

Abstract
Lipid droplets (LDs) are conserved, endoplasmic reticulum (ER)-derived organelles that act as a dynamic cellular repository for neutral lipids. Numerous studies have examined the composition of LD proteomes by using mass spectrometry to identify proteins present in biochemically isolated buoyant fractions that are enriched in LDs. Although many bona fide LD proteins were identified, high levels of non-LD proteins that contaminate buoyant fractions complicate the detection of true LD proteins. To overcome this problem, we recently developed a proximity-labeling proteomic method to define high-confidence LD proteomes. Moreover, employing this approach, we discovered that ER-associated degradation impacts the composition of LD proteomes by targeting select LD proteins for clearance by the 26S proteasome as they transit between the ER and LDs. These findings implicate the ER as a site of LD protein degradation and underscore the high degree of crosstalk between ER and LDs.
Keywords: organelle contacts, APEX, APEX2, ubiquitin, metabolic disease, obesity
Lipid droplets (LDs) are endoplasmic reticulum (ER)-derived neutral lipid (e.g., triacylglycerol or sterol esters) storage organelles present in most organisms and mammalian tissues (Walther, Chung, & Farese, 2017). LDs consist of a neutral lipid core that is encircled by a phospholipid monolayer decorated by an array of regulatory proteins. Storage of lipids within the interior of LDs prevents lipotoxic damage and provides a pool of fatty acids that can be mobilized for energy, membrane biosynthesis, and lipid-mediated signaling pathways. Consistent with their critical role in lipid metabolism, defects in LD biogenesis or catabolism have been implicated in the etiology of a wide variety of diseases (Krahmer, Farese, & Walther, 2013), including obesity, diabetes, atherosclerosis, lipodystrophy, and cachexia, highlighting the importance of understanding the mechanisms that underlie LD biogenesis and function.
LDs are regulated by a membrane-associated proteome that consists of integral and peripheral proteins (Bersuker & Olzmann, 2017). The unique ultrastructure of LDs constrains the possible protein topologies. Indeed, proteins are absent from the hydrophobic interior of the LD and the integral membrane LD proteins adopt monotopic conformations, embedding in the membrane via amphipathic helices and hydrophobic hairpins. Many studies have sought to define the composition of LD proteomes by employing mass spectrometry to analyze the proteins present in LD-enriched buoyant fractions isolated by density gradient centrifugation. However, the presence of contaminating organelles (e.g., ER and mitochondria) and the localization of some LD proteins to multiple organelles confounded the results from these studies. This is apparent by the frequent identification of abundant luminal and polytopic membrane ER proteins, which are topologically incompatible with the LD monolayer. To overcome this obstacle, we used a proximity labeling approach that employs LD-targeted mutant ascorbate peroxidase (APEX2), which mediates the temporally and spatially restricted biotinylation of LD proteins in living cells (Bersuker et al., 2018; Rhee et al., 2013). The biotinylated LD proteins can then be affinity purified and identified by mass spectrometry. This proximity labeling paradigm allowed us to identify high-confidence LD proteomes from two human cell lines and generate an LD proteomics resource (http://dropletproteome.org; Bersuker et al., 2018).
As anticipated, the number of proteins labeled by LD-targeted APEX2 was significantly lower than the number of proteins present in LD-enriched buoyant fractions. Importantly, the majority of the proteins identified in the buoyant fraction, but not by our proximity labeling approach, were contaminants (e.g., ER luminal proteins, ER chaperones, and polytopic ER membrane proteins). Our results also suggest that proximity labeling identifies a comprehensive LD proteome, as all 44 proteins in the buoyant fraction that were previously confirmed to localize to LDs using microscopy methods were labeled and identified. Moreover, the approach also identified many new LD proteins, and 11/13 candidate LD proteins were confirmed to localize to LDs by fluorescence microscopy. Thus, proximity labeling enables the selective and comprehensive identification of LD proteomes.
While all tissues have the capacity to make LDs, the composition of the LD proteome differs between cell types. To explore if LD-targeted APEX2 can be used to distinguish cell type-specific LD proteomes, we identified and compared high-confidence LD proteomes from two cultured human cell lines, U2OS osteosarcoma (152 proteins) and Huh7 hepatoma (77 proteins) cells. As expected, LDs from these cells shared much of the core lipid metabolism machinery, such as acyl-CoA synthetases, perilipin coat proteins, acyltransferases, and lipases. LDs from both cell types also contained proteins from several other functional categories, including membrane trafficking (nearly half of the known RAB proteins), ubiquitin-mediated protein degradation, and redox reactions. A notable difference was that LDs from Huh7 cells contained the LD fusion regulator CIDEB (Gao et al., 2017), which is likely responsible for the larger LDs in this cell type. The autophagy adaptor p62 was also selectively present in the Huh7 proteome, consistent with findings that LDs in liver cells are degraded by lipophagy (Martinez-Lopez & Singh, 2015).
The presence of a ubiquitin-related module in both cell types raises the possibility that these proteins constitute a ubiquitination apparatus that regulates LD proteins. This functional module included two soluble proteins, the E2 ubiquitin-conjugating enzyme Ube2g2 and the AAA ATPase valosin-containing protein (VCP), along with their respective membrane-embedded recruitment factors – UBXD8 (VCP recruitment), UBXD2 (VCP recruitment), and AUP1 (Ube2g2 recruitment). We noted the presence of the E3 ligase-recruitment factor SPG20, but its binding partners, the E3 ligases AIP4 and AIP5, were absent. VCP plays a principal role in the degradation of proteins from the early secretory pathway via ER-associated degradation (ERAD; Olzmann, Kopito, & Christianson, 2013). In this process, VCP extracts ubiquitinated substrates from the ER membrane, enabling their degradation by cytosolic 26S proteasomes (Figure 1). We reasoned that VCP inhibition would stabilize any LD proteins undergoing degradation through ubiquitin- and VCP-dependent pathways. Thus, we exploited the fast labeling time of APEX2 and quantitative proteomics to measure changes in LD protein abundance following pharmacological inhibition of VCP. This approach identified c18orf32, a small protein of unknown function on LDs, as a substrate of VCP. Further studies demonstrated that c18orf32 undergoes ubiquitin- and VCP-dependent degradation that is facilitated by canonical ER-resident ERAD components, including the dislocation factor derlin-1 and the E3 ligase gp78. Future studies will examine the extent to which ERAD regulates LD proteome remodeling in metabolic state shifts, such as during signaling-induced lipolytic degradation of LDs.
Figure 1.
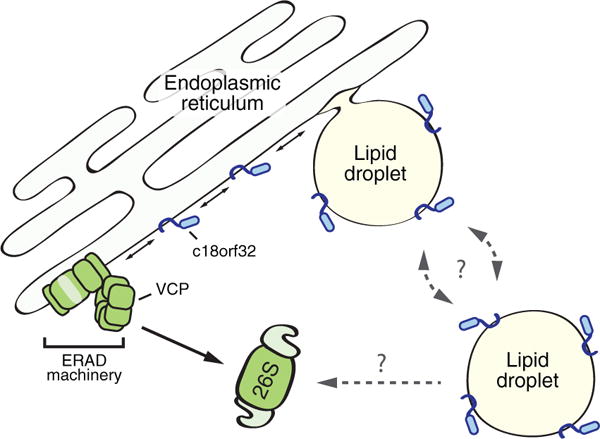
ERAD impacts the composition of the LD proteome. A subset of LD proteins inserts into the ER and traffics into nascent LDs (e.g., c18orf32). These LD proteins are accessible to ERAD machinery, which regulates their levels in the ER and flux into emerging LDs. Whether LD proteins are able to traffic back into the ER for degradation by ERAD, such as via ER-LD membrane bridges, is unknown. In addition, although several proteins associated with ubiquitin-dependent protein degradation are present on LDs, it remains unclear if LD proteins are ubiquitinated and degraded directly from the LD surface. ERAD = endoplasmic reticulum-associated degradation; VCP = valosin-containing protein.
Together, our data suggest that the ER functions as a platform for LD protein degradation (Figure 1) and underscore the high level of communication between these two organelles. We propose that LD proteins (e.g., c18orf32) that insert into the ER are potential substrates for degradation by ERAD. Thus, ERAD is able to regulate their steady-state levels in the ER and consequently the level of proteins that traffic into forming LDs. A recent study identified a similar ERAD mechanism of LD protein degradation in yeast (Ruggiano, Mora, Buxó, & Carvalho, 2016), indicating that this mode of LD protein quantity control is conserved. Another possibility is that LD proteins are degraded by traveling from LDs to the ER through membrane bridges that connect the two organelles (Wilfling et al., 2013). In support of this model, induction of LDs did not increase the stability of c18orf32, suggesting that LD-associated c18orf32 still has access to ERAD machinery. It remains possible that some proteins are degraded by ubiquitination machinery on the LD surface, which would provide a rapid posttranslational mechanism for controlling the composition of the LD proteome. In addition, although LDs are not generally required for ERAD, certain ERAD substrates appear to be degraded from ER subdomains proximal to LDs (i.e., HMG-CoA reductase) or from the LD surface (i.e., lipi-dated Apolipoprotein B-100) (Bersuker & Olzmann, 2017). Why HMG-CoA reductase and lipidated Apolipoprotein B-100 undergo ERAD at these specialized ER-LD subdomains is unclear. This spatial relationship may provide a mechanism to couple the regulation of lipid storage with cholesterol biosynthesis and secretion of lipoprotein particles. Future studies will identify potential substrates of LD-associated ubiquitination machinery and determine the role of this ubiquitination complex in LD function.
Acknowledgments
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by grants from the National Institutes of Health (R01GM112948 to J. A. O.) and the American Heart Association (16GRNT30870005 to J. A. O.).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Bersuker K, Olzmann JA. Establishing the lipid droplet proteome: Mechanisms of lipid droplet protein targeting and degradation. Biochimica et Biophysica Acta. 2017;1862(10 Pt B):1166–1177. doi: 10.1016/j.bbalip.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Peterson CWH, To M, Sahl SJ, Savikhin V, Grossman EA, Olzmann JA. A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Developmental Cell. 2018;44(1):97–112.e7. doi: 10.1016/j.devcel.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Chen FJ, Zhou L, Su L, Xu D, Xu L, Li P. Control of lipid droplet fusion and growth by CIDE family proteins. Biochimica et Biophysica Acta. 2017;1862(10 Pt B):1197–1204. doi: 10.1016/j.bbalip.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Krahmer N, Farese RV, Walther TC. Balancing the fat: Lipid droplets and human disease. EMBO Molecular Medicine. 2013;5(7):973–983. doi: 10.1002/emmm.201100671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez N, Singh R. Autophagy and lipid droplets in the liver. Annual Review of Nutrition. 2015;35(1):215–237. doi: 10.1146/annurev-nutr-071813-105336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339(6125):1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Mora G, Buxó L, Carvalho P. Spatial control of lipid droplet proteins by the ERAD ubiquitin ligase Doa10. The EMBO Journal. 2016;35(15):1644–1655. doi: 10.15252/embj.201593106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harbor Perspectives in Biology. 2013;5(9) doi: 10.1101/cshperspect.a013185. pii: a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther TC, Chung J, Farese RV. Lipid droplet biogenesis. Annual Review of Cell and Developmental Biology. 2017;33:491–510. doi: 10.1146/annurev-cellbio-100616-060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Walther TC. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Developmental Cell. 2013;24(4):384–399. doi: 10.1016/j.devcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]