEPH + METH data from a prior study (Angoa-Perez et al., 2013) are included for comparison (reprinted with permission from John Wiley and Sons, Inc.) These data are not statistically analyzed in the present figure, but METH elicited significant toxic effects on all three measures in the prior study, while MEPH significantly enhanced each marker of toxicity when co-administered with METH. *p < 0.05 compared to controls. Data are presented as mean + SEM. N = 5–16 mice per group.
3.2. Effects of increased DA on drug-induced glial reactivity in the striatum
Treatment with 4 MM alone did not alter the expression of Iba-1 with regard to controls, although there was a non-significant trend toward an increase. However, in combination with L-DOPA or METH, there was a significant increase in microglial activation over controls (Fig. 5A; F(7,77) = 11.6; p < 0.0001), which post hoc analysis also revealed was significantly elevated compared to 4 MM treatment alone. 4 MM + METH-treated mice trended higher than the Iba-1 increases evoked in animals treated with METH alone, but this did not reach significance. Pargyline did not significantly alter Iba-1 expression in combination with 4 MM. Treatment with MeCa alone significantly elevated Iba-1 levels compared to controls, as did its combination with L-DOPA or METH (Fig. 5B; F(7,87) = 5.310 p < 0.0001). Neither L-DOPA + MeCa nor METH + MeCa reached significance in comparison with MeCa alone, although L-DOPA + MeCa trended higher than MeCa alone. METH + MeCa showed no significant elevation over microglial activation elicited by METH treatment alone. Treatment with MEPH alone did not modify the Iba-1 expression found in controls. However, both METH and METH + MEPH treatment significantly increased microglial activation compared to controls as well as to MEPH, as revealed by one-way ANOVA (Fig. 5C; F(7,86) = 7.48; p < 0.0001). There was no significant difference between METH and METH + MEPH groups. Neither L-DOPA nor pargyline in combination with MEPH produced significant changes in Iba-1 levels. Also, L-DOPA or pargyline treatment alone failed to significantly increase Iba-1 expression compared to controls. Representative photomicrographs of striatal Iba-1 staining are presented in Fig. 5D.
Fig. 5.

Effects of MEPH, 4 MM, and MeCa ± L-DOPA, PARG, or METH on striatal Iba-1 immunoreactivity. Levels of Iba-1 immunofluorescence are shown in the upper panels (arbitrary units) for 4 MM (A), MeCa (B) and MEPH (C) + SEMs. Representative photomicrographs (20X) of each treatment condition stained against Iba-1 (red) and DAPI (counterstain; blue) are shown in the lower panels (D). *p < 0.05 **p < 0.01 ***p < 0.001 ****p < 0.0001 compared to controls. #p < 0.05 ##p < 0.01 ###p < 0.001 compared to MEPH/4MM/MeCa. Data are presented as mean + SEM. N = 3–5 mice per group.
Administration of 4 MM alone produced an increase in GFAP levels (Fig. 6A; F(7,82) = 18.6; p < 0.0001). Further post hoc comparisons revealed that the combination of 4 MM with either L-DOPA, pargyline, or METH also resulted in significantly elevated GFAP expression. These combined treatments trended higher than 4 MM alone, but only METH + 4 MM significantly enhanced the effects of 4 MM on GFAP levels. METH + 4 MM was also significantly elevated compared to the increase in GFAP elicited by METH alone. As with Iba-1, MeCa alone evoked a significant elevation in GFAP expression compared to controls, as did animals given MeCa with L-DOPA, pargyline, or METH (Fig. 6B; F(7,88) = 15.89 p < 0.0001). L-DOPA + MeCa-treated animals evoked a significant increase in GFAP when compared to MeCa alone. METH + MeCa treatment did not enhance GFAP expression compared with the increases evoked by METH or MeCa treatment alone. GFAP expression after MEPH administration was not different from controls, while it was increased following METH and METH + MEPH administration (Fig. 6C; F(7,88) = 17.97; p < 0.0001). METH + MEPH had enhanced GFAP expression compared to METH or MEPH alone. Neither L-DOPA nor pargyline produced any changes in GFAP in combination with MEPH. Also, L-DOPA or pargyline treatment alone failed to significantly increase GFAP expression compared to controls. Representative photomicrographs of striatal GFAP staining are presented in Fig. 6D.
Fig. 6.

Effects of MEPH, 4 MM, and MeCa ± L-DOPA, PARG, or METH on striatal GFAP immunoreactivity. Levels of GFAP immunofluorescence (mean + SEM) are shown in the upper panels (arbitrary units) for 4 MM (A), MeCa (B) and MEPH (C) + SEMs. Representative photomicrographs (20X) of each treatment condition stained against GFAP (green) and DAPI (counterstain; blue) are shown in the lower panels (D). *p < 0.05 **p < 0.01 ***p < 0.001 ****p < 0.0001 compared to controls. #p < 0.05 ####p < 0.0001 compared to MEPH/4MM/ MeCa. @ p < 0.05 @@ p < 0.01 denotes comparison to METH. Data are presented as mean + SEM. N = 3–5 mice per group.
3.3. Effect of increased DA on drug-induced changes in core body temperature
Significant changes in body temperatures were elicited by binge treatment with 4 MM, METH, and/or DA enhancers. A 2-way ANOVA revealed a significant effect of time (Fig. 7A; F(30,990) = 9.934, p < 0.0001) and treatment (F(7,33) = 4.506, p < 0.01), as well as a significant time X treatment interaction (F(210,990) = 10.35, p < 0.0001). 4 MM, both alone and in combination with L-DOPA, pargyline, or METH, elicited significant hyperthermia peaking around 2°C above controls, although L-DOPA administration caused an initial transient hypothermia that recovered within 40 min. Furthermore, though significant, the hyperthermia observed in L-DOPA + 4 MM-treated animals was not as prolonged as in other 4 MM-treated groups. METH alone was found to elicit significant hyperthermia of roughly the same magnitude as 4 MM.
Fig. 7.

Effects of MEPH, 4 MM, and MeCa ± L-DOPA, PARG, or METH on core body temperature. Core body temperatures of mice treated with 4 MM (A), MeCa (B), MEPH (C), ± L-DOPA, pargyline (PARG), or METH were recorded every 20 min. MEPH, 4 MM, MeCa, and METH were administered at time points 40, 160, 280, and 400 min. L-DOPA and PARG were administered 20 min prior to time point 0, and L-DOPA was also administered at time points 100, 220, and 340 min. Data are presented as mean temperature in C. SEMs were <5% of the mean for each group and are omitted for clarity. L-DOPA, PARG, and METH controls were run with each experiment and included for analysis, but each is included on only one graph for clarity. Significance versus controls (p < 0.05) is indicated with open symbols. Data for MEPH + METH from Angoa-Perez et al. (2013) were reprinted for comparison, with permission from John Wiley and Sons, Inc. These were not analyzed in the present figure, but induced a significant hyperthermia compared to controls in the original publication. N = 4–9 mice per group.
Binge MeCa administration, with or without L-DOPA, pargyline, or METH, caused profound hypothermia following each injection. A 2-way ANOVA revealed a significant effect of time (Fig. 7B; F(30,960) = 31.13, p < 0.0001), as well as a significant time X treatment interaction (F(210,960) = 11.76, p < 0.0001). All MeCa-treated mice showed a similar 3–4°C reductions in body temperature following drug administration, which recovered to baseline at the time of the next injection, with the exception of L-DOPA + MeCa animals. These animals showed an initial, transient hypothermia following L-DOPA administration, but a less extreme hypothermia following the second and third injections of MeCa.
There were also significant alterations in body temperature elicited by binge treatment with MEPH alone or in combination with L-DOPA or pargyline. A 2-way ANOVA revealed a significant effect of time (Fig. 7C; F(30,780) = 8.131, p < 0.0001) and treatment (F(5,26) = 4.04, p < 0.01), as well as a significant time X treatment interaction (F(150,780) = 7.192, p < 0.0001). Animals that received MEPH alone showed a reduction in core temperature after the first 2 injections, followed by a significant hyperthermia after the last 2 injections. Pargyline elicited no significant changes compared to control animals and did not alter the course of MEPH-induced temperature effects. L-DOPA treatment, with or without MEPH, caused an immediate, transient reduction in temperature, as it did with 4 MM and MeCa. Likewise, this recovered after 40 min. While the hyperthermia elicited by MEPH was reduced by L-DOPA treatment, it still achieved significance compared to controls.
4. Discussion
The present report sought to evaluate the hypothesis that the extent of DA release during binge treatment with the amphetamine-related compounds METH, MEPH, 4 MM, and MeCa determines the observed differences in dopaminergic neurotoxicity observed between these closely related structures. Specifically, it was proposed that MEPH lacks dopaminergic toxicity because it cannot increase the cytosolic pool of releasable DA from vesicular stores in a VMAT2-dependent manner and has lesser DA release via the DAT, compared to the releasing effects of the neurotoxic compounds METH, and to a less robust extent, MeCa (Eshleman et al., 2013). The studies in this report combined MEPH and the structurally related compounds, 4 MM and MeCa, with 3 treatments known to increase the pool of releasable DA: the DA synthesis precursor, L-DOPA; the inhibitor of MAO-B DA metabolism, pargyline; or a mildly toxic dose of METH, a well-known releaser of vesicular DA into the cytosol. According to our hypothesis, the increase in releasable DA from these treatments was expected to confer enhanced toxicity to MEPH, 4 MM, and MeCa, as assessed by reductions in dopaminergic markers and increases in glial activation.
The levels of dopaminergic toxicity elicited by 4 MM, MeCa, or MEPH alone were in agreement with prior studies (Angoa-Perez et al., 2012, 2013; Anneken et al., 2015; Anneken et al., 2017; Baumann et al., 2012; den Hollander et al., 2013; Gygi et al., 1996, 1997; Motbey et al., 2013; Sparago et al., 1996). 4 MM evoked DA and DAT depletions, MeCa caused significant reductions in all dopaminergic markers, while MEPH treatment did not elicit any toxic effects (see Figs. 2–4). In agreement with our hypothesis, the dopaminergic toxicity of 4 MM was greatly enhanced by all 3 treatments that increased the pool of releasable DA. Likewise, the toxicity evoked by MeCa was enhanced by combination with L-DOPA, although it was not altered by either pargyline or METH. MEPH, in combination with METH, has been previously reported to show greatly enhanced dopaminergic depletions (Angoa-Perez et al., 2013). However, unlike the intermediate compounds, MEPH elicited no dopaminergic toxicity when combined with either L-DOPA or pargyline.
Pretreatment with L-DOPA or pargyline is a well-established method for increasing the drug-releasable pool of DA. L-DOPA, as the precursor to DA synthesis, has been shown by multiple groups to rapidly increase striatal DA concentrations by 50–70% (Gianutsos et al., 1983; Kuhn et al., 2008; Thibaut et al., 1996; Thomas et al., 2008, 2009). Furthermore, a microdialysis study by Colado et al. (1999) reported increased efflux of striatal DA when L-DOPA was administered in combination with MDMA, compared to MDMA alone. In a similar manner, the inhibitor of MAO-B DA metabolism, pargyline, has been reported to rapidly increase striatal DA content by 10–20% (Gianutsos et al., 1983; Kuhn et al., 2008; Thomas et al., 2008, 2009). It has also been shown to significantly augment striatal DA release when given with amphetamine (Butcher et al., 1988). These data demonstrate that the DA-enhancing treatments utilized in the present study increase both striatal DA concentrations and DA efflux stimulated by amphetamines.
While each of the enhancement treatments in this study has been demonstrated to augment neurotoxicity through their actions on the dopaminergic system (Angoa-Perez et al., 2013; Anneken et al., 2015; Kita et al., 1995; Kuhn et al., 2008; Thomas et al., 2008, 2009), they act through different mechanisms, and thus may have differing enhancement potential. As previously mentioned, L-DOPA increases DA in the striatum through increased synthesis, while pargyline does so through the prevention of metabolism by MAO. METH causes release of vesicular DA from VMAT2, increasing the pool of drug-releasable DA in the cytosol (Eshleman et al., 2013). As L-DOPA causes a direct increase in newly synthesized DA, while both pargyline and METH merely alter the existing pools of DA, it would be reasonable to expect that L-DOPA would be the most effective enhancer of toxicity. Indeed, both 4 MM and MeCa toxicity were enhanced significantly by L-DOPA, while only 4 MM was impacted by pargyline or METH.
As glial reactivity typically accompanies METH-elicited neuro-toxicity (Guilarte et al., 2003; Pubill et al., 2003; Thomas et al., 2004b), the impact of each drug treatment regimen on Iba-1 (microglia) and GFAP (astrocytes) expression was also assessed. In the present study, levels of Iba-1 and GFAP generally correlated well with the extent of dopaminergic depletion observed in each treatment group (see Figs. 5 and 6). Furthermore, the DA-enhancing treatments that increased toxicity tended to increase glial reactivity. In agreement with its robust enhancement of dopaminergic depletions, combination with L-DOPA seemed to evoke the strongest glial activation in combined treatment for both 4 MM and MeCa.
The inability of L-DOPA or pargyline to impart dopaminergic toxicity to MEPH was surprising, given the similarity of its toxic profile to 4 MM, and the observed responsiveness of 4 MM to the dopamine-enhancing compounds in this study. In our prior investigation, only the highest dose in the range tested for 4 MM (2.5–40 mg/kg) elicited significant DA and DAT depletions, and thus closely resembled the lack of toxicity seen in MEPH (Anneken et al., 2017). This lead to the conclusion that the ring substitution in MEPH had a greater reductive impact on toxicity than the β-keto substitution, especially since the overall magnitude of MeCa toxicity was closer to METH. However, the enhancement of 4 MM toxicity by L-DOPA and pargyline is a characteristic shared by METH but not MEPH (Guillot et al., 2008; Kita et al., 1995; Kuhn et al., 2008; Thomas et al., 2008, 2009). Thus, it seems that the relationship between individual substituents and dopaminergic neurotoxicity in these compounds is more complex than had been originally assumed.
As it has been well-established that increases (Xie et al., 2000) or decreases (Ali et al., 1994; Miller and O'Callaghan, 1994) in associated hyperthermia can profoundly alter METH toxicity, the final experiment of this study measured the effects of an increased pool of releasable DA on the body temperature effects of 4 MM, MeCa, and MEPH, to assess any potential impact on toxicity. When given alone, these compounds had remarkably divergent effects on core body temperatures, in agreement with previous reports (Angoa-Perez et al., 2012, 2013; Anneken et al., 2015; Anneken et al., 2017; Grecco and Sprague, 2016; Lopez-Arnau et al., 2015; Rockhold et al., 1997; Shortall et al., 2013, 2016; Wright et al., 2012). While 4 MM induced hyperthermia, MeCa produced profound, prolonged hypothermia after each injection. MEPH-treated animals showed an initial hypothermia following the first 2 injections, followed by prolonged hyperthermia after the final 2 injections (see Fig. 7). Interestingly, L-DOPA itself induced an immediate, transient hypothermia lasting for 20–40 min after the first injection, an effect also seen in combination with 4 MM, MeCa, and MEPH. While combination with L-DOPA diminished the duration of either hyperthermia, in 4 MM and MEPH, or hypothermia, in MeCa, these effects still achieved significance compared to controls. Although it is well known that induced hypothermia can abrogate the toxicity of amphetamines (Ali et al., 1994), it is unlikely that the transient hypothermia of L-DOPA interfered with any potential MEPH toxicity in this study. L-DOPA is known to enhance METH dopaminergic neurotoxicity despite this initial reduction in body temperature (Thomas et al., 2008), and was in fact successful in enhancing both 4 MM and MeCa toxicity. Finally, combination with pargyline or METH did not alter the temperature effects for any of the drugs. This agrees with our prior study that reported no change in MEPH hyperthermia due to combined treatment with METH (Angoa-Perez et al., 2013).
It is clear that the structural substituents in these compounds greatly impact the body temperature effects of each drug, though the mechanism of these effects is still unclear. In view of the generally minimal impact of increased DA on the drug-induced temperature effects in this study, it is unlikely that the dopaminergic system alone mediates these observed differences. A recent investigation found evidence for adrenergic modulation of MEPH hyperthermia in rodents (Zona et al., 2016), while another group prevented the initial hypothermic effects of MEPH using 5-HT depletion and 5HT1A receptor antagonists (Shortall et al., 2016). It would seem the observed divergence in body temperature effects among bath salts involves multiple neurotransmitter pathways, but further study of MeCa and 4 MM body temperature effects is needed to confirm this. Importantly, although hyperthermia has long been regarded as essential for METH toxicity, there was no clear relationship between temperature and dopaminergic depletions in the present study. The generally hyperthermic MEPH exhibited no toxicity and did not increase glial reactivity, while the profoundly hypothermic MeCa depleted DA, DAT and TH in striatum and resulted in significant increases in glial reactivity alone and when given with enhanced releasable DA. This dissociation supports the view of hyperthermia as merely a contributing factor in METH toxicity, rather than a causal one.
Taken together, the data presented in this study point to an as yet unidentified determining factor that triggers the neurotoxic cascade following METH administration, which is present in both MeCa and 4 MM but lacking in MEPH. The hypothesis at the outset of the study was that the observed differences in neurotoxicity were due to the two structural substituents in MEPH interfering, to varying degrees, with the drug's ability to release DA from vesicular stores via VMAT2 and then via the DAT, and that increasing the pool of drug-releasable DA would confer METH-like toxicity to MEPH, as METH elicits neurotoxicity and releases neurotransmitter from VMAT2 and DAT in greater amounts than MEPH (Eshleman et al., 2013). The results of the study have demonstrated that, while increased DA can modulate the severity of toxicity evoked by the intermediate structures 4 MM and MeCa as well as METH, it is not the crucial missing element that can account for the lack of dopaminergic toxicity in MEPH. Future investigations should focus on alternative, non-dopaminergic pathways that may differ between MEPH, METH, and their closely related intermediates.
One such pathway yet to be assessed may lie outside of the CNS. A recent series of studies has identified hyperthermia-dependent liver toxicity, and the resulting increase in systemic ammonia, as mediators of METH neurotoxicity through glutamate excitotoxicity (Halpin et al., 2013, 2014b; Halpin and Yamamoto, 2012). To date, only METH has been evaluated for these peripheral, though neurotoxic, effects. It will be valuable to assess baths salts and their intermediates for ammonia-mediated toxicity, especially as these compounds have varying body temperature effects that can impact liver damage and ammonia production.
Despite a body of literature devoted to explaining the lack of MEPH dopaminergic neurotoxicity compared to METH, the key mechanism(s) responsible remains elusive. Identifying this mechanism will improve our understanding of METH-related neurotoxicity, and may lead to an effective treatment to prevent or repair lasting damage from METH exposure.
Acknowledgments
Funding
Funding for this study was provided by the National Institute on Drug Abuse (R21-DA039667] and the Veterans Administration [I01-RX000458).
The authors would like to acknowledge the NIDA Research Resources Drug Supply Program for providing mephedrone and methcathinone for this study. We would also like to acknowledge Dr. Roxanne Vaughan for generously providing DAT antibodies for Western blot analysis.
Footnotes
Abbreviations: 4 MM, 4-methylmethamphetamine; 5-HT, serotonin; ANOVA, analysis of variance; DA, dopamine; DAT, dopamine transporter; GAPDH, glyceral-dehyde 3-phosphate dehydrogenase; GFAP, glial fibrillary acid protein; HPLC, high performance liquid chromatography; Iba-1, ionized calcium-binding adapter molecule 1; MeCa, methcathinone; MEPH, mephedrone; METH, methamphetamine; TH, tyrosine hydroxylase; VMAT2, vesicular monoamine transporter-2.
Declaration of interests
The authors have no conflicts of interest concerning this work.
References
- Ali SF, Newport GD, Holson RR, Slikker W, Jr, Bowyer JF. Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- Angoa-Perez M, Kane MJ, Briggs DI, Francescutti DM, Sykes CE, Shah MM, Thomas DM, Kuhn DM. Mephedrone does not damage dopamine nerve endings of the striatum, but enhances the neurotoxicity of methamphetamine, amphetamine, and MDMA. J Neurochem. 2013;125:102–110. doi: 10.1111/jnc.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angoa-Perez M, Kane MJ, Francescutti DM, Sykes KE, Shah MM, Mohammed AM, Thomas DM, Kuhn DM. Mephedrone, an abused psychoactive component of 'bath salts' and methamphetamine congener, does not cause neurotoxicity to dopamine nerve endings of the striatum. J Neurochem. 2012;120:1097–1107. doi: 10.1111/j.1471-4159.2011.07632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anneken JH, Angoa-Perez M, Kuhn DM. 3,4-Methylenedioxypyrovalerone prevents while methylone enhances methamphetamine-induced damage to dopamine nerve endings: beta-ketoamphetamine modulation of neurotoxicity by the dopamine transporter. J Neurochem. 2015;133:211–222. doi: 10.1111/jnc.13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anneken JH, Angoa-Perez M, Sati GC, Crich D, Kuhn DM. Dissecting the influence of two structural substituents on the differential neurotoxic effects of acute methamphetamine and mephedrone treatment on dopamine nerve endings with the use of 4-methylmethamphetamine and methcathinone. J Pharmacol Exp Ther. 2017;360:417–423. doi: 10.1124/jpet.116.237768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Ayestas MA, Jr, Partilla JS, Sink JR, Shulgin AT, Daley PF, Brandt SD, Rothman RB, Ruoho AE, Cozzi NV. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37:1192–1203. doi: 10.1038/npp.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SP, Fairbrother IS, Kelly JS, Arbuthnott GW. Amphetamine-induced dopamine release in the rat striatum: an in vivo microdialysis study. J Neurochem. 1988;50:346–355. doi: 10.1111/j.1471-4159.1988.tb02919.x. [DOI] [PubMed] [Google Scholar]
- Cameron K, Kolanos R, Vekariya R, De Felice L, Glennon RA. Mephedrone and methylenedioxypyrovalerone (MDPV), major constituents of “bath salts,” produce opposite effects at the human dopamine transporter. Psychopharmacol (Berl) 2013;227:493–499. doi: 10.1007/s00213-013-2967-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colado MI, O'Shea E, Granados R, Esteban B, Martin AB, Green AR. Studies on the role of dopamine in the degeneration of 5-HT nerve endings in the brain of Dark Agouti rats following 3,4-methylenedioxymethamphetamine (MDMA or 'ecstasy') administration. Br J Pharmacol. 1999;126:911–924. doi: 10.1038/sj.bjp.0702373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creehan KM, Vandewater SA, Taffe MA. Intravenous self-administration of mephedrone, methylone and MDMA in female rats. Neuropharmacology. 2015;92:90–97. doi: 10.1016/j.neuropharm.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Blakey K, Rands-Trevor K. GC-MS and GC-IRD analysis of 2-, 3- and 4-methylmethamphetamine and 2-, 3- and 4-methylamphetamine. Forensic Sci Int. 2012;220:67–73. doi: 10.1016/j.forsciint.2012.01.028. [DOI] [PubMed] [Google Scholar]
- den Hollander B, Rozov S, Linden AM, Uusi-Oukari M, Ojanpera I, Korpi ER. Long-term cognitive and neurochemical effects of “bath salt” designer drugs methylone and mephedrone. Pharmacol Biochem Behav. 2013;103:501–509. doi: 10.1016/j.pbb.2012.10.006. [DOI] [PubMed] [Google Scholar]
- den Hollander B, Sundstrom M, Pelander A, Ojanpera I, Mervaala E, Korpi ER, Kankuri E. Keto amphetamine toxicity-focus on the redox reactivity of the cathinone designer drug mephedrone. Toxicol Sci. 2014;141:120–131. doi: 10.1093/toxsci/kfu108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman AJ, Wolfrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A. Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol. 2013;85:1803–1815. doi: 10.1016/j.bcp.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianutsos G, Carlson GM, Godfrey JG. Drug-induced changes in motor activity after selective MAO inhibition. Pharmacol Biochem Behav. 1983;19:263–268. doi: 10.1016/0091-3057(83)90050-3. [DOI] [PubMed] [Google Scholar]
- Golembiowska K, Jurczak A, Kaminska K, Noworyta-Sokolowska K, Gorska A. Effect of some psychoactive drugs used as 'legal highs' on brain neurotransmitters. Neurotox Res. 2016;29:394–407. doi: 10.1007/s12640-015-9569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grecco GG, Sprague JE. Impact of functional group modifications on designer phenethylamine induced hyperthermia. Chem Res Toxicol. 2016;29:871–878. doi: 10.1021/acs.chemrestox.6b00030. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, Nihei MK, McGlothan JL, Howard AS. Methamphetamine-induced deficits of brain monoaminergic neuronal markers: distal axotomy or neuronal plasticity. Neuroscience. 2003;122:499–513. doi: 10.1016/s0306-4522(03)00476-7. [DOI] [PubMed] [Google Scholar]
- Guillot TS, Shepherd KR, Richardson JR, Wang MZ, Li Y, Emson PC, Miller GW. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J Neurochem. 2008;106:2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi MP, Fleckenstein AE, Gibb JW, Hanson GR. Role of endogenous dopamine in the neurochemical deficits induced by methcathinone. J Pharmacol Exp Ther. 1997;283:1350–1355. [PubMed] [Google Scholar]
- Gygi MP, Gibb JW, Hanson GR. Methcathinone: an initial study of its effects on monoaminergic systems. J Pharmacol Exp Ther. 1996;276:1066–1072. [PubMed] [Google Scholar]
- Hadlock GC, Webb KM, McFadden LM, Chu PW, Ellis JD, Allen SC, Andrenyak DM, Vieira-Brock PL, German CL, Conrad KM, Hoonakker AJ, Gibb JW, Wilkins DG, Hanson GR, Fleckenstein AE. 4-Methylmethcathinone (mephedrone): neuropharmacological effects of a designer stimulant of abuse. J Pharmacol Exp Ther. 2011;339:530–536. doi: 10.1124/jpet.111.184119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, Collins SA, Yamamoto BK. Neurotoxicity of methamphetamine and 3,4-methylenedioxymethamphetamine. Life Sci. 2014a;97:37–44. doi: 10.1016/j.lfs.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, Gunning WT, Yamamoto BK. Methamphetamine causes acute hyperthermia-dependent liver damage. Pharmacol Res Perspect. 2013;1:e00008. doi: 10.1002/prp2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, Northrop NA, Yamamoto BK. Ammonia mediates methamphetamine-induced increases in glutamate and excitotoxicity. Neuropsychopharmacology. 2014b;39:1031–1038. doi: 10.1038/npp.2013.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, Yamamoto BK. Peripheral ammonia as a mediator of methamphetamine neurotoxicity. J Neurosci. 2012;32:13155–13163. doi: 10.1523/JNEUROSCI.2530-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harun R, Hare KM, Brough ME, Munoz MJ, Grassi CM, Torres GE, Grace AA, Wagner AK. Fast-scan cyclic voltammetry demonstrates that L-DOPA produces dose-dependent regionally selective, bimodal effects on striatal dopamine kinetics in vivo. J Neurochem. 2015;136:1270–1283. doi: 10.1111/jnc.13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenhull J, Murphy KG, Paterson S. Mephedrone use is increasing in London. Lancet. 2016;387:1719–1720. doi: 10.1016/S0140-6736(16)30258-6. [DOI] [PubMed] [Google Scholar]
- Karlsson L, Andersson M, Kronstrand R, Kugelberg FC. Mephedrone, methylone and 3,4-methylenedioxypyrovalerone (MDPV) induce conditioned place preference in mice. Basic Clin Pharmacol Toxicol. 2014;115:411–416. doi: 10.1111/bcpt.12253. [DOI] [PubMed] [Google Scholar]
- Kita T, Wagner GC, Philbert MA, King LA, Lowndes HE. Effects of pargyline and pyrogallol on the methamphetamine-induced dopamine depletion. Mol Chem Neuropathol. 1995;24:31–41. doi: 10.1007/BF03160110. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Billingsley ML. Tyrosine hydroxylase: purification from PC-12 cells, characterization and production of antibodies. Neurochem Int. 1987;11:463–475. doi: 10.1016/0197-0186(87)90036-2. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine disposition in the presynaptic process regulates the severity of methamphetamine-induced neurotoxicity. Ann N Y Acad Sci. 2008;1139:118–126. doi: 10.1196/annals.1432.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisek R, Xu W, Yuvasheva E, Chiu YT, Reitz AB, Liu-Chen LY, Rawls SM. Mephedrone ('bath salt') elicits conditioned place preference and dopamine-sensitive motor activation. Drug Alcohol Depend. 2012;126:257–262. doi: 10.1016/j.drugalcdep.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Arnau R, Martinez-Clemente J, Pubill D, Escubedo E, Camarasa J. Comparative neuropharmacology of three psychostimulant cathinone derivatives: butylone, mephedrone and methylone. Br J Pharmacol. 2012;167:407–420. doi: 10.1111/j.1476-5381.2012.01998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Arnau R, Martinez-Clemente J, Rodrigo T, Pubill D, Camarasa J, Escubedo E. Neuronal changes and oxidative stress in adolescent rats after repeated exposure to mephedrone. Toxicol Appl Pharmacol. 2015;286:27–35. doi: 10.1016/j.taap.2015.03.015. [DOI] [PubMed] [Google Scholar]
- Martinez-Clemente J, Lopez-Arnau R, Abad S, Pubill D, Escubedo E, Camarasa J. Dose and time-dependent selective neurotoxicity induced by mephedrone in mice. PLoS One. 2014;9:e99002. doi: 10.1371/journal.pone.0099002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusich JA, Grant KR, Blough BE, Wiley JL. Effects of synthetic cathinones contained in “bath salts” on motor behavior and a functional observational battery in mice. Neurotoxicology. 2012;33:1305–1313. doi: 10.1016/j.neuro.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DB, O'Callaghan JP. Environment-, drug- and stress-induced alterations in body temperature affect the neurotoxicity of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:752–760. [PubMed] [Google Scholar]
- Moratalla R, Khairnar A, Simola N, Granado N, Garcia-Montes JR, Porceddu PF, Tizabi Y, Costa G, Morelli M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: main mechanisms. Prog Neurobiol. 2015;155:149–170. doi: 10.1016/j.pneurobio.2015.09.011. [DOI] [PubMed] [Google Scholar]
- Motbey CP, Clemens KJ, Apetz N, Winstock AR, Ramsey J, Li KM, Wyatt N, Callaghan PD, Bowen MT, Cornish JL, McGregor IS. High levels of intravenous mephedrone (4-methylmethcathinone) self-administration in rats: neural consequences and comparison with methamphetamine. J Psychopharmacol. 2013;27:823–836. doi: 10.1177/0269881113490325. [DOI] [PubMed] [Google Scholar]
- Motbey CP, Hunt GE, Bowen MT, Artiss S, McGregor IS. Mephedrone (4-methylmethcathinone, 'meow'): acute behavioural effects and distribution of Fos expression in adolescent rats. Addict Biol. 2012;17:409–422. doi: 10.1111/j.1369-1600.2011.00384.x. [DOI] [PubMed] [Google Scholar]
- Nguyen JD, Aarde SM, Cole M, Vandewater SA, Grant Y, Taffe MA. Locomotor stimulant and rewarding effects of inhaling methamphetamine, MDPV, and mephedrone via electronic cigarette-type technology. Neuropsychopharmacology. 2016;41:2759–2771. doi: 10.1038/npp.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaseit E, Perez-Mana C, Mateus JA, Pujadas M, Fonseca F, Torrens M, Olesti E, de la Torre R, Farre M. Human pharmacology of mephedrone in comparison with MDMA. Neuropsychopharmacology. 2016;41:2704–2713. doi: 10.1038/npp.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pubill D, Canudas AM, Pallas M, Camins A, Camarasa J, Escubedo E. Different glial response to methamphetamine- and methylenedioxymethamphetamine-induced neurotoxicity. Naunyn Schmiedeb Arch Pharmacol. 2003;367:490–499. doi: 10.1007/s00210-003-0747-y. [DOI] [PubMed] [Google Scholar]
- Rockhold RW, Carlton FB, Jr, Corkern R, Derouen L, Bennett JG, Hume AS. Methcathinone intoxication in the rat: abrogation by dextrorphan. Ann Emerg Med. 1997;29:383–391. doi: 10.1016/s0196-0644(97)70351-2. [DOI] [PubMed] [Google Scholar]
- Shortall SE, Green AR, Swift KM, Fone KC, King MV. Differential effects of cathinone compounds and MDMA on body temperature in the rat, and pharmacological characterization of mephedrone-induced hypothermia. Br J Pharmacol. 2013;168:966–977. doi: 10.1111/j.1476-5381.2012.02236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortall SE, Spicer CH, Ebling FJ, Green AR, Fone KC, King MV. Contribution of serotonin and dopamine to changes in core body temperature and locomotor activity in rats following repeated administration of mephedrone. Addict Biol. 2016;21:1127–1139. doi: 10.1111/adb.12283. [DOI] [PubMed] [Google Scholar]
- Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME. Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol. 2013;168:458–470. doi: 10.1111/j.1476-5381.2012.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparago M, Wlos J, Yuan J, Hatzidimitriou G, Tolliver J, Dal Cason TA, Katz J, Ricaurte G. Neurotoxic and pharmacologic studies on enantiomers of the N-methylated analog of cathinone (methcathinone): a new drug of abuse. J Pharmacol Exp Ther. 1996;279:1043–1052. [PubMed] [Google Scholar]
- Suyama JA, Sakloth F, Kolanos R, Glennon RA, Lazenka MF, Negus SS, Banks ML. Abuse-related neurochemical effects of para-substituted methcathinone analogs in rats: microdialysis studies of nucleus accumbens dopamine and serotonin. J Pharmacol Exp Ther. 2016;356:182–190. doi: 10.1124/jpet.115.229559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibaut F, Bonnet JJ, Vaugeois JM, Costentin J. Pharmacological modifications of dopamine transmission do not influence the striatal in vivo binding of [3H]mazindol or [3H]cocaine in mice. Neurosci Lett. 1996;205:145–148. doi: 10.1016/0304-3940(96)12399-5. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM. Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett. 2004a;367:349–354. doi: 10.1016/j.neulet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J Neurochem. 2008;105:605–616. doi: 10.1111/j.1471-4159.2007.05155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. Increases in cytoplasmic dopamine compromise the normal resistance of the nucleus accumbens to methamphetamine neurotoxicity. J Neurochem. 2009;109:1745–1755. doi: 10.1111/j.1471-4159.2009.06094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004b;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Wright MJ, Jr, Angrish D, Aarde SM, Barlow DJ, Buczynski MW, Creehan KM, Vandewater SA, Parsons LH, Houseknecht KL, Dickerson TJ, Taffe MA. Effect of ambient temperature on the thermoregulatory and locomotor stimulant effects of 4-methylmethcathinone in Wistar and Sprague-Dawley rats. PLoS One. 2012;7:e44652. doi: 10.1371/journal.pone.0044652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, McCann UD, Kim S, Yuan J, Ricaurte GA. Effect of temperature on dopamine transporter function and intracellular accumulation of methamphetamine: implications for methamphetamine-induced dopaminergic neurotoxicity. J Neurosci. 2000;20:7838–7845. doi: 10.1523/JNEUROSCI.20-20-07838.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Raudensky J. The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J Neuroimmune Pharmacol. 2008;3:203–217. doi: 10.1007/s11481-008-9121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zona LC, Grecco GG, Sprague JE. Cooling down the bath salts: carvedilol attenuation of methylone and mephedrone mediated hyperthermia. Toxicol Lett. 2016;263:11–15. doi: 10.1016/j.toxlet.2016.10.012. [DOI] [PubMed] [Google Scholar]