

Summary
The modulator of retrovirus infection (MRI or CYREN) is a 30 kDa protein with a conserved N-terminal Ku-binding motif (KBM) and a C-terminal XLF-like motif (XLM). We show that MRI is intrinsically disordered and interacts with many DNA damage response (DDR) proteins, including the kinases ATM and DNA-PKcs and the classical non-homologous end joining (cNHEJ) factors Ku70, Ku80, XRCC4, XLF, PAXX, and XRCC4. MRI forms large multimeric complexes that depend on its N- and C-termini and localizes to DNA double-strand breaks (DSBs), where it promotes the retention of DDR factors. Mice deficient in MRI and XLF exhibit embryonic lethality at a stage similar to those deficient in the core cNHEJ factors XRCC4 or DNA Ligase IV. Moreover, MRI is required for cNHEJ-mediated DSB repair in XLF-deficient lymphocytes. We propose that MRI is an adaptor that, through multivalent interactions, increases the avidity of DDR factors to DSB-associated chromatin to promote cNHEJ.
Keywords: Non-homologous end joining, V(D)J recombination, DNA double-strand break repair, MRI, adaptor protein
eTOC Blurb
Hung et al. demonstrate that MRI is a disordered protein that functions in DSB repair and is essential for cNHEJ in the absence of XLF. MRI interacts with cNHEJ and DDR signaling factors at its termini and promotes the avidity of these proteins for chromatin in response to DNA damage.
Introduction
In mammalian cells, DNA double-strand breaks (DSBs) activate a DNA damage response (DDR) to repair lesions by two main pathways: non-homologous end joining (NHEJ) and homologous recombination (HR) (Chang et al., 2017; Ciccia and Elledge, 2010). HR functions to repair DSBs in the S and G2 phases of the cell cycle, using the sister chromatid as a template for precise repair. In contrast, NHEJ functions during all phases of the cell cycle and is the primary pathway of DSB repair in G1-phase cells. A core set of proteins is essential for DSB repair by classical NHEJ (cNHEJ) (Chang et al., 2017). This core set includes the Ku70/Ku80 (Ku) heterodimer, which binds to broken DNA ends; DNA Ligase IV, which ligates broken DNA ends; and XRCC4, which is essential for the stability and activity of DNA Ligase IV and, with XLF, bridges DNA ends during cNHEJ (Andres et al., 2007; Brouwer et al., 2016; Chang et al., 2017; Hammel et al., 2010; Reid et al., 2015; Ropars et al., 2011). NHEJ that occurs in the absence of one or more of these core factors is referred to as alternative NHEJ (aNHEJ) (Chang et al., 2017). In G1-phase cells, DDR and DSB repair by NHEJ are orchestrated by DNA-PKcs and ATM, two related serine/threonine kinases (Blackford and Jackson, 2017). DNA-PKcs is recruited and activated at DSBs by Ku, whereas ATM is recruited and activated by the MRN complex, composed of Mre11, Rad50 and Nbs1 (Paull, 2015; Stracker and Petrini, 2011).
The generation of DNA DSBs and their repair by NHEJ are required in two physiological processes in lymphocytes: immunoglobulin (Ig) class switch recombination (CSR), which generates antibodies of different isotypes; and V(D)J recombination, which assembles antigen receptor genes (Chaudhuri et al., 2007; Helmink and Sleckman, 2012). Ig CSR occurs in mature B lymphocytes and involves the generation of DSBs by activation-induced cytidine deaminase (AID) and repair of these DSBs by either cNHEJ or aNHEJ (Chaudhuri et al., 2007; Yan et al., 2007). V(D)J recombination occurs only in G1-phase developing lymphocytes and is initiated when the RAG-1 and RAG-2 proteins (RAG endonuclease) introduce DNA DSBs between two recombining variable (V), diversity (D) or joining (J) gene segments and their flanking recombination signal (RS) sequences (Fugmann et al., 2000). RAG cleavage leads to the formation of two hairpin-sealed coding ends (CEs) and two blunt signal ends (SEs). cNHEJ joins the CEs to form a coding join (CJ) and the SEs to form a signal join (SJ) (Helmink and Sleckman, 2012). In addition to the core cNHEJ proteins, other proteins have important functions during cNHEJ but are not essential for the repair of all RAG DSBs. For example, ATM deficiency leads to a partial block in V(D)J recombination due to its role in promoting the stability of post-cleavage RAG DSB complexes (Bredemeyer et al., 2006; Helmink and Sleckman, 2012). The Artemis endonuclease is required to open hairpin-sealed CEs before they can be joined and thus is critical for CJ, but not SJ, formation (Chang et al., 2017; Ma et al., 2002). While neither DNA-PKcs nor ATM are individually required for SJ formation, the loss of both proteins leads to a complete block in SJ formation, suggesting that these two kinases have essential overlapping activities in cNHEJ (Gapud et al., 2011; Zha et al., 2011b). Thus, some cNHEJ factors can have redundant repair activities for a subset of DSBs.
XRCC4 and the XRCC4-like factor (XLF) form a complex that aligns and bridges broken ends during cNHEJ (Andres et al., 2007; Brouwer et al., 2016; Hammel et al., 2010; Reid et al., 2015; Ropars et al., 2011). XLF functions during cNHEJ in mouse embryonic fibroblasts (MEFs) and embryonic stem cells, but is not required for the cNHEJ-mediated repair of RAG DSBs in lymphocytes, where perhaps other proteins can function to align and bridge broken DNA ends generated by RAG cleavage (Ahnesorg et al., 2006; Li et al., 2008). Neither XLF, 53BP1, H2AX, nor PAXX is individually required for the cNHEJ-mediated repair of RAG DSBs in lymphocytes; however, the combined deficiency of XLF with either 53BP1, H2AX, or PAXX leads to severe defects in RAG DSB repair (Abramowski et al., 2017; Hung et al., 2017; Kumar et al., 2016; Lescale et al., 2016; Liu et al., 2012; Liu et al., 2017; Oksenych et al., 2012; Tadi et al., 2016; Zha et al., 2011a). Thus, XLF deficiency in lymphocytes can reveal the cNHEJ activities of other proteins during V(D)J recombination.
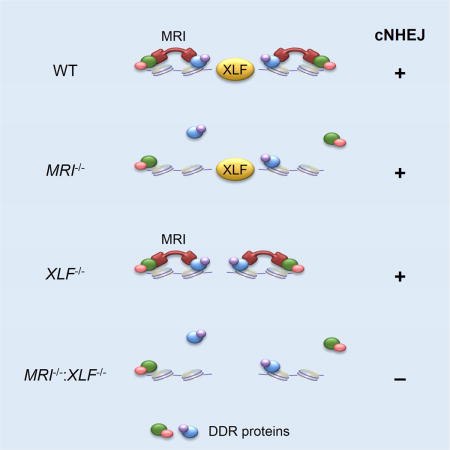
The modulator of retrovirus infection (MRI), initially identified in a screen for proteins that promote retroviral infection, is a 30 kDa protein with an N-terminal Ku-binding motif (KBM) and a C-terminal XLF-like motif (XLM) also found in the C-termini of XLF and PAXX (Agarwal et al., 2006; Grundy et al., 2016; Slavoff et al., 2014). MRI was shown to bind to Ku and stimulate NHEJ in vitro (Slavoff et al., 2014). However, MRI binding to Ku was found to inhibit cNHEJ-mediated chromatid fusion upon telomere deprotection in vivo, suggesting that MRI inhibits cNHEJ at the S and G2 phases of the cell cycle, and MRI was named the cell cycle regulator of NHEJ (CYREN) to reflect this activity (Arnoult et al., 2017). Here we show that in G1-phase XLF-deficient lymphocytes, MRI is required for cNHEJ-mediated repair of DSBs generated by RAG and the I-PpoI endonuclease. MRI is an intrinsically disordered protein that associates with a broad array of cNHEJ and DDR proteins through either its conserved N-terminal or C-terminal domains, and promotes the association of these proteins with chromatin at DNA DSBs. We propose that MRI is an adaptor that promotes cNHEJ in G1-phase cells through multivalent interactions that increase the avidity of DDR proteins for DSB-associated chromatin.
Results
MRI-deficient MEFs are sensitive to ionizing radiation
MRI+/− mice were generated with an MRI− allele containing a LacZ cassette, which replaced the entire protein-coding region of MRI (Fig. S1A and S1B). Intercrossing MRI+/− mice yielded MRI+/+, MRI+/− and MRI−/− mice in the expected Mendelian ratios (Fig. S1C). MRI−/− mice were fertile and did not exhibit any gross abnormalities (data not shown). MEFs were generated from wild-type (WT) and MRI−/− mice. A hamster monoclonal antibody (13E10.E12.C10) recognizing murine MRI was developed. This antibody recognized a single 30 kDa band in whole-cell lysates from WT MEFs, but did not detect the presence of any immunoreactive proteins in MRI−/− MEFs (Fig. 1A). MRI−/− MEFs exhibited increased sensitivity to ionizing radiation (IR) when compared to WT MEFs, but a lower level of sensitivity to IR than XLF−/− MEFs (Fig. 1B and S2A). MRI-GFP and Ku80-RFP C-terminal fusion proteins were expressed in MRI−/− MEFs, and time-lapse confocal microscopy revealed that MRI-GFP was rapidly recruited to sites of laser-induced DNA damage and displayed kinetics similar to those observed for Ku80-RFP recruitment (Fig. 1C and S2B). MRI-GFP remained associated with DNA damage sites for at least 45 minutes (Fig. S2C). Robust MRI-GFP localization was also observed upon laser-induced DNA damage of serum starved G1-phase MRI−/− MEFs (Fig. S2D). We conclude that, like the core cNHEJ factor Ku, MRI localizes to DNA damage sites in MEFs, and MRI deficiency in MEFs leads to IR sensitivity, a common hallmark of cNHEJ deficiency.
Figure 1. MRI functions in cNHEJ.
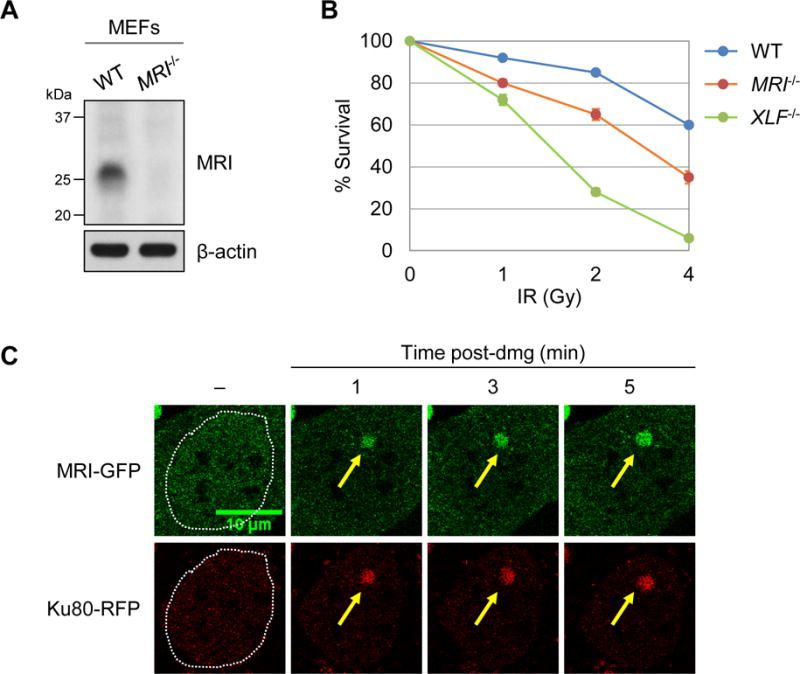
(A) Western blot analyses of MRI in MEFs derived from WT and MRI−/− mice. β-actin is shown as a protein loading control. (B) Percent survival of WT (line SZ), MRI−/− (line M61.2), and XLF−/− (line X-SZ) MEFs four days after exposure to the indicated doses of IR. Data are mean ± SEM (n = 3). (C) Representative time-lapse micrographs of MRI-GFP and Ku80-RFP recruitment to a site of laser-induced DNA damage (designated by a yellow arrow) in MRI−/− MEFs at the indicated times post-damage (minutes, min). Dotted lines mark the nuclear boundaries. See also Figures S1-S3.
MRI-deficient B cells exhibit defects in Ig CSR
Loss of proteins required for cNHEJ leads to diminished B and T lymphocyte development due to defects in V(D)J recombination. When compared to MRI+/+ mice, MRI−/− mice did not exhibit defects in B cell development in the bone marrow (Fig. S1D and S1E) or in T cell development in the thymus (Fig. S1F and S1G). MRI+/+ and MRI−/− mice showed similar numbers of mature splenic B and T cells (Fig. S1H and S1I). Naïve MRI−/− splenic B cells exhibited modest defects in Ig CSR in vitro, that are not due to a requirement for MRI in promoting AID expression or cell proliferation suggesting that MRI may function in the repair of Ig CSR DSBs by cNHEJ or aNHEJ (Fig. S3A-F).
MRI−/−:XLF−/− mice exhibit embryonic lethality
Intercrossing of MRI+/−:XLF+/− and MRI−/−:XLF+/−mice produced no viable MRI−/−:XLF−/− offspring (Fig. 2A and Table S1). Analyses of day E14.5 and E16.5 embryos from MRI−/−:XLF+/− intercrosses revealed MRI−/−:XLF−/− embryos at near Mendelian ratios, but these embryos were significantly smaller in size (Fig. 2B and Table S2). Lig4−/−, XRCC4−/−, and PAXX−/−:XLF−/− mice also exhibit embryonic lethality at a similar developmental stage, and cell lines with these genotypes all exhibit severe cNHEJ defects (Abramowski et al., 2017; Balmus et al., 2016; Frank et al., 1998; Frank et al., 2000; Gao et al., 1998; Hung et al., 2017; Kumar et al., 2016; Lescale et al., 2016; Liu et al., 2017; Tadi et al., 2016). Moreover, like those embryos, MRI−/−:XLF−/− embryos exhibited widespread neuronal apoptosis within the cortex and ganglionic eminences, as shown by cleaved caspase 3 immunostaining (Fig. 2C). Together, these data support the notion that combined deficiencies in MRI and XLF result in lethality due to defects in cNHEJ.
Figure 2. MRI−/−:XLF−/− mice exhibit embryonic lethality.
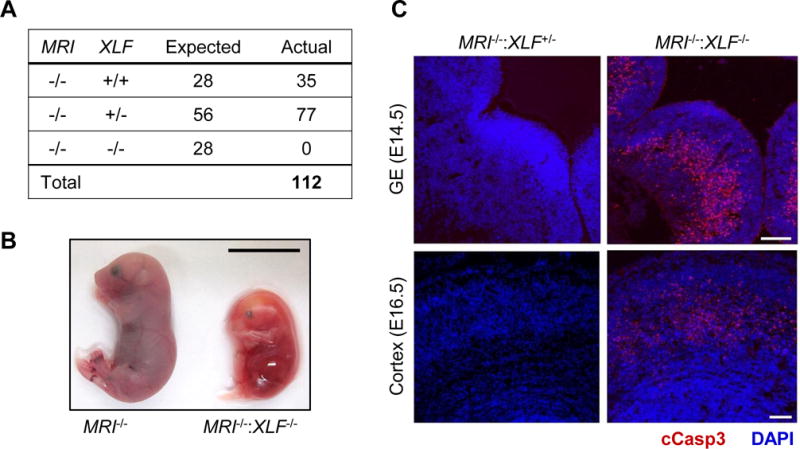
(A) Number of live births produced from intercrossing MRI−/−:XLF+/− mice. (B) MRI−/− and MRI−/−:XLF−/− littermates at day E16.5. Scale bar, 1 cm. (C) Micrographs of the ganglionic eminence (GE, top row) and cortex (bottom row) in MRI−/−:XLF+/− and MRI−/−:XLF−/− embryos at days E14.5 and E16.5, respectively, stained with DAPI (blue) and anti-cleaved caspase 3 antibody (red). Scale bar, 100 m. See also Tables S1 and S2.
MRI−/−:XLF−/− abl pre-B cells exhibit cNHEJ defects
Abelson murine leukemia virus-transformed pre-B cell-lines, hereafter referred to as abl pre-B cells, were generated from MRI−/− mice. V(D)J recombination can be assayed in abl pre-B cells with chromosomally integrated retroviral recombination substrates that contain a single pair of RS sequences (Bredemeyer et al., 2006). The pMG-INV retroviral V(D)J recombination substrate was introduced into WT, MRI−/−, and XLF−/− abl pre-B cells (Fig. 3A) (Hung et al., 2017). V(D)J recombination of pMG-INV occurs by inversion, placing an anti-sense GFP cDNA in the sense orientation and leading to GFP expression in cells that have generated RAG DSBs and successfully repaired them by cNHEJ (Fig. 3A). CRISPR/Cas9 was used to generate MRI−/−:XLF−/− abl pre-B cells by inactivating both MRI alleles in XLF−/− abl pre-B cells with a single MRI-targeting guide RNA. Loss of MRI expression in these cells was confirmed by western blotting (Fig. S4A).
Figure 3. Combined MRI and XLF deficiency blocks cNHEJ-mediated repair of RAG DSBs.
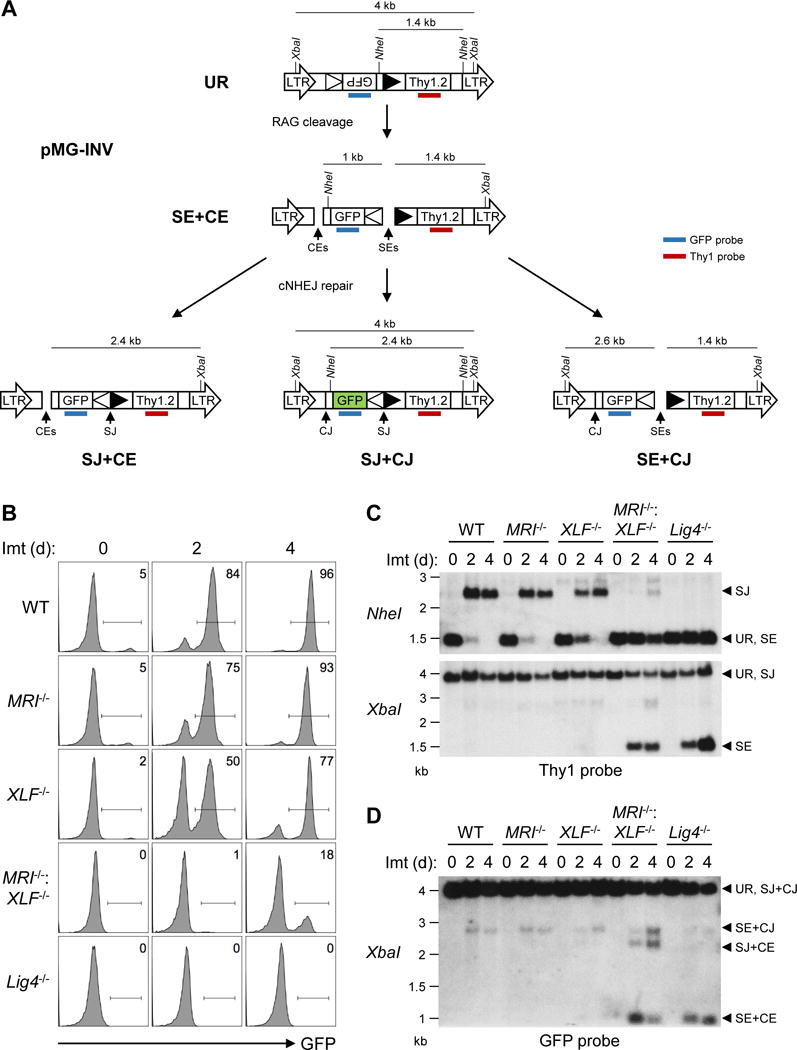
(A) Schematic of the unrearranged pMG-INV V(D)J recombination substrate (UR), its SEs and CEs intermediates and the resulting SJ and CJs. The long-terminal repeats (LTR), NheI and XbaI restriction sites, RSs (open and filled triangles), GFP cDNA, Thy1.2 cDNA, and Thy1 (red) and GFP (blue) probes are shown. (B) Flow cytometric analyses of GFP expression in WT (line M51.1-22), MRI−/− (line M66.1-24), XLF−/− (line XA3-8), MRI−/−:XLF−/− (line MX-19), and Lig4−/− (line B25-3) abl pre-B cells that were treated with imatinib for the indicated lengths of time (days, d). (C) Southern blot analyses of genomic DNA from cells in (B) that were digested with NheI (top) or XbaI (bottom) and hybridized to the Thy1 probe. Bands that correspond to UR, SJs, and SEs are indicated. (D) Southern blot analyses of genomic DNA from cells in (B) that were digested with XbaI and hybridized to the GFP probe. Bands that correspond to UR, SJs and CJs (SJ+CJ), both SEs and CEs (SE+CE), and either SEs or CEs (SE+CJ or SJ+CE) are indicated. Molecular weights (kilobases, kb) are also shown. See also Figures S4-S6.
Treatment of abl pre-B cells with the abl kinase inhibitor imatinib leads to G1 cell cycle arrest and RAG induction. Similar levels of pMG-INV rearrangement were observed in WT, MRI−/−, and XLF−/− abl pre-B cells, as indicated by flow cytometric analyses of GFP expression (Fig. 3B and S4B). Southern blot analyses revealed robust pMG-INV SJ and CJ formation without evidence of unrepaired SEs and CEs in WT, MRI−/−, and XLF−/− abl pre-B cells (Fig. 3C, 3D, S4C, and S4D). Thus, like XLF, MRI is not essential for cNHEJ-mediated RAG DSB repair in abl pre-B cells. However, MRI−/−:XLF−/− abl pre-B cells exhibited a severe block in V(D)J recombination, as evidenced by the reduced fraction of GFP-expressing cells after imatinib treatment (Fig. 3B and S4B). Moreover, Southern blot analysis revealed an accumulation of unrepaired RAG DSBs (pMG-INV SEs and CEs) and a reduction in pMG-INV CJ and SJ formation in MRI−/−:XLF−/− abl pre-B cells, similar to what is observed in abl pre-B cells deficient in the core cNHEJ factor DNA Ligase IV (Lig4−/−) (Fig. 3C, 3D, S4C, and S4D). At later time points (four days) after RAG induction, low levels of pMG-INV SJ and CJ formation were observed in MRI−/−:XLF−/− abl pre-B cells (Fig. 3C, 3D, S4C and S4D). Thus, MRI−/−:XLF−/− abl pre-B cells exhibit severely compromised DNA end joining kinetics during V(D)J recombination.
We next assessed MRI activity in the cNHEJ-mediated repair of chromosomal DSBs generated by an estrogen receptor-I-PpoI endonuclease fusion protein (ER-I-PpoI). Abl pre-B cells were arrested in G1 with the CDK4/6 inhibitor palbociclib, and treated with 4-hydroxytamoxifen (4-OHT) to induce ER-I-PpoI translocation to the nucleus (Fig. S5A) (Fry et al., 2004; Liu et al., 2017). DSB generation and imprecise repair by cNHEJ results in loss of the I-PpoI site (Fig. S5B). PCR amplicons spanning an I-PpoI site in untreated WT abl pre-B cells were completely digested by I-PpoI (Fig. S5C). However, after 24 hours in 4-OHT, ~50% of the amplicons could no longer be digested by I-Ppo-I, indicating that at least half of the I-PpoI sites had been cut and imprecisely repaired by cNHEJ (Fig. S5C). XLF−/− and MRI−/− abl pre-B cells exhibited levels of imprecise cNHEJ-mediated joining comparable to that of WT abl pre-B cells (Fig. S5C). In contrast, similar to Lig4−/− abl pre-B cells, MRI−/−:XLF−/− abl pre-B cells demonstrated minimal loss of I-PpoI sites after cleavage, indicative of a defect in cNHEJ (Fig. S5C). Indeed, Southern blot analyses revealed an increase of I-PpoI DSBs in MRI−/−:XLF−/− and Lig4−/− abl pre-B cells, as compared to WT, MRI−/−, and XLF−/− abl pre-B cells (Fig. S5D and E). Thus, cNHEJ-mediated repair of I-PpoI DSBs is also compromised in G1-phase abl pre-B cells that are deficient in both MRI and XLF.
To assess cNHEJ in G2-phase cells, in which DSBs can be repaired by cNHEJ or HR, we treated abl pre-B cells with the CDK1 inhibitor RO-3306 (Fig. S5A) (Vassilev et al., 2006). Similar levels of cNHEJ-mediated repair of I-PpoI DSBs were observed in G2-phase WT, MRI−/−, and XLF−/− abl pre-B cells, as evidenced by loss of I-PpoI sites after 4-OHT treatment (Fig. S5F). As was observed for G1-phase cells, G2-phase MRI−/−:XLF−/− abl pre-B cells did not exhibit a significant loss of I-PpoI sites and contained an increased accumulation of I-PpoI DSBs, indicative of a defect in cNHEJ (Fig. S5F and G). Thus, MRI deficiency does not lead to demonstrable alterations in the cNHEJ-mediated repair of I-PpoI DSBs in G1- or G2-phase abl pre-B cells; however, the combined deficiency of MRI and XLF compromises cNHEJ in both G1 and G2.
MRI and XLF have distinct cNHEJ activities
If MRI and XLF have overlapping cNHEJ activities, we reasoned that the requirements for efficient cNHEJ should be similar between MRI−/− and XLF−/− abl pre-B cells. Inhibition of ATM kinase activity with KU55933 causes a severe block in cNHEJ and V(D)J recombination in XLF−/− abl pre-B cells (Fig. S6A) (Zha et al., 2011a). However, treatment of MRI−/− abl pre-B cells with KU55933 leads to a block in V(D)J recombination of similar magnitude to that observed in WT abl pre-B cells (Fig. S6A). Indeed, while lymphocyte development is significantly impaired in XLF−/−:ATM−/− mice, B and T lymphocyte developmental defects in MRI−/−:ATM−/− mice are similar to those of ATM−/− mice (data not shown) (Zha et al., 2011a). Combined deficiency of XLF and PAXX profoundly impairs cNHEJ and V(D)J recombination in lymphocytes (Abramowski et al., 2017; Balmus et al., 2016; Frank et al., 1998; Frank et al., 2000; Gao et al., 1998; Liu et al., 2017). CRISPR/Cas9 was used to excise the entire PAXX gene in MRI−/− abl pre-B cells to generate MRI−/−:PAXX−/− abl pre-B cells that do not express PAXX (Fig. S6B-D). Analysis of GFP expression shows that imatinib treatment of MRI−/−:PAXX−/− abl pre-B cells containing pMG-INV leads to V(D)J recombination at similar levels to those observed in WT, MRI−/−, and PAXX−/− abl pre-B cells (Fig. S6E). In contrast, V(D)J recombination is severely impaired in XLF−/−:PAXX−/− abl pre-B cells (Fig. S6E). Moreover, while unrepaired pMG-INV RAG DSBs (SE and CE) are readily observed in XLF−/−:PAXX−/− abl pre-B cells, there is no evidence of these unrepaired DSBs in MRI−/−:PAXX−/− abl pre-B cells (Fig. S6F and S6G). Thus, while cNHEJ in XLF−/− abl pre-B cells relies on ATM and PAXX, the same dependencies are not observed in MRI−/− abl pre-B cells, suggesting that XLF and MRI have unique cNHEJ functions.
MRI has adaptor features
The N-terminal KBM and C-terminal XLM of MRI are conserved across species, but the central region of MRI is relatively non-conserved (Fig. S7A). The amino acid composition of the central region of MRI suggests that it is an intrinsically disordered protein, which could nucleate heterogeneous protein complexes (Wright and Dyson, 2015). To test this, recombinant mouse MRI protein was generated in E. coli and subjected to hydrogen-deuterium exchange (HDX)-mass spectrometry (HDX-MS) (Keppel et al., 2011; Marciano et al., 2014; Zhang et al., 2014). HDX-MS is used to study protein structure and dynamics with HDX rates varying depending on the relative solvent exposure, folding kinetics, and structural dynamics of different regions of the protein (Johnson et al., 2016; Konermann et al., 2011). Protein regions that are intrinsically disordered exhibit high levels of HDX over time due to increased solvent exposure, whereas structured regions, such as α-helices and β-sheets, are less exposed (Balasubramaniam and Komives, 2013; Goswami et al., 2013; Konermann et al., 2011).
We employed HDX-MS to derive a peptide-level analysis of MRI solvent accessibility with pepsin digestion yielding peptides with an 85% MRI sequence coverage (Fig. S7B). The resulting peptides exhibited high percentages of deuterium uptake, with all peptides undergoing 50-100% average percent deuterium uptake (Fig. S7B). Many of these peptides displayed near maximal deuterium uptake at the earliest time point (10 seconds) suggesting that MRI is intrinsically disordered in solution (Fig. S7C). Notably, MRI N-terminal peptides, including the residues 1-25 comprising the KBM, were relatively more protected from HDX than the rest of MRI, suggestive of a propensity to form secondary structure (Fig. S7B and C). Indeed, while circular dichroism (CD) spectroscopy also demonstrates that MRI is an intrinsically disordered protein, the addition of trifluoroethanol (TFE), a helix-inducing crowding agent, resulted in CD spectra suggestive that MRI has the potential to form helical structures (Fig. S7D) (Lopes et al., 2014). Finally, size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) revealed that, in solution, MRI can exist as a monomer, dimer, or multimer (Fig. S7E). Together, these data demonstrate that MRI is an intrinsically disordered protein with conserved N- and C-terminal domains that exists as monomers or multimers in solution. We reasoned that these properties of MRI make it particularly well-suited to nucleate multi-protein complexes as an adaptor during DDR and cNHEJ-mediated DSB repair.
MRI binds distinct DDR proteins at its N- and C-termini
Mass spectrometry was used to identify proteins in WT abl pre-B cells that bind to FLAG-HA-tagged versions of full-length MRI, MRI with a deletion of the 17 N-terminal amino acids comprising the KBM (MRIΔN), and MRI with a deletion of the 15 C-terminal amino acids comprising the XLM (MRIΔC) (Fig. 4A, 4B and S7A). These analyses revealed that MRI associates with proteins with diverse DDR functions that preferentially interact with either the N-terminus (KBM) or C-terminus (XLM) of MRI (Fig. 4C and Table S3). Immunoprecipitation with the 13E10.E12.C10 anti-MRI monoclonal antibody confirmed that endogenous MRI associates with DNA-PKcs, ATM, Ku70, Mre11, Nbs1, Rad50, XLF and PAXX (Fig. 4D). FLAG-HA-tagged MRI, MRIΔC, MRIΔN or MRIΔNΔC were expressed in WT abl pre-B cells and immunoprecipitated with anti-HA. Members of the DNA-PK complex – Ku70, Ku80 and DNA-PKcs – as well as XLF, PAXX, and XRCC4 all interact with MRI and MRIΔC, but do not interact with MRIΔN or MRIΔNΔC, indicating that the association of these proteins with MRI depends on the N-terminal KBM (Fig. 4E). Conversely, KAP-1, ATM, and components of the MRN complex - Mre11, Nbs1 and Rad50 - all interact with MRI and MRIΔN, but not MRIΔC or MRIΔNΔC, indicating that the binding of these proteins to MRI depends on the C-terminal XLM (Fig. 4E). ATM and its DSB sensor MRN and DNA-PKcs and its DSB sensor Ku associate with MRI, but ATR and its DSB sensor ATRIP do not associate with MRI (Fig. 4F). MRI, XLF, and PAXX all have conserved C-terminal XLM domains; however, while the MRI XLM promotes the association with MRN and ATM, neither XLF nor PAXX associates with ATM or MRN (Fig. 4F). We conclude that MRI has unique properties that enable it to associate with functionally diverse DDR proteins in a manner that depends on either the N-terminal KBM or the C-terminal XLM.
Figure 4. Diverse DDR proteins interact with the MRI N- and C-termini.
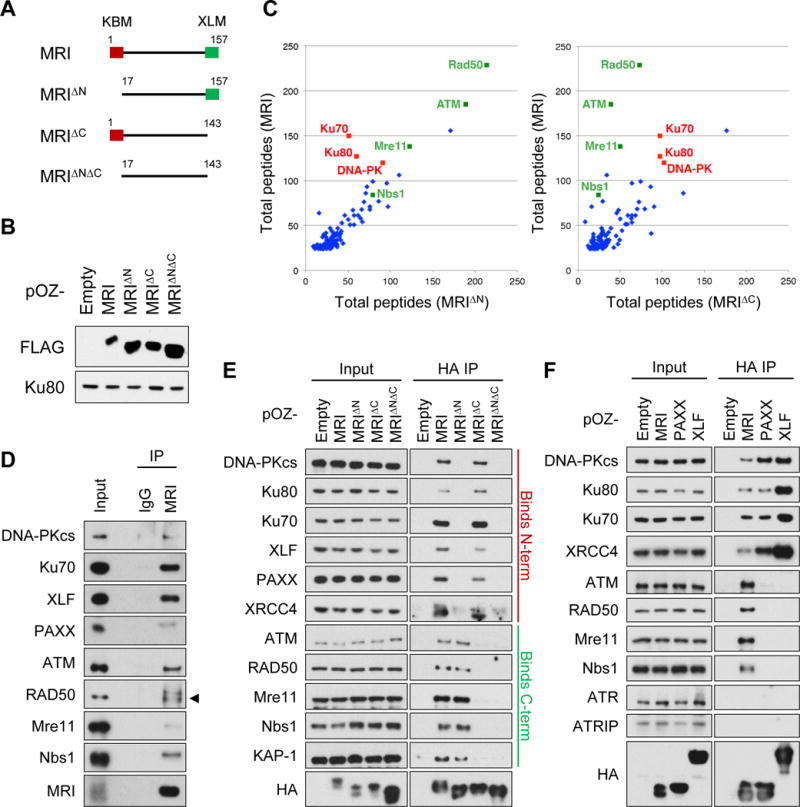
(A) Schematic of the MRI, MRIΔN, MRIΔC, and MRIΔNΔC proteins. The KBM (red box) and XLM (green box) are indicated. (B) Western blot analyses of retrovirally expressed FLAG-HA-tagged MRI, MRIΔN, MRIΔC, and MRIΔNΔC in MRI−/−:XLF−/− abl pre-B cells using anti-FLAG. Ku80 is shown as a protein loading control. (C) Scatter plots comparing the total numbers of MRI-associated peptides in WT abl pre-B cells expressing MRI versus MRIΔN (left plot) or MRIΔC (right plot), as determined by mass spectrometry. (D) Western blot analyses of DDR proteins that co-immunoprecipitated with endogenous MRI in WT abl pre-B cell nuclear extracts using the 13E10.E12.C10 anti-MRI antibody. (E) Western blot analyses of DDR proteins that co-immunoprecipitated with FLAG-HA-tagged MRI, MRIΔN, MRIΔC, and MRIΔNΔC in WT abl pre-B cell nuclear extracts using anti-HA. (F) Western blot analyses of DDR proteins that co-immunoprecipitated with FLAG-HA-tagged MRI, PAXX, and XLF in WT abl pre-B cell nuclear extracts using anti-HA. See also Figure S7 and Table S3.
MRI protein complexes
That MRI functions as an adaptor, forming higher order protein complexes with DDR proteins, is evidenced in several ways. First, FLAG-HA-tagged MRI was expressed in Ku70−/− abl pre-B cells that express neither Ku70 nor Ku80, which normally associate with the N-terminus of MRI (Fig. 5A). The association of MRI with DNA-PKcs, XLF, PAXX and XRCC4 is lost in the absence of Ku, demonstrating that the association of these proteins through the N-terminus of MRI depends on Ku (Fig. 5A). Loss of Ku did not alter the association of ATM, Mre11, Rad50, Nbs1, or KAP-1, which depend on the C-terminus for binding to MRI (Fig. 5A). Thus, the N-terminus of MRI can nucleate complexes containing Ku and at least one other protein (DNA-PKcs, XLF, PAXX or XRCC4). Next, we carried out sequential immunoprecipitations to determine whether MRI complexes form with proteins bound at both the N- and C-terminus (Fig. 5B). FLAG-tagged Ku80 and HA-tagged MRI were expressed in MRI−/− abl pre-B cells, followed by immunoprecipitation with anti-HA and elution of complexes with the HA peptide (Fig. 5B). Immunoprecipitation of these complexes with anti-FLAG, but not an IgG isotype control, recovered HA-tagged MRI; FLAG-tagged Ku80 and Ku70, which associates with the N-terminus of MRI; and ATM, which associates with the C-terminus (Fig. 5B). We find that in vitro human ATM binds directly to human MRI and MRIΔN, but not MRIΔC, demonstrating that ATM can directly bind to the C-terminus of MRI (Fig. 5C). Thus, MRI can form complexes with Ku associated at the N-terminus and ATM at the C-terminus. Third, size-exclusion chromatography revealed that, in abl pre-B cells, MRI exists in fractions that range in size from 100 kDa to over 1 MDa (Fig. 5D). The 30 kDa MRI protein was most abundant in 200 kDa fractions that contained Ku and in fractions greater than 1 MDa that contained Ku, Mre11, ATM, and DNA-PKcs (Fig. 5D). We conclude that MRI is an adaptor that forms large multimeric complexes containing DDR proteins bound at the N-terminus, C-terminus, or both.
Figure 5. MRI forms higher order protein complexes.
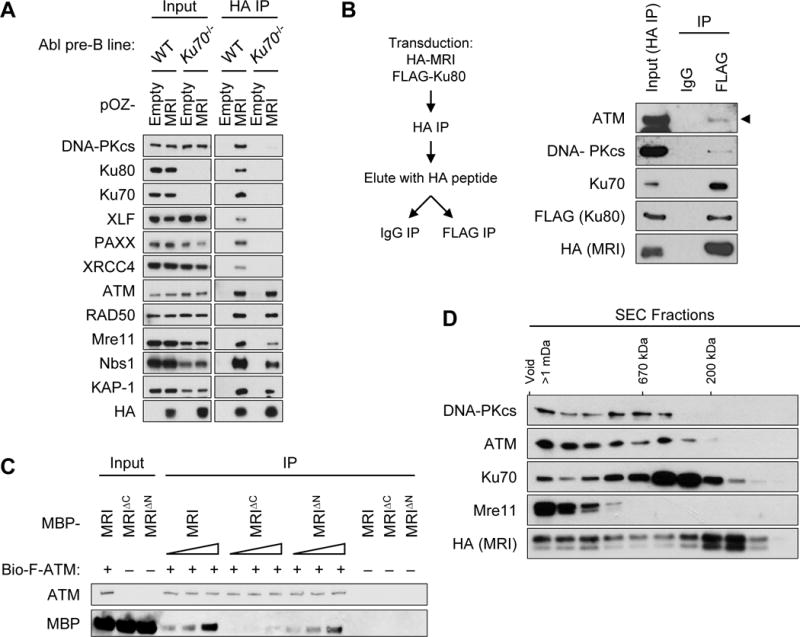
(A) Western blot analyses of DDR proteins that co-immunoprecipitated with FLAG-HA-tagged MRI in WT and Ku70−/− abl pre-B cell nuclear extracts using anti-HA. (B) Schematic of sequential immunoprecipitations of MRI followed by Ku80 in cells expressing HA-MRI and FLAG-Ku80 (left). Western blot analyses of ATM, DNA-PKcs, Ku70, FLAG-Ku80, and HA-MRI from MRI−/− abl pre-B cell nuclear extract after first immunoprecipitation with anti-HA (Input HA IP) and following second immunoprecipitation with anti-FLAG or an IgG isotype control (right). (C) Western blot analyses of purified recombinant biotin-FLAG-ATM (bio-F-ATM) co-immunoprecipitated with 50 nM, 100 nM, or 200 nM of MBP-tagged human MRI, MRIΔN, and MRIΔC proteins. (D) Western blot analyses of DDR proteins that associated with FLAG-purified MRI in different-sized fractions separated by a sucrose gradient column.
MRI cNHEJ functions depend on the KBM and XLM
The N-terminus (KBM) and C-terminus (XLM) of MRI bind to proteins with distinct cNHEJ functions. To assess the requirement for the KBM and XLM in cNHEJ, we assayed V(D)J recombination in MRI−/−:XLF−/− abl pre-B cells reconstituted with XLF, MRI, MRIΔN, MRIΔC, or MRIΔNΔC. Expression of XLF or MRI restored V(D)J recombination of pMG-INV in MRI−/−:XLF−/− abl pre-B cells, as indicated by GFP expression after imatinib treatment (Fig. 6A). In contrast, expression of MRIΔN or MRIΔC partially rescued recombination, while expression of MRIΔNΔC failed to augment V(D)J recombination in MRI−/−:XLF−/− abl pre-B cells (Fig. 6A). Southern blot analyses revealed that defects in V(D)J recombination in MRI−/−:XLF−/− abl pre-B cells expressing MRIΔN, MRIΔC, or MRIΔNΔC were due to defects in the cNHEJ-mediated repair of RAG DSBs generated at pMG-INV, as evidenced by the accumulation of unrepaired SEs and CEs after imatinib treatment (Fig. 6B and 6C). We conclude that in G1-phase lymphocytes the N-terminal KBM and C-terminal XLM of MRI have independent functions in cNHEJ that likely reflect the activity of distinct DDR factors bound at these two domains.
Figure 6. The N- and C-termini of MRI are both required for its function in cNHEJ.
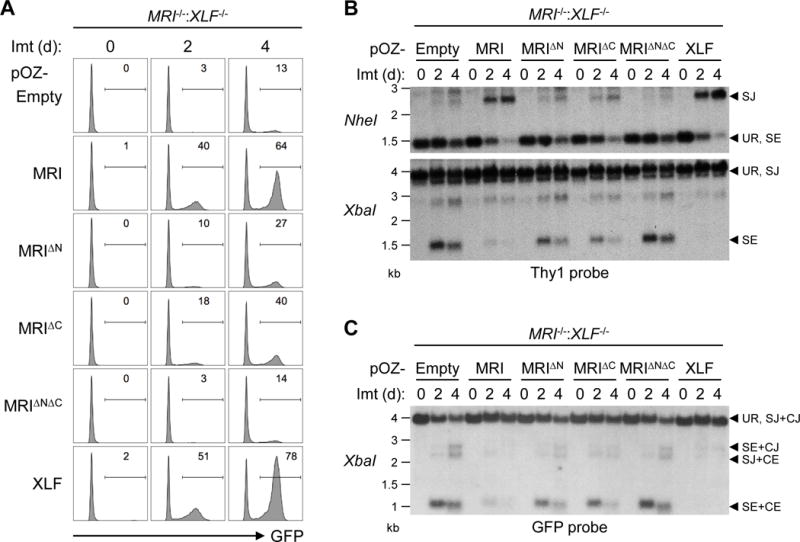
(A) Flow cytometric analyses of GFP expression in MRI−/−:XLF−/− abl pre-B cells retrovirally transduced with MRI, MRIΔN, MRIΔC, MRIΔNΔC, or XLF and treated with imatinib for the indicated lengths of time (days, d). (B) Southern blot analyses of genomic DNA from cells in (A) that were digested with NheI (top) or XbaI (bottom) and hybridized to the Thy1 probe. (C) Southern blot analyses of genomic DNA from cells in (A) that were digested with XbaI and hybridized to the GFP probe. Bands corresponding to different pMG-INV arrangements are indicated as described in Figure 3.
MRI promotes association of DDR factors with chromatin
MRI associates with many DDR proteins, such as Ku and MRN, which are recruited to DNA DSBs soon after their generation. Thus, MRI could function in the recruitment or retention of these DDR proteins at chromatin associated with DNA DSBs. To test this, we isolated chromatin from irradiated and non-irradiated WT and MRI−/− abl pre-B cells arrested in G1 by imatinib treatment (Fig. 7A). Western blot analysis revealed robust accumulation of DNA-PKcs, Ku70, XRCC4, Mre11, Rad50, Nbs1, XLF, and PAXX in the chromatin fraction after irradiation of WT abl pre-B cells (Fig. 7A). In contrast, all of these proteins accumulated at lower levels in the chromatin fraction of G1-phase MRI−/− abl pre-B cells after irradiation (Fig. 7A). The recruitment of MDC1 to chromatin after irradiation was not affected by the loss of MRI (Fig. 7A). Ku70-GFP association with laser-induced DNA damage sites was diminished in MRI−/− MEFs when compared to WT MEFs (Fig. 7B). We conclude that MRI promotes the recruitment or retention of DDR factors at DNA DSB-associated chromatin. Notably, this includes Ku70 and XRCC4, which are required for cNHEJ. Moreover, as XLF−/−:PAXX−/− abl pre-B cells exhibit a severe cNHEJ defect, it is also conceivable that the diminished association of PAXX with DSB-associated chromatin in the absence of MRI contributes to the cNHEJ defect in MRI−/−:XLF−/− abl pre-B cells.
Figure 7. MRI promotes DDR protein localization at DSB-associated chromatin.
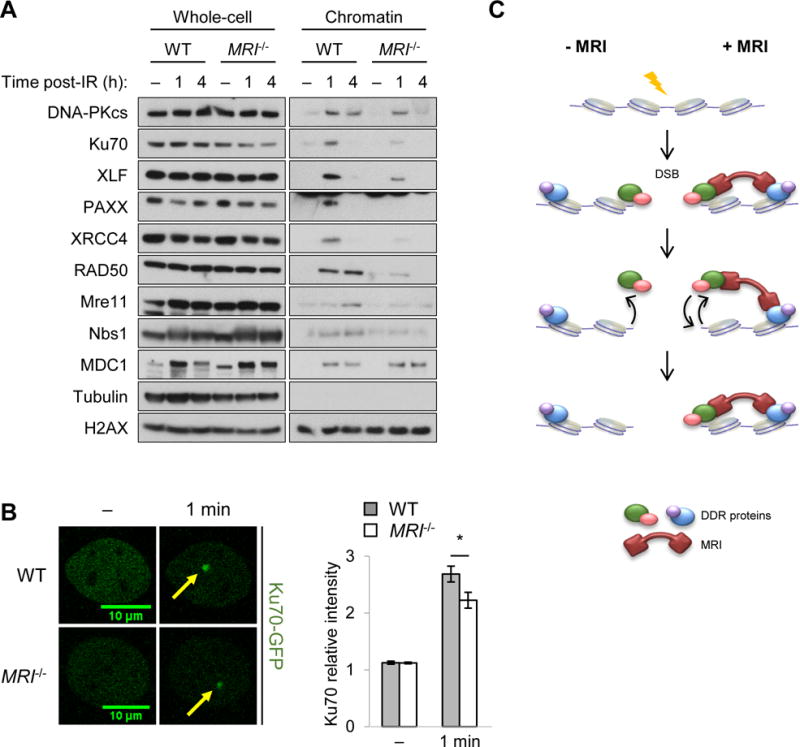
(A) Western blot analyses of DDR proteins in the whole-cell extracts and chromatin fractions of G1-arrested (with imatinib) WT and MRI−/− abl pre-B cells at the indicated times (hours, h) following treatment with 10 Gy of IR or no IR (−). (B) Representative micrographs of GFP-Ku70 recruitment to a laser-induced DNA damage site (designated by a yellow arrow) in WT and MRI−/− MEFs prior to damage (−) or 1 minute post-damage (1 min). The relative fluorescent intensities of GFP-Ku70 foci in these cells were then quantified (bottom histogram). Data are mean ± SEM. >20 cells of each genotype were analyzed in two independent experiments. * p <0.05. (C) Model for MRI-induced increase in the avidity of DDR factors with chromatin at DSBs as described in the discussion.
Discussion
Here we show that MRI is a small intrinsically disordered adaptor protein which nucleates heterogeneous complexes that function during DNA DSB repair by cNHEJ in G1- and G2-phase cells. MRI has conserved N-terminal (KBM) and C-terminal (XLM) domains that mediate association with distinct sets of proteins with diverse functions in DDR signaling and cNHEJ. Both of these domains are required for the optimal activity of MRI in cNHEJ-mediated DSB repair. Through its multivalent activity, MRI promotes the avidity of DDR and cNHEJ factors to chromatin associated with DNA DSBs.
Several lines of evidence demonstrate that MRI promotes cNHEJ. MRI is required for cNHEJ-dependent repair of RAG DSBs in XLF-deficient G1-phase abl pre-B cells and for cNHEJ-mediated repair of I-PpoI DSBs in G1- and G2-phase XLF-deficient cells (Fig. 3, S4, and S5). MRI−/−:XLF−/− mice exhibit early embryonic lethality associated with apoptosis of cortical and subcortical neurons, similar to mice deficient in the core cNHEJ factors XRCC4 or DNA Ligase IV, as well as mice with a combined deficiency of XLF and PAXX (Fig. 2) (Abramowski et al., 2017; Balmus et al., 2016; Frank et al., 1998; Frank et al., 2000; Gao et al., 1998; Liu et al., 2017). MRI-deficient MEFs are sensitive to IR, and MRI associates with sites of laser-induced DNA damage in MEFs with kinetics similar to those of the core cNHEJ factor Ku (Fig. 1C and S2). Mature MRI−/− B cells exhibit reduced Ig CSR, consistent with a function for MRI in the repair of CSR DSBs by either cNHEJ or aNHEJ (Fig. S3). MRI promotes the association of the core cNHEJ factors Ku and XRCC4 with DSB-associated chromatin in G1-phase lymphocytes (Fig. 7A). However, MRI has been shown to inhibit the cNHEJ-mediated fusion of deprotected telomeres in S- and G2-phase cells (Arnoult et al., 2017). Thus, MRI may have both positive and negative effects on cNHEJ that could depend on the type of DSB.
The requirement for MRI to mediate cNHEJ in XLF-deficient lymphocytes could reflect overlapping functions of MRI and XLF during cNHEJ. Indeed, XLF and MRI share a C-terminal (XLM) domain that could mediate similar cNHEJ functions. However, while the MRI XLM associates with ATM and MRN, the XLF XLM does not (Fig. 4F). Moreover, cNHEJ in XLF-deficient cells relies on proteins (ATM and PAXX) that are not required for cNHEJ in MRI-deficient cells beyond their requirement in WT cells, suggesting that MRI and XLF have distinct cNHEJ functions (Fig. S6). Loss of MRI or XLF activities may slow DNA end joining kinetics without leading to a detectable accumulation of unrepaired RAG DSBs. However, loss of both activities may result in a threshold reduction in joining kinetics that leads to a cNHEJ defect and the accumulation of unrepaired RAG DSBs. XLF and XRCC4 form filaments that align and bridge broken DNA ends during cNHEJ (Andres et al., 2007; Brouwer et al., 2016; Hammel et al., 2010; Reid et al., 2015; Ropars et al., 2011). MRI promotes the association of DDR proteins, including cNHEJ factors, with DSB-associated chromatin in G1-phase lymphocytes (Fig. 7A). In the absence of XLF, misaligned broken DNA ends may still be joined due to optimal localization of DDR factors at DSBs by MRI. In the absence of MRI, XLF aligns and bridges broken DNA ends, allowing them to be joined despite the reduced localization of DDR factors at DSBs. However, in the absence of both XLF and MRI, the combined defects in DNA end bridging and DDR factor localization may lead to a significant reduction in the efficiency of cNHEJ-mediated DSB repair and the accumulation of un-repaired DSBs. MRI could conceivably be critical for the repair of a small fraction of RAG DSBs. For example, some RAG DSBs may be modified in ways that require additional processing before they can be joined by cNHEJ. MRI adaptor function in retaining cNHEJ factors at DSBs could ensure efficient joining once these DNA ends are appropriately processed. In this regard, some IR-induced DSBs have DNA end structures that require additional processing before they can be joined and MRI−/− MEFS exhibit a mild sensitivity to IR, which could reflect the failure to repair a small fraction of DSBs (Fig. 1B and S2A) (Riballo et al., 2004).
MRI forms heterogeneous complexes with DDR proteins. In pre-B cells MRI associates with ATM, MRN, XLF and PAXX, but neither XLF nor PAXX associates with ATM or MRN (Fig. 4F). This suggests that some MRI complexes contain PAXX or XLF but not ATM or MRN, and others contain ATM or MRN but not PAXX or XLF. MRI can form a complex with Ku associated at its N-terminus and ATM at its C-terminus (Fig. 5B and 5C). This demonstrates that MRI can functionally link proteins bound in trans at its N- and C- termini. Ku association with the N-terminus of MRI is required for the association of DNA-PKcs, XLF, PAXX, and XRCC4 with MRI (Fig. 5A). Thus, MRI can form complexes that link two or more proteins in cis through one terminus. That deletion of the N-terminus or C-terminus each leads to a partial defect in V(D)J recombination in abl pre-B cells suggests that the cis adaptor functions of MRI may be most important for cNHEJ-mediated RAG DSB repair (Fig. 6).
How does MRI function during cNHEJ? The conserved N- and C-terminal domains coupled with the intrinsically disordered central domain make MRI particularly well-suited for adaptor protein function. Intrinsically disordered proteins generate protein-protein interaction networks due to their ability to adopt conformations that permit heterotypic and homotypic interactions (Wright and Dyson, 2015). Purified MRI in solution forms dimers and larger multimers, which in cells would further allow it to nucleate large protein complexes (Fig. S7E). As an adaptor protein, MRI may function to link enzymes and their substrates. In this regard, ATM and DNA-PKcs both associate with MRI and many of the other proteins that associate with MRI are regulated through phosphorylation by ATM and DNA-PKcs (Chen et al., 2007; Zhou et al., 2017).
We find that MRI promotes the association of several cNHEJ factors with DSB-associated chromatin in G1-phase lymphocytes (Fig. 7A). MRN and Ku are rapidly recruited to newly generated DSBs and, if bound to MRI, could recruit other proteins in the MRI complex to the DSB. The binding of multiple proteins by MRI at the DSB could increase the avidity of these proteins for the DSB-associated chromatin (Fig. 7C). For example, if a DDR factor monovalently bound to DSB-associated chromatin were to dissociate, it would be lost by diffusion (Fig. 7C, left). However, the tethering of this DDR factor to chromatin at the DSB through its association with MRI and other MRI-bound DDR factors would promote its re-association with the DSB (Fig. 7C, right). Given the multivalent and diverse nature of its binding, MRI is particularly well suited for increasing the avidity of DDR proteins to DSB-associated chromatin. Intrinsically disordered proteins have been implicated in liquid-liquid phase separations that are important in many cellular processes, including DNA repair (Altmeyer et al., 2015; Uversky, 2017). Thus, it is conceivable that MRI may promote liquid-liquid phase separations that function to retain DDR factors at DSB-associated chromatin. Finally, we speculate that MRI may be important in clearing DDR factors from DSB-associated chromatin after repair is complete. In this regard, signals that lead to loss of MRI from DSB-associated chromatin would promote the dissociation of DDR factors bound by MRI at the DSB.
STAR Methods
Contact for Resource and Reagent Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Barry P. Sleckman (bas2022@med.cornell.edu).
Experimental Model and Subject Details
Mice
MRI+/− mice were generated by in vitro fertilization using cryopreserved MRI−/− sperm obtained from the KOMP Repository (https://www.komp.org/ProductSheet.php?cloneID=862138) and intercrossed to produce MRI−/− mice, which were maintained on a C57BL/6 genetic background. Genotypes were confirmed by PCR analyses, as shown in Figures S1A and B, with the WT allele (MRI+) yielding a 1.1-kb amplicon and the targeted knockout allele (MRI−) yielding a 400-bp amplicon (see Key Resources Table for primer sequences). For lymphocyte development analyses, 6-week old WT and MRI−/− littermates (n = 5, five females and five males) were used.
Cell-line generation and culture
WT (lines M51.1-22 and M63.1-7), MRI−/− (lines M46.3-19 and M66.1-24), XLF−/− (lines XA3-8 and XB1-5), and Lig4−/− (lines A20-115 and B25-3) abl pre-B cell-lines were generated by co-centrifuging (1,800 RPM, 90 minutes) bone marrow cells from 3-5-week old mice harboring Eμ-Bcl2 transgenes (which prevent cell death in response to persistent DSBs) with the pMSCV-v-abl retrovirus and 5 μg/mL polybrene (Santa Cruz), as previously described (Bredemeyer et al., 2006). Stably transformed abl pre-B cells were then transduced with the retroviral pMSCV-RSS-GFP-INV-Thy1.2 (pMG-INV) recombination substrate, and clones containing a single integrant were obtained by limiting dilution (Hung et al., 2017). To induce V(D)J recombination, cells were treated with 3 μM imatinib (Novartis) for the indicated lengths of time at a density of 1 × 106 cells/mL. For ATM inhibition, cells were treated with 15 μM KU55933 (Selleckchem) at a density of 1 × 106 cells/mL.
To generate MRI−/−:XLF−/− abl pre-B cells (lines MX-4 and MX-19), XLF−/− abl pre-B cells were transduced with a lentiviral vector pCW-Cas9 (Addgene) containing a tetracycline-inducible FLAG-Cas9 cDNA and selected in 2 μg/mL puromycin (Invivogen) before being subcloned by limiting dilution. Clones were treated with 2 μg/mL doxycycline (Thermo Fisher) for two days, and those that exhibited robust Cas9 expression by anti-FLAG western blotting were further transduced with a lentiviral vector pKLV-BFP (Addgene) containing a gRNA that targets exon 3 of the mouse MRI gene. These cells were treated with 2 μg/mL doxycycline for one week, sorted for BFP expression, and subcloned again by limiting dilution. MRI knockout clones were identified by western blotting, PCR sequencing of MRI exon 3, and significantly reduced GFP expression after imatinib treatment. MRI−/−:PAXX−/− abl pre-B cells (line MP-80) were generated using a previously described strategy (Liu et al., 2017). A pair of gRNAs that target sequences flanking the entire mouse PAXX gene were cloned into the Cas9 vector pX330 (Addgene), and the plasmids were electroporated into MRI−/− abl pre-B cells using the Nucleofector 2b device (human B cell kit, program X-001, Lonza). After four days, the cells were subcloned by limiting dilution, and PAXX knockout clones were identified by PCR analyses and western blotting, as shown in Fig. S6B-D. See Key Resources Table for the list of gRNA and primer sequences.
WT (lines SZ and WT-1), MRI−/− (lines M61.2 and M61.7), and XLF−/− (lines X-AB and X-SZ) MEFs were generated from E14.5 or 15.5 mice and immortalized by transfection with pBABE-neo-SV40 (Addgene), after which they were selected in 400 μg/mL G418 (Thermo Fisher).
All cell-lines were grown in media consisting of DMEM (no L-glutamine, Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Sigma), 100 U/mL penicillin/streptomycin (Gibco), 1 mM sodium pyruvate (Gibco), 2 mM L-glutamine (Gibco), 1X nonessential amino acids (Gibco), and 55 μM DMSO at 37°C, unless speci fied otherwise. The abl pre-B cell-lines M51.1-22, M46.3-19, and M66.1-24 were derived from female mice, whereas line M63.1-7 originated from a male mouse. The sexes of the XLF−/− and Lig4−/− abl pre-B cell-lines, which had been generated years prior to the initiation of this project, are not known. The sexes of the MEF cell-lines are also undetermined, as they could not easily be identified. Genotypes of all cell-lines used were authenticated by PCR analyses.
Primary B cell cultures
Naive CD43- B cells were purified from the spleens of 6-8-week old WT and MRI−/− mice (n = 6, six females and six males) using a MACS negative selection kit (Miltenyi Biotec). They were then seeded into 96-well plates at a density of 4 × 105 cells/mL and stimulated with either 25 μg/mL LPS (Sigma) or 1 μg/μL anti-CD40 (eBioscience) plus 20 ng/mL IL-4 (Peprotech) for up to four days. These cells were grown in RPMI (Gibco) supplemented with 10% FBS, 100 U/mL penicillin/streptomycin, and 55 μM DMSO at 37°C.
Method Details
MEF radiosensitivity assay
MEFs were seeded in 48-well plates at a density of 2 × 103 cells/well and irradiated using a RS 2000 X-ray irradiator (Rad Source Technologies). After four days, cell survival was determined by incubating the cells with 10% v/v PrestoBlue reagent (Thermo Fisher) for two hours at 37°C and measuring the absorbances at 560 nm and 590 nm (reference wavelength) on a Multiskan Ascent microplate spectrophotometer (Thermo Fisher), in accordance with the manufacturer’s instructions.
Laser micro-irradiation and imaging
MRI−/− MEFs were stably transduced with a lentiviral vector pLV (Addgene) containing a MRI-GFP fusion protein, then transiently transfected with 1.5 μg Ku80-RFP using Lipofectamine 2000 (Thermo Fisher) and imaged after 48 hours. To obtain reliable recruitment of GFP-Ku70 with the 405 nm laser, the transfected cells were sensitized with 10 μM BrdU for 24 hours. To arrest cells in G1, MRI−/− MEFs expressing MRI-GFP were serum starved in DMEM with 0.5% FBS (Hyclone) for 72 hours prior to BrdU sensitization and imaging. Live cell imaging was carried out on a Nikon Ti Eclipse inverted microscope (Nikon, Inc.) equipped with an A1 RMP confocal microscope system (Nikon, Inc.) and Lu-N3 Laser Units (Nikon, Inc.). Laser micro-irradiation manipulation and time-lapse imaging were performed with the NIS Element High Content Analysis software (Nikon, Inc.) using a 405 nm laser at an energy level adequate for Ku70 accumulation. Relative intensities at laser-damaged sites were calculated as the ratio of the mean intensity at each of the micro-irradiated sites to the corresponding mean intensity of the nucleus as background (considering the entire nucleus). >20 individual cells were analyzed for each data point.
Brain immunohistochemistry
E14.5 and E16.5 mouse embryos were dissected in ice-cold phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde/PBS solution overnight at 4°C. Brain tissues were rinsed with PBS and cryoprotected using 30% sucrose/PBS solution overnight at 4°C. They were then embedded in TissueTek (Sakura Finetek), flash-frozen on dry ice, and stored at −80°C before sectioning. Brain tissues were cryosectioned at 18 μm and processed using PBS with 1.5% donkey serum and 0.35% Triton X-100 for all subsequent steps, except for the washing steps when only PBS was used. Sections were blocked for 30 minutes, followed by incubation with primary rabbit anti-cleaved caspase 3 (1:200, Cell Signaling) overnight at 4°C. After three more washes with PBS, secondary Cy3 donkey anti-rabbit IgG (1:1,000, Jackson ImmunoResearch) was applied for one hour at room temperature. Nuclear counterstaining was performed with 100 ng/mL DAPI solution. An additional three washes were performed with PBS, and the slides were coverslipped using VectaShield mounting medium (Vector Laboratories). Fluorescent images were captured using a Fluoview FV3000 Olympus confocal laser scanning microscope (Olympus, Inc.). The following mice were analyzed: E14.5 MRI−/−:XLF+/− (n = 2) and MRI−/−:XLF−/− (n = 2) littermates and E16.5 MRI−/−:XLF+/− (n = 1) and MRI−/−:XLF−/− (n = 2) littermates, with five coronal sections observed per animal for a total of 35 sections.
Flow cytometric analyses
For analyses of lymphocyte development or CSR, single-cell suspensions derived from bone marrow, thymus, or spleen were incubated with Fc block (BD Pharmingen) at a density of 1 × 106 cells/mL on ice for 15 minutes prior to staining with the indicated fluorescent antibodies (1:500) at 4°C for 30 minutes. For cell proliferati on assays, purified splenic CD43- B cells were incubated with 10 μM CFSE (eBioscience) at a density of 1 × 106 cells/mL in the dark at 37°C for 10 minutes and quenched with FBS on ice for five minutes prior to stimulation with LPS or anti-CD40 plus IL-4. For cell cycle analyses, cells were fixed in Cytofix/Cytoperm solution (BD Biosciences) at 4°C for 15 minutes, washed with PBS, frozen in 10% DMSO/FBS at −80°C overnight, thawed, re-fixed in Cytofix/Cytoperm solution at 4°C for 5 minutes, washed again with PBS, and incubated with 7-AAD (BD Pharmingen) at room temperature for 10 minutes. Data were acquired on a LSR II flow cytometer (BD Biosciences) and analyzed using FlowJo software (FlowJo, LLC).
I-PpoI cutting and repair assay
Abl pre-B cells were transduced with a retrovirus vector pBMN-HA-ER-I-PpoI-hCD8 containing an I-PpoI and estrogen receptor fusion protein. One day after infection, the cells were treated with either 2 μM palbociclib (Selleckchem) or 9 μM RO-3306 (Millipore) at a density of 1 × 106 cells/mL for ~16 hours to induce G1 or G2 arrest, respectively, and then with 200 nM 4-OHT (Sigma) to stimulate ER-I-PpoI activity. Genomic DNA from these cells were harvested at the indicated lengths of time following 4-OHT treatment, and amplicons spanning an I-PpoI site on chromosome 1 were generated by PCR and digested with I-PpoI enzyme at 37°C overnight (see Key Resources Table for primer sequences). Intensities of the DNA bands corresponding to cut and uncut amplicons were quantified using ImageJ software.
Southern blot analyses
For analyses of V(D)J recombination, 10 μg of genomic DNA from abl pre-B cells containing pMG-INV were digested with NheI or XbaI and hybridized to a 32P-labeled Thy1 or GFP probe, as previously described (Bredemeyer et al., 2006; Hung et al., 2017). The Thy1 probe is an 800-bp Thy1.1 cDNA (which differs from Thy1.2 by only a single amino acid residue) fragment, while the GFP probe is a 700-bp GFP cDNA fragment.
For analyses of I-PpoI cutting and repair, 10 μg of genomic DNA from cells expressing ER-I-PpoI were digested with HindIII and hybridized to a 32P-labeled probe generated from a 500-bp DNA fragment that is homologous to a sequence 3′ of an I-PpoI site on chromosome 1.
Retroviral MRI cDNA expression
The murine MRI coding sequence was amplified by PCR from the cDNA clone BC000168 and ligated into a retroviral vector pOZ-FH-N downstream of a FLAG-HA tag and between a XhoI site and a NotI site. MRI mutants (MRIΔN, MRIΔC, and MRIΔNΔC) were generated by overlapping PCRs of the MRI cDNA (see Table S4 for primer sequences). Three days after transduction, cells containing the plasmid were sorted by human CD25 expression.
Protein immunoprecipitation
For nuclear extraction and immunoprecipitation, cells were lysed in cytoplasmic extraction (CE) buffer (10 mM Tris-HCl pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 0.05% Triton X-100) with a protease inhibitor cocktail (Sigma) on ice for 15 minutes. Following centrifugation (3,000 RPM, five minutes), the nuclear pellet was washed with CE buffer and incubated in 0.5X pellet volume of nuclease buffer (20 mM Tris-HCl pH 7.4, 1.5 mM MgCl2, 25% v/v glycerol) with 5 U/μL benzonase (Sigma) on ice for one hour to digest the genomic DNA. 1X pellet volume of nuclear extraction (NE) buffer (20 mM Tris-HCl pH 7.4, 500 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% v/v glycerol) was added, and the pellet was ground using a dounce homogenizer (Sigma) before incubation with gentle rotation at 4°C for one hour. The lysate was clarified by centrifugation (14,800 RPM, 30 minutes), and the supernatant was dialyzed in BC100 buffer (20 mM Tris-HCl pH 7.4, 100 mM KCl, 0.2 mM EDTA, 20% v/v glycerol) at 4°C overnight using the Pur-A-Lyzer Maxi 6000 kit (Sigma). The dialyzed supernatant was clarified once again by centrifugation (14,800 RPM, 30 minutes) and incubated with 30 μL bed volume of EZView Red HA or FLAG affinity gel beads (Sigma) on a rotator at 4°C for four hours. The beads were washed four times with TAP buffer (50 mM Tris-HCl pH 7.4, 100 mM KCl, 5 mM MgCl2, 0.2 mM EDTA, 10% v/v glycerol, 0.1% Triton X-100) and eluted by boiling in LDS sample buffer (Thermo Fisher) prior to SDS-PAGE and western blot analyses. For immunoprecipitation of endogenous MRI, nuclear extracts were first incubated with 5 μg nonspecific Armenian hamster IgG (BioLegend) or anti-MRI (13E10.E12.C10) at 4°C overnight and then with Protein L magnetic beads (Thermo Fisher) at 4°C for two hours.
For proteomic analyses, the anti-FLAG beads were eluted by incubating with 0.4 mg/mL FLAG peptide (Sigma) in TAP buffer on a rotator at 4°C f or one hour. The MRI-containing complexes were visualized by SDS-PAGE and silver staining, then precipitated with trichloroacetic acid (TCA). Associated proteins were identified by LC-MS/MS at the Taplin Mass Spectrometry Facility (Harvard Medical School) using an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher) and SEQUEST software (Eng, McCormack, and Yates, 1994).
For sequential immunoprecipitation, nuclear extracts from WT abl pre-B cells expressing HA-tagged MRI and FLAG-tagged Ku80 were incubated with EZView Red HA affinity gel beads at 4°C for four hours, after which the beads were washed four times with TAP buffer and eluted with 0.5 mg/mL HA peptide (Sigma) in TAP buffer at 4°C for one hour. The HA eluate was incubated with 4 μg mouse IgG1 (Santa Cruz) or mouse anti-FLAG (Sigma) at 4°C overnight and then with 30 μL bed volume of Dynabeads Protein G (Thermo Fisher) at 4°C for two hours. The beads were washed four times with TAP buffer and boiled in LDS sample buffer for SDS-PAGE and western blot analyses.
Size exclusion chromatography (SEC)
SEC was performed using an AKTA Pure FPLC on a Superose 6 Increase 10/300 GL column (GE Healthcare). Anti-FLAG-immunoprecipitated MRI complexes (500 μL) were loaded onto the pre-equilibrated column and eluted with TAP buffer at a flow rate of 0.5 mL/minute. 0.5 mL fractions were collected and concentrated using StrataClean resin (Agilent) before SDS-PAGE and western blot analyses.
Chromatin fractionation
Cells were first pre-extracted in CSK buffer (100 mM NaCl, 10 mM PIPES pH 6.8, 3 mM MgCl2, 300 mM sucrose) with 0.05% Triton X-100 on ice for 15 minutes. After centrifugation (1,500 × g, five minutes), the chromatin pellet was washed with PBS and incubated in CSK buffer with 0.5% Triton X-100 and 0.1 μg/mL RNase A (Sigma) at room temperature for 10 minutes and on ice for 20 minutes. After centrifugation (1,500 × g, five minutes), the pellet was washed with PBS and incubated in CSK buffer with 5 U/μL benzonase on ice for one hour. The chromatin fraction was boiled in LDS sample buffer and then analyzed by SDS-PAGE and western blotting.
MRI protein expression and purification
The coding regions of the mouse MRI and human MRI genes were codon-optimized for expression in E. coli (GenScript) and used as templates to subclone the coding regions into a modified pET15b vector (Novagen). Peptide truncations were generated by overlapping PCRs and validated by sequencing. Mouse MRI and human MRI proteins were then expressed in BL21(DE3) E. coli cells (Novagen), cultured in Luria Broth media at 37°C, induced at an OD600 (optical density at 600 nm) of 0.6 with 0.5 mM IPTG, and grown for 12-15 hours at 18°C. Cells were harvested, resuspended in lysis bu ffer (20 mM Tris-HCl pH 7.5, 250 mM NaCl, 5 mM 2-mercaptoethanol), and lysed using an EmulsiFlex-C5 homogenizer (Avestin), after which the resulting lysate was clarified by centrifugation (47,000 × g at 4°C, 40 minutes). Proteins were purified using a series of affinity and ion-exchange chromatographic columns (GE Healthcare). Following TEV protease digestion to separate the maltose-binding protein (MBP) fusion, the resulting sample was purified further by sequential ion-exchange and SEC. Protein purity was assessed by Coomassie staining of SDS-PAGE and mass spectrometry.
MRI-ATM co-immunoprecipitation
40 nM recombinant biotin-FLAG-tagged human ATM was incubated with MBP-tagged human MRI, MRIΔN, or MRIΔC (50, 100, or 200 nM) and 100 ng bovine serum albumin (BSA, New England Biolabs) in A buffer (25 mM Tris-HCl pH 8.0, 100 mM NaCl, 10% v/v glycerol) at room temperature for 15 minutes in a final volume of 30 μL. 2 μL of Dynabeads M-280 Streptavidin (Thermo Fisher) were pre-washed with 1 mg/mL BSA and 1% CHAPS (Sigma). The samples were then added to the bead mixture with 0.1% CHAPS in A buffer and incubated on ice for 15 minutes. After three washes with 2 μg/mL BSA in A buffer with 0.1% CHAPS, proteins bound to the beads were resolved by SDS-PAGE using NuPAGE 4-12% Bis-Tris (Thermo Fisher), followed by western blotting with anti-ATM (Santa Cruz) or anti-MBP (GeneTex).
Hydrogen-deuterium exchange mass spectrometry (HDX-MS)
MRI samples were buffer-exchanged with PBS pH 7.4. Deuterium labeling was initiated by diluting samples (50 μM, 2 μL) 10-fold with either D2O buffer or H2O buffer for measuring no-deuterium control samples. At eight different time intervals (10, 30, 60, 120, 360, 900, 3600, and 14400 seconds), the labeling reaction was quenched by rapidly adjusting the pH to 2.5 with 30 μL of quench buffer (3 M urea, 0.6% trifluoroacetic acid, H2O) at 4°C. The protein mixture was then immediately injected into a custom-built HDX device and passed through a column containing immobilized pepsin (2 mm × 20 mm) at a flow rate of 100 μL/minute in 0.1% formic acid, and the resulting peptic peptides were captured on a ZORBAX Eclipse XDB C8 column (2.1 mm × 15 mm, Agilent) for desalting (three minutes). The C8 column was then switched in-line with a Hypersil Gold C18 column (2.1 mm × 50 mm, Thermo Fisher), and a linear gradient (4-40% acetonitrile, 0.1% formic acid, 50 μL/minute flow rate for five minutes) was used to separate the peptides and direct them to a LTQ FT-ICR mass spectrometer (Thermo Fisher) equipped with an electrospray ionization source. Valves, columns, and tubing for protein digestion and peptide separation were submerged in an ice-water bath to minimize back-exchange.
The resulting data were processed and peptides identified by exact mass analysis and LC-MS/MS using Mascot (Matrix Science). The raw HDX spectra and peptide sets were submitted to HDX Workbench for calculation and data visualization in a fully automated fashion (Pascal et al., 2012). Peptides for each run were assessed based on their relative representation and statistical validation. Only the top six peptides from each MS scan were included in the final analysis. Deuterium uptake at each time point was calculated by subtracting the centroid of the isotopic distribution of the undeuterated peptide from that of the deuterated peptide. Relative deuterium uptake was plotted versus labeling time to afford kinetic curves.
Circular dichroism (CD)
CD wavelength scans were taken using a Chirascan CD spectrometer (Applied Photophysics). The changes in molar ellipticity of 10 μM MRI protein samples were monitored at 4°C. Samples were prepared in 25 mM Na3PO4 pH 7, 150 mM NaCl, and 5 mM 2-mercaptoethanol buffer containing 0-50% 2,2,2-trifluorethanol (TFE). All experiments were performed in triplicate.
Multi-angle light scattering-size exclusion chromatography (MALS-SEC)
Standard MALS experiments were carried out on a Superdex 200 10/300 GL column (GE Healthcare) connected in-line to a Dawn Heleos II MALS detector (Wyatt Technologies). 100 μL samples (2 mg/mL) were injected at a flow rate of 0.3 mL/minute into a column equilibrated in 10 mM HEPES pH 7.5, 150 mM NaCl, and 2 mM tris(2-carboxyethyl)phosphine (TCEP) buffer. Molecular weights and standard deviations were determined using Astra software package version 6.1 (Wyatt Technologies). All experiments were performed at room temperature and in triplicate.
Hybridoma generation
Armenian hamsters (Cytogen Research and Development) were immunized s.c. with 50 μg each of four KLH-conjugated peptides (GenScript), which cover the entire length of the mouse MRI protein, emulsified in Freund’s adjuvant (CFA then IFA at two-week intervals), followed by one injection of peptides with Alhydrogel (Invivogen) (Sheehan K et al. 1989). Immune serum showed positive titers by ELISA using peptide-coated plates and also by western blot analyses of recombinant protein. Three days prior to fusion, animals were boosted i.v. with 25 μg each of the KLH-conjugated peptides solubilized in endotoxin-free PBS. Immune splenocytes were fused to the P3X63Ag8.653 murine myeloma line using standard procedures. Cultures were screened initially by ELISA using individual peptides and then by western blotting of WT and MRI−/− abl pre-B cell lysates. Antigen-reactive wells were subcloned through two rounds of limiting dilution to ensure clonal populations. Isotype analysis were performed using cassettes provided by Antagen Pharmaceuticals. Monoclonal antibodies were purified and concentrated by Protein A affinity chromatography using standard methods (Sheehan K et al. 1989). See Key Resources Table for sequences of the immunization peptides (named GAP121, GAP122, GAP123, and GAP124).
Quantification and Statistical Analysis
Statistical analyses were performed using Microsoft Excel and StatPlus (AnalystSoft). Results are shown as mean ± standard error of the mean (SEM). Comparisons between two sets of data were determined using unpaired two-tailed Student’s t-tests assuming unequal variances. Numbers of experimental replicates (n) and statistical significance (defined at a minimum as a p-value <0.05) are reported in the figure legends.
Data and Software Availability
All original image files and the full list of MRI IP-MS results had been deposited on Mendeley Data (http://dx.doi.org/10.17632/2wdg6wf9fx.1).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Armenian hamster anti-MRI | This paper | Clone 13E10.E12.C10 |
| Rabbit anti-cytoskeletal actin | Bethyl Laboratories | Cat#A300-485A |
| Mouse anti-FLAG | Sigma | Cat#F1804; clone M2 |
| Rabbit anti-FLAG | Sigma | Cat#F7425 |
| Rabbit anti-HA | Santa Cruz | Cat#sc-805 |
| Mouse anti-DNA-PKcs | Thermo Fisher | Cat#MS-423-P |
| Rabbit anti-Ku80 | Cell Signaling | Cat#2753S |
| Rabbit anti-Ku70 | Cell Signaling | Cat#4588S |
| Rabbit anti-XLF | Bethyl Laboratories | Cat#A300-730A |
| Rabbit anti-C9orf142 (PAXX) | Abcam | Cat#ab126353 |
| Goat anti-XRCC4 | Santa Cruz | Cat#sc-8285 |
| Mouse-anti-ATM | Sigma | Cat#A1106; clone MAT3-4G10/8 |
| Mouse anti-RAD50 | Abcam | Cat#ab89; clone 2C6 |
| Rabbit anti-Mre11 | Novus Biologicals | Cat#NB100-142 |
| Rabbit anti-Nbs1 | Abcam | Cat#ab23996 |
| Rabbit anti-KAP-1 | GeneTex | Cat#GTX102226 |
| Rabbit anti-ATR | Novus Biologicals | Cat#NB100-323 |
| Rabbit anti-ATRIP | Sigma | Cat#SAB4503325 |
| Mouse anti-α tubulin | Sigma | Cat#T5168; clone B-1-2 |
| Rabbit anti-H2AX | Millipore | Cat#07-627 |
| Rabbit anti-AID | Chaudhuri et al., 2003 | N/A |
| Mouse IgG1 | Santa Cruz | Cat#sc-3877 |
| Armenian hamster IgG | BioLegend | Cat#400940 |
| EZView Red mouse anti-HA | Sigma | Cat#E6779; clone HA-7 |
| EZView Red mouse anti-FLAG | Sigma | Cat#F2426; clone M2 |
| Rabbit anti-cleaved caspase 3 | Cell Signaling | Cat#9661 |
| Cy3 donkey anti-rabbit IgG | Jackson Immunoresearch | Cat#711-165-152 |
| Pacific Blue rat anti-B220 | BioLegend | Cat#103227; clone RA3-6B2 |
| APC rat anti-CD43 | BioLegend | Cat#143208; clone S11 |
| PE/Cy7 rat anti-IgM | BioLegend | Cat#406514; clone RMM-1 |
| Pacific Blue rat anti-CD4 | BioLegend | Cat#100531; clone RM4-5 |
| PE/Cy7 rat anti-CD8a | BioLegend | Cat#100722; clone 53-6.7 |
| FITC rat anti-CD3ε | BioLegend | Cat#100306; clone 145-2C11 |
| APC Armenian hamster anti-TCRβ | eBioscience | Cat#17-5961-82; clone H57-597 |
| PE rat anti-IgG2b | BioLegend | Cat#406707; clone RMG2b-1 |
| APC rat anti-IgG1 | BioLegend | Cat#406609; clone RMG1-1 |
| Rat anti-CD16/CD32 (Fc block) | BD Pharmingen | Cat#553141 |
| Mouse anti-ATM | Santa Cruz | Cat#sc-135663; clone 1B10 |
| Mouse anti-MBP | GeneTex | Cat#GTX50060; clone 10C12 |
| Armenian hamster anti-CD40 | eBioscience | Cat#16-0402-81; HM40-3 |
| Bacterial and Virus Strains | ||
| pMSCV-v-abl | Bredemeyer et al., 2003 | N/A |
| pMSCV-RSS-GFP-INV-Thy1.2 (pMG-INV) | Hung et al., 2017 | N/A |
| pCW-Cas9 | Addgene | Cat#50661 |
| pKLV-U6gRNA-EF(BbsI)-PGKpuro2ABFP | Addgene | Cat#62348 |
| pLV-eGFP | Addgene | Cat#36083 |
| pOZ-FH-N | Nakatani and Ogryzko, 2003 | N/A |
| pBMN-FLAG-Ku80-hCD4 | Jiang et al., 2015 | N/A |
| pBMN-HA-ER-I-PpoI-hCD8 | Liu et al., 2017 | N/A |
| BL21(DE3) | Novagen | Cat#69450 |
| Biological Samples | ||
| 3110062M04Riktm1.1(KOMP)Vlcg mouse sperm | KOMP Repository | Clone 12667B-H5 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Imatinib | Novartis | N/A |
| KU55933 | Selleckchem | Cat#S1092 |
| Polybrene | Santa Cruz | Cat#sc-134220 |
| Doxycycline | Thermo Fisher | Cat#BP26531 |
| Puromycin | Invivogen | Cat#ant-pr-1 |
| Lipofectamine 2000 | Thermo Fisher | Cat#11668500 |
| Lipopolysaccharides from Escherichia coli O111:B4 | Sigma | Cat#L3024 |
| Recombinant murine interleukin-4 | Peprotech | Cat#214-14 |
| CFSE | eBioscience | Cat#65-0850-84 |
| Benzonase | Sigma | Cat#E1014 |
| FLAG peptide | Sigma | Cat#F3290 |
| HA peptide | Sigma | Cat#I2149 |
| RNase A | Sigma | Cat#R6513 |
| Palbociclib | Selleckchem | Cat#S1116 |
| RO-3306 | Millipore | Cat#217721 |
| 4-hydroxytamoxifen | Sigma | Cat#H7904 |
| Cytofix/Cytoperm solution | BD Biosciences | Cat#554722 |
| 7-AAD | BD Pharmingen | Cat#559925 |
| I-PpoI restriction enzyme | Promega | Cat# R7031 |
| Recombinant MBP-tagged murine and human MRI peptides | This paper | N/A |
| Recombinant biotin-FLAG-tagged human ATM peptide | This paper | N/A |
| Immunization peptide for generating hybridomas: LKSKTKTRVLPSWMTAPVDERKVC | This paper | GAP121 |
| Immunization peptide for generating hybridomas: KQTAAWAQRVGAATRAPATEC | This paper | GAP122 |
| Immunization peptide for generating hybridomas: GRKQEKPWEQRSLEATDKLQC | This paper | GAP123 |
| Immunization peptide for generating hybridomas: EEKEEEDALKYVREIFFS | This paper | GAP124 |
| Critical Commercial Assays | ||
| TissueTek | Sakura Finetek | Cat#4583 |
| Human B cell Nucleofector kit | Lonza | Cat#VAPA-1001 |
| PrestoBlue cell viability reagent | Thermo Fisher | Cat#A13262 |
| Mouse B cell isolation kit | Miltenyi Biotec | Cat#130-090-862 |
| Pur-A-Lyzer Maxi 6000 dialysis kit | Sigma | Cat#PURX60015 |
| Protein L magnetic beads | Thermo Fisher | Cat#88849 |
| Dynabeads Protein G | Thermo Fisher | Cat#10003D |
| Dynabeads M-280 streptavidin | Thermo Fisher | Cat#11205D |
| Superose 6 Increase 10/300 GL column | GE Healthcare | Cat#29091596 |
| StrataClean resin | Agilent | Cat#400714 |
| Deposited Data | ||
| Original data files for this paper | Mendeley Data | http://dx.doi.org/10.17632/2wdg6wf9fx.1 |
| Mass spectrometry data of MRI-interacting peptides | This paper; Mendeley Data (full list) | Table S3; http://dx.doi.org/10.17632/2wdg6wf9fx.1 |
| Experimental Models: Cell Lines | ||
| Mouse: WT abl pre-B cells | This paper | Lines M51.1-22, M63.1-7 |
| Mouse: MRI−/−abl pre-B cells | This paper | Lines M46.3-19, M66.1-24 |
| Mouse: XLF−/− abl pre-B cells | This paper | Lines XA3-8, XB1-5 |
| Mouse: Lig4−/− abl pre-B cells | This paper | Lines A20-115, B25-3 |
| Mouse: MRI−/−:XLF−/− abl pre-B cells | This paper | Lines MX-4, MX-19 |
| Mouse: PAXX−/− abl pre-B cells | Hung et al., 2017 | Line P2 |
| Mouse: MRI−/−:PAXX−/− abl pre-B cells | This paper | Line MP80 |
| Mouse: XLF−/−:PAXX−/− abl pre-B cells | Hung et al., 2017 | Line XP3 |
| Mouse: WT MEFs | This paper | Lines SZ, WT-1 |
| Mouse: MRI−/− MEFs | This paper | Lines M61.2, M61.7 |
| Mouse: XLF−/− MEFs | This paper | Lines X-AB, X-SZ |
| Experimental Models: Organisms/Strains | ||
| Mouse: MRI−/−: C57BL/6NTac-3110062M04Riktm1.1(KOMP)Vlcg | This paper | N/A |
| Mouse: XLF−/−: 129S6/SvEvTac-Nhej1tm1Fwa | Li et al., 2008 | N/A |
| Oligonucleotides | ||
| MRI gRNA sequence: 5′-GAAATGGTAGACGTGGCAC-3′ | This paper | N/A |
| PAXX gRNA sequence 1: 5′-CTAAGGTGTTCGCTCGGCGG-3′ | Liu et al., 2017; this paper (see Fig. S6B) | N/A |
| PAXX gRNA sequence 2: 5′-GCAGTTTATTTGACGGAGAA-3′ | Liu et al., 2017; this paper (see Fig. S6B) | N/A |
| Fwd primer sequence: genotyping the MRI allele: 5′-CCTTTCTTCCTTCCTTTGGG-3′ (P1) | This paper (see Fig. S1A and B) | N/A |
| Rev primer sequence: genotyping the MRI+ allele: 5′-CAGGCCAGAGCCCAGTTTG-3′ (P2) | This paper (see Fig. S1A and B) | N/A |
| Rev primer sequence: genotyping the MRI− allele: 5′-GTCTGTCCTAGCTTCCTCACTG-3′ (P3) | This paper (see Fig. S1A and B) | N/A |
| Fwd primer sequence: PCR analyses of the I-PpoI site: 5′-TCGTCCTTCCTTTTTCCTGAC-3′ (P1) | Liu et al., 2017; this paper (see Fig. S5B) | N/A |
| Rev primer sequence: PCR analyses of the I-PpoI site: 5′-TGGTTTCGCTGGATAGTAGGT-3′ (P2) | Liu et al., 2017; this paper (see Fig. S5B) | N/A |
| Fwd primer sequence: PCR sequencing of MRI exon 3: 5′-GTGCCCTGCCCCTGGACC-3′ | This paper | N/A |
| Rev primer sequence: PCR sequencing of MRI exon 3: 5′-AAAAACATGGCTGGAAAGGTGAGG-3′ | This paper | N/A |
| Fwd primer sequence: PCR screening of PAXX deletion: 5′-ATTGAAGAGCGGCAGATATGT-3′ (P1) | Liu et al., 2017; this paper (see Fig. S6B and C) | N/A |
| Rev primer sequence: PCR screening of PAXX deletion: 5′-AGCCAGAATCAACACAGTAGGT-3′ (P2) | Liu et al., 2017; this paper (see Fig. S6B and C) | N/A |
| Primer sequences used for generating MRI mutants are listed in Table S4 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pBABE-neo-SV40 | Addgene | Cat#1780 |
| Plasmid: pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | Cat#42230 |
| cDNAs: murine MRI, MRIΔN, MRIΔC, and MRIΔNΔC | This paper | CCDS19997 |
| Plasmid: pET15b | Novagen | Cat#69661 |
| Software and Algorithms | ||
| ImageJ | NIH | N/A |
| NIS Element High Content Analysis | Nikon, Inc. | N/A |
| Mass analysis/LC-MS/MS: Mascot | Matrix Science | N/A |
| HDX Workbench | Pascal et al., 2012 | N/A |
| FlowJo | FlowJo, LLC | N/A |
| StatPlus | AnalystSoft | N/A |
| Other | ||
| LSRII flow cytometer | BD Bioscience | N/A |
| Ti Eclipse inverted microscope + A1 RMP confocal system + Lu-N3 laser units | Nikon, Inc. | N/A |
| Fluoview FV3000 confocal laser scanning microscope | Olympus, Inc. | N/A |
| Multiskan Ascent microplate spectrophotometer | N/A | |
| RS 2000 X-ray irradiator | Rad Source Technologies | N/A |
| Nucleofector 2b device | Lonza | Cat#AAB-1001 |
| LTQ-FTICR mass spectrometer | Thermo Fisher | Cat#13437 |
| Chirascan CD spectrometer | Applied Photophysics | N/A |
Supplementary Material
Highlights.
MRI is a small disordered protein that promotes efficient cNHEJ
Combined MRI and XLF deficiency leads to a severe defect in cNHEJ
MRI forms multimeric complexes with diverse DDR factors at both its termini
MRI enhances the association of DDR factors with chromatin at DSBs
Acknowledgments
This work was supported by NIH grants AI047829 (B.P.S.), AI074953 (B.P.S.), AI120943 (G.K.A.), CA193318 (N.M.), GM59413 (J.JH.P.), and CA184187 (S.Z.) as well as the Alvin Siteman Cancer Research Fund (N.M.). The mass spectrometry research was supported in part by NIH grant P41GM103422 (M.L.G.). We thank Drs. H. Rohrs, and D. Leung for discussion and guidance with the characterization of MRI by biophysical methods. We thank S. Z. Duan for assistance with immunohistochemistry. We thank Dr. Kathy Sheehan and the Washington University School of Medicine, Department of Pathology and Immunology Hybridoma Center for monoclonal antibody development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: D.P. carried out and interpreted the analysis of CNS defects. T.E.J. and T.P. designed, carried out, and interpreted the analysis of in vitro binding of ATM to MRI. B.J.L. and S.Z. designed, carried out, and interpreted the analysis of MRI and Ku association at sites of laser damage in MEFs. A.L.B. performed IP-MS experiments of MRI complexes, with assistance from N.M. A.K.B. and N.M. designed, carried out, and interpreted the size exclusion chromatography analyses. B.J., P.A. M.L.G., and G. K. A. generated recombinant proteins and designed, carried out, and interpreted the HDX-MX and SEC-MALS. W.T.Y. and J.C. designed the Ig CSR experiments. J.JH.P. was involved in the interpretation of the biochemical analyses. B.P.S., J.K.T., and A.L.B. were involved in the design and interpretation of all experiments. P.J.H. was involved in the design and interpretation of all experiments and conducting all experiments except those listed above. B.C., S.D., and I. H. assisted P.J.H. in conducting some experiments.
Declaration of Interests: The authors declare no competing interests.
References
- Abramowski V, Etienne O, Elsaid R, Yang J, Berland A, Kermasson L, Roch B, Musilli S, Moussu JP, Lipson-Ruffert K, et al. PAXX and Xlf interplay revealed by impaired CNS development and immunodeficiency of double KO mice. Cell Death Differ. 2017;25:444–452. doi: 10.1038/cdd.2017.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Harada J, Schreifels J, Lech P, Nikolai B, Yamaguchi T, Chanda SK, Somia NV. Isolation, characterization, and genetic complementation of a cellular mutant resistant to retroviral infection. Proc Natl Acad Sci U S A. 2006;103:15933–15938. doi: 10.1073/pnas.0602674103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grofte M, Rask MB, Streicher W, Jungmichel S, Nielsen ML, et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose) Nat Commun. 2015;6:8088. doi: 10.1038/ncomms9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres SN, Modesti M, Tsai CJ, Chu G, Junop MS. Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Mol Cell. 2007;28:1093–1101. doi: 10.1016/j.molcel.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Arnoult N, Correia A, Ma J, Merlo A, Garcia-Gomez S, Maric M, Tognetti M, Benner CW, Boulton SJ, Saghatelian A, et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature. 2017;549:548–552. doi: 10.1038/nature24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam D, Komives EA. Hydrogen-exchange mass spectrometry for the study of intrinsic disorder in proteins. Biochim Biophys Acta. 2013;1834:1202–1209. doi: 10.1016/j.bbapap.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmus G, Barros AC, Wijnhoven PW, Lescale C, Hasse HL, Boroviak K, le Sage C, Doe B, Speak AO, Galli A, et al. Synthetic lethality between PAXX and XLF in mammalian development. Genes Dev. 2016;30:2152–2157. doi: 10.1101/gad.290510.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- Brouwer I, Sitters G, Candelli A, Heerema SJ, Heller I, de Melo AJ, Zhang H, Normanno D, Modesti M, Peterman EJ, et al. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature. 2016;535:566–569. doi: 10.1038/nature18643. [DOI] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Lobrich M, Shiloh Y, Chen DJ. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582–6587. doi: 10.1074/jbc.M611605200. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng HL, Davidson L, Kangaloo L, Alt FW. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396:173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol. 2000;18:495–527. doi: 10.1146/annurev.immunol.18.1.495. [DOI] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- Gapud EJ, Dorsett Y, Yin B, Callen E, Bredemeyer A, Mahowald GK, Omi KQ, Walker LM, Bednarski JJ, McKinnon PJ, et al. Ataxia telangiectasia mutated (Atm) and DNA-PKcs kinases have overlapping activities during chromosomal signal joint formation. Proc Natl Acad Sci U S A. 2011;108:2022–2027. doi: 10.1073/pnas.1013295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami D, Devarakonda S, Chalmers MJ, Pascal BD, Spiegelman BM, Griffin PR. Time window expansion for HDX analysis of an intrinsically disordered protein. J Am Soc Mass Spectrom. 2013;24:1584–1592. doi: 10.1007/s13361-013-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy GJ, Rulten SL, Arribas-Bosacoma R, Davidson K, Kozik Z, Oliver AW, Pearl LH, Caldecott KW. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat Commun. 2016;7:11242. doi: 10.1038/ncomms11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285:1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Sleckman BP. The Response to and Repair of RAG-Mediated DNA Double-Strand Breaks. Annu Rev Immunol. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung PJ, Chen BR, George R, Liberman C, Morales AJ, Colon-Ortiz P, Tyler JK, Sleckman BP, Bredemeyer AL. Deficiency of XLF and PAXX prevents DNA double-strand break repair by non-homologous end joining in lymphocytes. Cell Cycle. 2017;16:286–295. doi: 10.1080/15384101.2016.1253640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B, Li J, Adhikari J, Edwards MR, Zhang H, Schwarz T, Leung DW, Basler CF, Gross ML, Amarasinghe GK. Dimerization Controls Marburg Virus VP24-dependent Modulation of Host Antioxidative Stress Responses. J Mol Biol. 2016;428:3483–3494. doi: 10.1016/j.jmb.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppel TR, Howard BA, Weis DD. Mapping unstructured regions and synergistic folding in intrinsically disordered proteins with amide H/D exchange mass spectrometry. Biochemistry. 2011;50:8722–8732. doi: 10.1021/bi200875p. [DOI] [PubMed] [Google Scholar]
- Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- Kumar V, Alt FW, Frock RL. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc Natl Acad Sci U S A. 2016;113:10619–10624. doi: 10.1073/pnas.1611882113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescale C, Lenden Hasse H, Blackford AN, Balmus G, Bianchi JJ, Yu W, Bacoccina L, Jarade A, Clouin C, Sivapalan R, et al. Specific Roles of XRCC4 Paralogs PAXX and XLF during V(D)J Recombination. Cell Rep. 2016;16:2967–2979. doi: 10.1016/j.celrep.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Alt FW, Cheng HL, Brush JW, Goff PH, Murphy MM, Franco S, Zhang Y, Zha S. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol Cell. 2008;31:631–640. doi: 10.1016/j.molcel.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang W, Dubois RL, Yamamoto K, Wolner Z, Zha S. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc Natl Acad Sci U S A. 2012;109:3903–3908. doi: 10.1073/pnas.1120160109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Shao Z, Jiang W, Lee BJ, Zha S. PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat Commun. 2017;8:13816. doi: 10.1038/ncomms13816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes JL, Miles AJ, Whitmore L, Wallace BA. Distinct circular dichroism spectroscopic signatures of polyproline II and unordered secondary structures: applications in secondary structure analyses. Protein Sci. 2014;23:1765–1772. doi: 10.1002/pro.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- Marciano DP, Dharmarajan V, Griffin PR. HDX-MS guided drug discovery: small molecules and biopharmaceuticals. Curr Opin Struct Biol. 2014;28:105–111. doi: 10.1016/j.sbi.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenych V, Alt FW, Kumar V, Schwer B, Wesemann DR, Hansen E, Patel H, Su A, Guo C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A. 2012;109:2455–2460. doi: 10.1073/pnas.1121458109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT. Mechanisms of ATM Activation. Annu Rev Biochem. 2015;84:711–738. doi: 10.1146/annurev-biochem-060614-034335. [DOI] [PubMed] [Google Scholar]
- Reid DA, Keegan S, Leo-Macias A, Watanabe G, Strande NT, Chang HH, Oksuz BA, Fenyo D, Lieber MR, Ramsden DA, et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proc Natl Acad Sci U S A. 2015;112:E2575–2584. doi: 10.1073/pnas.1420115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Ropars V, Drevet P, Legrand P, Baconnais S, Amram J, Faure G, Marquez JA, Pietrement O, Guerois R, Callebaut I, et al. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proc Natl Acad Sci U S A. 2011;108:12663–12668. doi: 10.1073/pnas.1100758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavoff SA, Heo J, Budnik BA, Hanakahi LA, Saghatelian A. A human short open reading frame (sORF)-encoded polypeptide that stimulates DNA end joining. J Biol Chem. 2014;289:10950–10957. doi: 10.1074/jbc.C113.533968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadi SK, Tellier-Lebegue C, Nemoz C, Drevet P, Audebert S, Roy S, Meek K, Charbonnier JB, Modesti M. PAXX Is an Accessory c-NHEJ Factor that Associates with Ku70 and Has Overlapping Functions with XLF. Cell Rep. 2016;17:541–555. doi: 10.1016/j.celrep.2016.09.026. [DOI] [PubMed] [Google Scholar]
- Uversky VN. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol. 2017;44:18–30. doi: 10.1016/j.sbi.2016.10.015. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A. 2006;103:10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Zha S, Guo C, Boboila C, Oksenych V, Cheng HL, Zhang Y, Wesemann DR, Yuen G, Patel H, Goff PH, et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 2011a;469:250–254. doi: 10.1038/nature09604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Jiang W, Fujiwara Y, Patel H, Goff PH, Brush JW, Dubois RL, Alt FW. Ataxia telangiectasia-mutated protein and DNA-dependent protein kinase have complementary V(D)J recombination functions. Proc Natl Acad Sci U S A. 2011b;108:2028–2033. doi: 10.1073/pnas.1019293108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cui W, Gross ML. Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies. FEBS Lett. 2014;588:308–317. doi: 10.1016/j.febslet.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Lee JH, Jiang W, Crowe JL, Zha S, Paull TT. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol Cell. 2017;65:91–104. doi: 10.1016/j.molcel.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.