

 A series of chalcone-linked pyrazolo[1,5-a]pyrimidines was synthesized and evaluated for antiproliferative activity.
A series of chalcone-linked pyrazolo[1,5-a]pyrimidines was synthesized and evaluated for antiproliferative activity.
Abstract
A series of pyrazolo[1,5-a]pyrimidines substituted at C5 with 1-phenylprop-2-en-1-one (6a–q) and 3-phenylprop-2-en-1-one (7a–k) was synthesized and evaluated for antiproliferative activity. Among them, 6h was found to be the most active compound against the MDA-MB-231 cell line with an IC50 of 2.6 μM . The antiproliferative activity of this series of compounds ranged from 2.6 to 34.9 μM against A549 (lung cancer), MDA-MB-231 (breast cancer) and DU-145 (prostate cancer) cell lines. FACS analysis revealed that these hybrids arrest the cell cycle at the subG1 phase. Western blot analysis and an immunofluorescence assay showed the inhibition of the EGFR and STAT3 axis, which plays an important role in cell survival and apoptosis. Western blot and RT-PCR analyses that displayed an increase in apoptotic proteins such as p53, p21 and Bax and a decrease in the anti-apoptotic proteins Bcl-2 and procaspase-9 confirmed the ability of these hybrids to trigger cell death by apoptosis. Molecular docking studies described the binding of these hybrids to the ATP binding site of EGFR.
Introduction
Cancer has become a leading cause of mortality which solely accounted for 8.5 million deaths (17% of total deaths) in 2012 and it is estimated that it will cause 13.1 million deaths in 2030.1,2 Despite advances in diagnosis and treatment, the burden of cancer is increasing every year. Despite the fact that enormous effort is being put into the development of novel molecules and newer chemotherapeutic strategies to treat different types of cancer, this remains a key concern across the globe.3,4
Protein tyrosine kinase inhibitors are of tremendous interest due to their potential as therapeutic agents for the treatment of a variety of diseases, particularly cancer.5–7 Among the protein tyrosine kinases, the epidermal growth factor receptor (EGFR) has emerged as a key and validated target for the development of anticancer agents.8–10 Over ten drugs, including erlotinib111, gefitinib, lapatinib, icotinib, vandetanib etc., which act on EGFR have been approved by the United States Food and Drug Administration (US FDA) in the past two decades and several molecules, such as BMS-690514 (ref. 12 and 13), are currently in clinical trials. EGFR transduces signals across the membrane that are important for cellular functions, such as proliferation,10,14 angiogenesis, invasiveness, decreased apoptosis and differentiation,15 through which it regulates the development and progression of several types of cancer. EGFR is overexpressed in many solid tumors and has been associated with poor prognosis.16–18 It plays a central role in the development of non-small-cell lung cancer (NSCLC),19,20 as well as in breast cancer.21,22 Therefore, the molecules which inhibit EGFR are effective in the treatment of cancer. Since EGFR has become a fascinating and validated target for developing new anticancer agents, the exploration of EGFR with newly synthesized hybrids is likely to be a fruitful research area.
Chalcones (1,3-diarylprop-2-en-1-one) represent one of the largest classes of plant metabolites that are important precursors in the biosynthesis of flavonoids and related compounds.23 They remain attractive scaffolds for researchers due to their abundance in plants, ease of synthesis, availability to introduce diversity in the core structure by various methods and ability to transfer drug-like properties to the synthesized compounds. The derivatives of chalcones are known to possess a broad spectrum of biological activities, such as antidiabetic, antihypertensive, antihistaminic, antiretroviral, anti-inflammatory, antioxidant, antibacterial, antituberculosis, and anticancer.24–26 Moreover, chalcones have been successfully employed as tubulin inhibitors (2)27 and EGFR inhibitors (3 and 4)28,29 in the development of anticancer agents, which highlights the importance of the chalcone pharmacophore in the development of therapeutic agents. Similarly, pyrazolo[1,5-a]pyrimidine derivatives demonstrate a wide variety of promising biological activities, such as antitumor,30,31 anxiolytic,32,33 antimicrobial,34,35 antifungal, antiobesity and anti-inflammatory.36 They are effectively employed in the development of tyrosine kinase inhibitors (CDK).30,37,38 Compound SCH727965 (5)39 with a pyrazolo[1,5-a]pyrimidine scaffold in it was found to be a selective CDK inhibitor and is undergoing clinical trials. Also, our previous efforts towards the synthesis of a pyrazolo[1,5-a]pyrimidine scaffold coupled with anthranilamide40 and aminobenzothiazole37 led to several compounds with promising anticancer activity, which further substantiates the potential of the pyrazolo[1,5-a]pyrimidine scaffold.
The amalgamation of two pharmacophores into a single chemical entity is an effective and commonly used strategy for finding novel potential molecules, the resulting molecules being known as hybrids or chimeric molecules.41,42 The selection of two pharmacophores is based on their anticipated additive or synergistic effect. Hybrids are designed to synergise biological activity, minimize resistance, affect multiple targets and/or reduce known side effects.43–45 Thus, owing to the importance of chalcone, pyrazolo[1,5-a]pyrimidine pharmacophores and our interest in the synthesis of biologically active molecules, herein we have combined both pharmacophores into a single chemical entity by anticipating their improved pharmacological activity (Fig. 1). The resulting series of chalcone-linked pyrazolo[1,5-a]pyrimidines was evaluated for antiproliferative activity.
Fig. 1. Chemical structures of EGFR inhibitors: 1 (erlotinib), chalcone scaffold containing compounds 2 (MDL-27048), 3 (butein) and 4 (flavokavain B), pyrazolo[1,5-a]pyrimidine containing scaffold compound 5 (SCH-727965) and designed chalcone-linked pyrazolo[1,5-a]pyrimidines (6a–q and 7a–k).

Results and discussion
Chemistry
Synthesis of 6a–q hybrids
6a–q were synthesized as shown in Scheme 1. Different acetophenones 8a–c were oxalylated by diethyl oxalate in the presence of sodium ethoxide in ethanol to give 9a–c. These were condensed with 3-amino-5-phenyl-1H-pyrazole in the presence of a catalytic amount of conc. HCl in ethanol to yield ethyl pyrazolo[1,5-a]pyrimidine-5-carboxylates 10a–c. The ester functionality of the ethyl pyrazolo[1,5-a]pyrimidine-5-carboxylates was then reduced with DIBAL-H in dichloromethane to give the corresponding pyrazolo[1,5-a]pyrimidine-5-carbaldehydes 11a–c. Different acetophenones 12a–g were condensed with the pyrazolo[1,5-a]pyrimidine-5-carbaldehydes to afford the corresponding chalcone-linked pyrazolo[1,5-a]pyrimidines (6a–q).
Scheme 1. Synthesis of pyrazolo[1,5-a]pyrimidines (6a–q); reagents and conditions: a) diethyl oxalate, NaOEt, EtOH, rt, 12 h, (yield 80–85%); b) 3-amino-5-phenyl-pyrazole, conc. HCl (cat.), EtOH, reflux, 2–4 h, (yield 90–95%); c) DIBAL-H, CH2Cl2, –78 °C, 2 h, (yield 78–80%); d) Ba(OH)2, MeOH, rt, 4–6 h, (yield 71–91%).

Synthesis of 7a–k hybrids
7a–k were synthesized as shown in Scheme 2. Substituted pyrazolo[1,5-a]pyrimidine-5-carbaldehydes 11a and b were subjected to Weinreb amide in the presence of trimethylaluminum to give the corresponding amides (13a and b) which were further subjected to a Grignard reaction with methylmagnesium bromide to give the corresponding 1-(pyrazolo[1,5-a]pyrimidin-5-yl)ethan-1-ones (14a and b). Different aldehydes (15a–f) were condensed with the 1-(pyrazolo[1,5-a]pyrimidin-5-yl)ethan-1-ones to give the chalcone-linked pyrazolo[1,5-a]pyrimidines (7a–k).
Scheme 2. Synthesis of pyrazolo[1,5-a]pyrimidines (7a–k); reagents and conditions: a) N,O-dimethylhydroxylamine hydrochloride, Al(CH3)3, dry dichloromethane, 0–25 °C, 2 h, (yield 77–79%); b) methylmagnesium bromide, dry tetrahydrofuran, 0 °C, 1 h, (yield 85–92%); c) barium hydroxide, methanol, rt, 6 h, (yield 79–92%).

Biology
Anti-proliferative activity
The MTT assay was performed to evaluate the cytotoxic effect of the newly synthesized chalcone-linked pyrazolo[1,5-a]pyrimidine hybrids against the selected cell lines viz. A549 (lung cancer), MDA-MB-231 (breast cancer) and DU-145 (prostate cancer) using erlotinib as a reference standard; the obtained results are summarized in Table 1. All of the hybrids in this series possess moderate to good antiproliferative potential against selected cell lines. Hybrid 6h with 3,4-dimethoxy substitutions on the C7 phenyl ring and phenylprop-2-en-1-one on the C5 position of the pyrazolo[1,5-a]pyrimidine scaffold was found to be the most active against the MDA-MB-231 (IC50 = 2.6 μM) cell line. The same hybrid showed effective potential against the A549 and DU-145 cell lines with IC50 values of 3.9 μM and 7.2 μM, respectively. In an SAR study, it was very interesting to evaluate the effect of the substitutions on the C5 phenylprop-2-en-1-one and on the C7 phenyl ring. It was observed that the 6a–q series has superior antiproliferative activity than the 7a–k series except for the DU-145 cell line. In both series, the only difference is in the orientation of the enone bond. In the case of the 6a–q series, the double bond is near to the pyrazolo[1,5-a]pyrimidine scaffold, whereas in the case of the 7a–k series the carbonyl group is near to the pyrazolo[1,5-a]pyrimidine scaffold. The probable reason for this activity difference might be due to the extra hydrogen bonds formed by the carbonyl group of the hybrids of the 6a–q series with the Cys773 amino acid of EGFR; similar hydrogen bonding was not possible for the hybrids of the 7a–k series (explained with docking poses in the molecular modeling section). Further structure–activity relationships are discussed for the 6a–q series. Hybrids with 4-methoxy and 3,4-dimethoxy substitutions on the C7 phenyl ring displayed superior antiproliferative potential with IC50 values ranging from 2.6 to 19.8 μM against the MDA-MB-231 cell line. Hybrids with 3,4,5-trimethoxy substitutions on the C7 phenyl ring showed lower antiproliferative potential with IC50 values ranging from 13.2 to 32.5 μM against the MDA-MB-231 cell line. The activity order for the C7 phenyl ring substitutions was 3,4-dimethoxy ≥ 4-methoxy > 3,4,5-trimethoxy. Furthermore, the hybrids with electron withdrawing substituents on the C5 phenylprop-2-en-1-one showed lower cytotoxic potential than the hybrids with electron donating substituents. Meta-,para-disubstituted hybrids have higher cytotoxic potentials than meta-, para-, meta-trisubstituted hybrids followed by unsubstituted hybrids. However, hybrids with only para-substitution exhibit the lowest cytotoxic potential, except for 6b. The activity order for the C5 phenylprop-2-en-1-one substituents was 3,4-dimethoxy > 3,4,5-trimethoxy > 3,4-dimethyl > no substitution > 4-methoxy > 4-chloro > 4-methyl. Thus it could be concluded that the 3,4-dimethoxy and 4-methoxy substitutions on the C7 phenyl ring were optimum for antiproliferative potential and the 3,4-disubstitution on the C5 phenylprop-2-en-1-one enhanced the activity. In contrast, the 3,4,5-trimethoxy substitution on the C7 phenyl ring was detrimental for the activity. Based on the results obtained with the MTT assay, three of the most potent hybrids (6b, 6h and 6i) were considered for further mechanistic studies.
Table 1. Cytotoxicity (IC50 values in μM) a of chalcone-linked pyrazolo[1,5-a]pyrimidines 6a–q and 7a–k on selected cell lines.
| b A549 | c MDA-MB-231 | d DU-145 | e HEK293 | |
| 6a | 8.6 ± 0.4 | 9.9 ± 0.2 | 13.7 ± 0.3 | 53.2 ± 1.9 |
| 6b | 2.9 ± 0.3 | 6.3 ± 0.3 | 8.5 ± 0.4 | 36.1 ± 0.9 |
| 6c | 7.4 ± 0.2 | 8.7 ± 0.4 | 16.4 ± 0.4 | 48.5 ± 0.5 |
| 6d | 10.7 ± 0.3 | 11.8 ± 0.4 | 10 ± 0.3 | 51.2 ± 0.6 |
| 6e | 17.2 ± 0.4 | 19.8 ± 0.3 | 26.8 ± 0.4 | 62.6 ± 0.4 |
| 6f | 9.3 ± 0.6 | 11.5 ± 0.2 | 12.1 ± 0.6 | 46.2 ± 0.7 |
| 6g | 11.9 ± 0.3 | 13.9 ± 0.5 | 14.6 ± 0.5 | 40.0 ± 0.3 |
| 6h | 3.9 ± 0.4 | 2.6 ± 0.6 | 7.2 ± 0.4 | 32.5 ± 0.7 |
| 6i | 7.2 ± 0.4 | 4.7 ± 0.3 | 8.3 ± 0.3 | 35 ± 1.2 |
| 6j | 15.4 ± 0.5 | 17.1 ± 0.4 | 21.3 ± 0.4 | 44.1 ± 0.8 |
| 6k | 18.2 ± 0.4 | 15.8 ± 0.4 | 34.3 ± 0.5 | ≥100 |
| 6l | 19.4 ± 0.3 | 23.9 ± 0.6 | 27.1 ± 0.3 | 50.4 ± 1.9 |
| 6m | 10.6 ± 0.4 | 13.5 ± 0.6 | 15.4 ± 0.6 | 72.5 ± 1.2 |
| 6n | 14.6 ± 0.5 | 13.2 ± 0.4 | 25.2 ± 0.6 | 79.8 ± 0.7 |
| 6o | 17.8 ± 0.3 | 18.3 ± 0.4 | 29.9 ± 0.4 | 81.7 ± 1.6 |
| 6p | 20.5 ± 0.6 | 24.3 ± 0.5 | 34.9 ± 0.4 | ≥100 |
| 6q | 15.5 ± 0.4 | 32.5 ± 0.4 | 29.3 ± 0.2 | ≥100 |
| 7a | 18.62 ± 1.04 | 23.32 ± 0.8 | 15.57 ± 1.65 | 64.1 ± 1.4 |
| 7b | 22.19 ± 2.86 | 22.59 ± 1.87 | 19.49 ± 1.31 | 57.4 ± 0.9 |
| 7c | 19.27 ± 1.91 | 21.98 ± 0.92 | 14.93 ± 0.78 | 50.7 ± 1.2 |
| 7d | 15.44 ± 2.11 | 16.73 ± 0.67 | 14.74 ± 1.41 | 46.8 ± 1 |
| 7e | 22.01 ± 2.15 | 23.74 ± 0.91 | 22.4 ± 1.87 | 39.1 ± 0.7 |
| 7f | 17.97 ± 0.57 | 19.15 ± 1.11 | 16.31 ± 1.41 | 44.8 ± 1.3 |
| 7g | 20.03 ± 2.71 | 18.64 ± 1.12 | 21.89 ± 1.03 | 56.1 ± 0.8 |
| 7h | 21.23 ± 1.91 | 16.5 ± 1.46 | 16.91 ± 0.53 | 46.6 ± 1 |
| 7i | 20.47 ± 1.87 | 14.19 ± 2.16 | 15.33 ± 0.9 | 41.0 ± 1.9 |
| 7j | 25.86 ± 2.8 | 18.37 ± 1.8 | 17.91 ± 1.2 | 41.0 ± 0.3 |
| 7k | 23.83 ± 2.4 | 21.62 ± 0.92 | 19.51 ± 2.4 | 53.9 ± 1.6 |
| Erlo | 10.39 ± 1.6 | 14.74 ± 2.5 | 18.4 ± 1.4 | 30.3 ± 2.1 |
aCell lines were treated with different concentrations of compounds. Cell viability was measured by employing the MTT assay. The concentration required for 50% inhibition of cell growth was calculated and the values represent the means ± S.D. of the three different experiments performed in triplicate.
bLung cancer.
cBreast cancer.
dProstate cancer.
eHuman embryonic kidney cells.
Effect on cell cycle distribution
Anticancer molecules exert their action either by arresting the cell cycle at a checkpoint or by inducing apoptosis, or by a combined effect of both cycle block and apoptosis. Furthermore, regulation of the cell cycle and apoptosis are considered to be effective strategies in the development of cancer therapeutics. To shed light on the effects of active hybrids 6b, 6h and 6i on cell cycle progression, cell cycle analysis was performed on A549 cells. These cells were treated with a 2 μM concentration of the test compounds for 24 h. Cell cycle analysis performed with PI staining showed accumulation of cells at the G2/M phase (Fig. 2). Untreated cells showed 20.4% of cells were in the G2/M phase. In contrast, cells treated with compounds 6b, 6h and 6i showed accumulation of 29.6%, 29.5% and 33.11% of cells, respectively. When erlotinib was used as a positive control, it showed 24.5% in the G2/M phase. Thus, the compounds in this investigation showed a significant effect in restricting A549 cells to the G2/M phase, which in itself is strong evidence of apoptotic cell death. Also, the effect of the compounds was validated in a non-cancerous HEK cell line, which revealed the distribution of treated cells was similar to that of the untreated cells (Fig. S1†).
Fig. 2. Flow cytometric analysis displaying G2/M arrest in cells post-treatment. A549 cells subjected to compound treatment for 24 h at 2 μM concentration were collected, fixed and stained for FACS analysis. Untreated cells were employed as a negative control.

Effect on the EGFR/STAT3 pathway
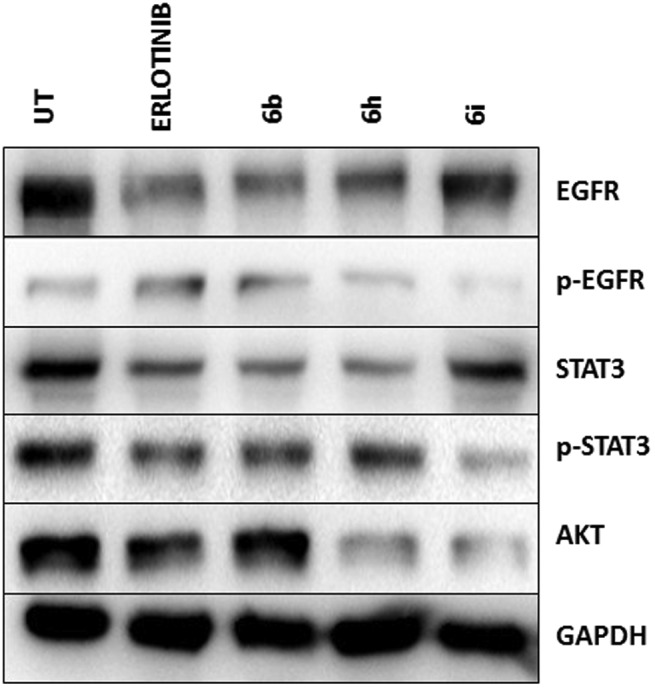
To investigate the pathway responsible for cell death and arrest, A549 cells were treated with hybrids 6b, 6h and 6i at a 2 μM concentration for 24 h, followed by total protein lysate extraction. The protein lysate was subjected to western blot analysis, which revealed elevated levels of EGFR and P-EGFR upon treatment with hybrids in comparison to the erlotinib-treated cells (Fig. 3). STAT3 is a well-known transcription factor responsible for cell survival and proliferation and is commonly over expressed in most cancer types. Interestingly, significant down-regulation of STAT3 and P-STAT3 upon treatment was observed. Furthermore, AKT, a key component of cellular survival pathways, was observed to be down-regulated. Both 6h and 6i showed significant down-regulation compared to the untreated cells. Among the compounds studied, 6b, 6h and 6i demonstrated a significant effect on the modulation of the EGFR/STAT3 axis and AKT pathway (Fig. 3). They also show a high degree of correlation with the erlotinib-treated cells. Also, a non-cancerous HEK cell line was employed to study the deleterious effects on normal cells and, much to our interest, there was no significant alteration in the expression of the EGFR/STAT3 pathway (Fig. S2A†).
Fig. 3. Identification of the affected signaling pathway using western blot analysis. A549 cells were treated with 6b, 6h and 6i at 2 μM final concentration and were collected 24 h post-treatment. The total protein lysate was isolated and analyzed by western blotting using the corresponding antibody. Compounds 6b and 6h revealed significantly reduced levels of p-EGFR, p-STAT3 and AKT. Erlotinib was utilized as a positive control, whereas untreated cells were considered as a negative control.
Effect on the co-localization of EGFR and STAT3
To further clarify the deregulation of the EGRF/STAT axis in the treated A549 cells, the cellular protein levels and the localization of the p-EGFR and STAT3 proteins were identified by an immunofluorescence method. After 24 h of treatment the cells were fixed, stained with EGFR/STAT3 tagged with a Cy3-conjugated secondary antibody fluorophore and analyzed with the aid of laser scanning confocal microscopy. Cells treated with hybrids 6b, 6h and 6i showed a significant down-regulation of EGFR as well as STAT3, in comparison to the positive control (erlotinib) treated cells (Fig. 4). This clearly shows the reduced co-localization of EGFR and STAT3 upon treatment with chalcone-linked pyrazolo[1,5-a]pyrimidine hybrids.
Fig. 4. Regulation of EGFR and STAT3 in cells 24 h post-treatment. A549 cells were fixed, stained and observed for 24 h post-treatment with compounds 6b, 6h and 6i at a 2 μM final concentration under a confocal microscope (FLOW VIEW FV 1000 series). The Cy3 dye was used to stain both EGFR and STAT3, whereas DAPI was used as a nuclear stain. Compounds 6h and 6i showed profound inhibitory effects on the levels of EGFR and STAT3.

Effect on pro-apoptotic and anti-apoptotic proteins
To understand the role of chalcone-linked pyrazolo[1,5-a]pyrimidine hybrids in regulating apoptosis, A549 cells were treated and the extracted protein lysate was subjected to analysis at both the transcriptional and translational levels of key apoptotic proteins like p53, p21, BAX, caspase-3 and Bcl-2.The test compounds significantly increased the levels of p53, p21 and BAX and decreased the expression of Bcl2 (Fig. 5a and b). The increase in the levels of many of these pro-apoptotic proteins also showed a concomitant decrease in the levels of the anti-apoptotic protein Bcl-2 upon treatment with the compounds. This regulation of apoptosis at both the transcriptional and translational levels shows the prominent effect of the chalcone-linked pyrazolo[1,5-a]pyrimidine hybrids in inducing apoptosis. To understand the effect on normal cells, a non-cancerous HEK cell line was studied with the same compounds and there was no significant alteration in the expression pattern compared to the untreated control cells (Fig. S2B†).
Fig. 5. Analysis of pro-apoptotic and anti-apoptotic proteins 24 h post-treatment. (a) The total lysate was isolated from A549 cells after 24 h of treatment with compounds 6b, 6h and 6i at 2 μM final concentrations and was analyzed by western blotting using a corresponding antibody. Compounds 6b and 6i show obvious up-regulation of apoptotic proteins including Bax, p53, p21 and caspase-3, whereas down-regulation of the anti-apoptotic protein Bcl2 was observed. Erlotinib was employed as a positive control and untreated cells were used as a negative control. (b) RNA was isolated from cells using the Trizol reagent and cDNA was synthesized. End point RT-PCR results showed p53 and Bax upregulation in cells treated with compounds 6b and 6h and 6i and a significant reduction in the levels of anti-apoptotic protein Bcl2.

Molecular docking studies
Molecular docking studies were performed to get insight into the interactions between the chalcone-linked pyrazolo[1,5-a]pyrimidine hybrids and EGFR. Molecular docking studies were carried out for all hybrids (6a–q and 7a–k). The docking pose of 6j showed that these hybrids bind well at the ATP binding site of EGFR (Fig. 6B). The C2 phenyl ring was buried in a specificity pocket where it was surrounded by the Val702, Ala719, Ile720, Lys721, Met742, Leu764, Ile765 and Thr766 amino acids. The superimposed pose of 6j with the co-crystal ligand erlotinib showed that the C2 phenyl ring of 6j was overlapping with the acetylphenyl group of erlotinib (Fig. 6C). The C7 phenyl ring is oriented towards the hinge region and surrounded by the Leu694, Val702, Ala719, Gln767, Leu768, Met769, Pro770, Gly772 and Leu820 amino acids. The 3-methoxy and 4-methoxy groups formed hydrogen bonds with the –NH group of Met769. This hydrogen bond is crucial for EGFR inhibitors and is present in all of the drug molecules and in ATP as well. The C5 phenylprop-2-en-1-one was oriented towards the ‘A loop’ and forms a close contact with the Cys773, Arg817 and Asn818 amino acids and the carbonyl carbon formed a hydrogen bond with Cys773. In the MTT assay it was observed that the 6a–q series has superior activity over the 7a–k series. In the superimposed pose for the hybrids of the 6a–q series, hydrogen bonding was observed between the carbonyl carbon and the Cys773 amino acid, whereas for the hybrids in the 7a–k series, such hydrogen bonding was not present (Fig. 6D). This is due to the difference in the geometrical orientation of the enone group in 6a–q and 7a–k and may be a reason for the superior activity of compounds 6a–q over 7a–k. The order of activity for the C7 phenyl substituents was 3,4-dimethoxy > 4-methoxy > 3,4,5-trimethoxy. From the docking studies it was observed that the 3,4-dimethoxy substituted hybrids formed two hydrogen bonds with Met769. On the other hand, the 4-methoxy substituted hybrids only formed one hydrogen bond (ESI† Fig. S3a). The superimposed pose of 3,4-dimethoxy and 4-methoxy explains the higher activity of the 3,4-dimethoxy substituted hybrids but it does not explain why the 3,4,5-trimethoxy substituted hybrids show the lowest activity. However, the fact that most of the drugs acting on EGFR have dialkoxy substitution supports the fact that dialkoxy substitution is optimum for the activity (the structures of drugs that have dialkoxy substitution are shown in the ESI† Fig. S4). The least active hybrid in the series is 6p with 3,4,5-trimethoxy substituents on both the C7 phenyl ring and the C5 phenylprop-2-en-1-one. The docking pose shows that the carbonyl group in 6p did not form a hydrogen bond with Cys773, which supports the low activity of 6p (ESI† Fig. S3b). The docking studies showed that these hybrids bind well in the ATP binding site of EGFR with the required hydrophobic interactions in the specificity pocket and hydrogen bonding with the Met769 amino acid in the hinge region. The docking studies explained the SAR of the series.
Fig. 6. Docking poses on EGFR. A) 2D structure of a chalcone-linked pyrazolo[1,5-a]pyrimidine with observed interactions with EGFR; B) binding pose of 6j (yellow color, EGFR is shown in green and hydrogen bonds are shown by the red dotted lines); C) superimposed pose of 6j and erlotinib; D) superimposed pose of 6j and 7a showing the difference in hydrogen bonding between the carbonyl carbons and Cys773.

Conclusion
A series of chalcone-linked pyrazolo[1,5-a]pyrimidines (6a–q and 7a–k) was synthesized and evaluated for cytotoxic activity. Most of the hybrids exhibited promising anti-proliferative potential with IC50 values ranging from 2.6 μM to 34.9 μM. In the series, 6b, 6h and 6i showed promising cytotoxicity and were advanced for further mechanistic studies. Cell cycle analysis showed a significant accumulation of cells at the G2/M phase of the cell cycle. Western blot analysis and an immunostaining assay suggested the down-regulation of EGFR, p-EGFR, STAT3 and p-STAT3, with a stronger down-regulation of the EGFR/ STAT3 axis. Western blot analysis and the RT-PCR results also showed the up-regulation of apoptotic proteins like p53, p21 and BAX, whereas down-regulation of anti-apoptotic proteins such as Bcl-2 and caspase-3 occurred at both the transcriptional and translational level. Our study establishes the role of chalcone-linked pyrazolo[1,5-a]pyrimidine hybrids in a lung carcinoma cell line, A459, in inducing cellular arrest and activating apoptosis. Molecular docking studies provided insight into the binding mode of these molecules at the ATP binding site of EGFR and shed light on the superior activity of compounds 6a–q in comparison to that of the 7a–k series. These studies suggest the potential of synthesized hybrid molecules for their development into promising anticancer agents.
Conflict of interest
The authors declare no competing interests.
Supplementary Material
Acknowledgments
The author C. B. acknowledges the Department of Pharmaceuticals (Ministry of Chemicals and Fertilizers, Govt. of India) for financial support & the CSIR-IICT, Hyderabad for providing facilities. G. K. R. thanks the UGC for his fellowship. This work was financially supported by the CSIR 12th FYP CSC0111 (SMiLE). The authors thank Y. Suresh for the flow cytometry and confocal microscopy studies.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00193b
References
- Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-Tieulent J., Jemal A. Ca-Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Bray F., Jemal A., Grey N., Ferlay J., Forman D. Lancet Oncol. 2012;13:790–801. doi: 10.1016/S1470-2045(12)70211-5. [DOI] [PubMed] [Google Scholar]
- Zugazagoitia J., Guedes C., Ponce S., Ferrer I., Molina-Pinelo S., Paz-Ares L. Clin. Ther. 2016;38:1551–1566. doi: 10.1016/j.clinthera.2016.03.026. [DOI] [PubMed] [Google Scholar]
- Biemar F., Foti M. Cancer Biol. Med. 2013;10:183. doi: 10.7497/j.issn.2095-3941.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes A., Zech B., Felber B., Klebl B., Müller G. Expert Opin. Drug Discovery. 2008;3:1427–1449. doi: 10.1517/17460440802580106. [DOI] [PubMed] [Google Scholar]
- Gschwind A., Fischer O. M., Ullrich A. Nat. Rev. Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Backes A., Zech B., Felber B., Klebl B., Müller G. Expert Opin. Drug Discovery. 2008;3:1409–1425. doi: 10.1517/17460440802579975. [DOI] [PubMed] [Google Scholar]
- Yewale C., Baradia D., Vhora I., Patil S., Misra A. Biomaterials. 2013;34:8690–8707. doi: 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]
- Warnault P., Yasri A., Coisy-Quivy M., Cheve G., Bories C., Fauvel B., Benhida R. Curr. Med. Chem. 2013;20:2043–2067. doi: 10.2174/0929867311320160001. [DOI] [PubMed] [Google Scholar]
- Speake G., Holloway B., Costello G. Curr. Opin. Pharmacol. 2005;5:343–349. doi: 10.1016/j.coph.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Dowell J., Minna J. D., Kirkpatrick P. Nat. Rev. Drug Discovery. 2005;4:13–14. doi: 10.1038/nrd1612. [DOI] [PubMed] [Google Scholar]
- Wong T. W., Lee F. Y., Yu C., Luo F. R., Oppenheimer S., Zhang H., Smykla R. A., Mastalerz H., Fink B. E., Hunt J. T. Clin. Cancer Res. 2006;12:6186–6193. doi: 10.1158/1078-0432.CCR-06-0642. [DOI] [PubMed] [Google Scholar]
- Yarden Y., Sliwkowski M. X. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Holbro T., Hynes N. E. Annu. Rev. Pharmacol. Toxicol. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- Olayioye M. A., Neve R. M., Lane H. A., Hynes N. E. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temam S., Kawaguchi H., El-Naggar A. K., Jelinek J., Tang H., Liu D. D., Lang W., Issa J.-P., Lee J. J., Mao L. J. Clin. Oncol. 2007;25:2164–2170. doi: 10.1200/JCO.2006.06.6605. [DOI] [PubMed] [Google Scholar]
- Tanaka E., Hashimoto Y., Ito T., Okumura T., Kan T., Watanabe G., Imamura M., Inazawa J., Shimada Y. Clin. Cancer Res. 2005;11:1827–1834. doi: 10.1158/1078-0432.CCR-04-1627. [DOI] [PubMed] [Google Scholar]
- Kurai M., Shiozawa T., Shih H.-C., Miyamoto T., Feng Y.-Z., Kashima H., Suzuki A., Konishi I. Hum. Pathol. 2005;36:1281–1288. doi: 10.1016/j.humpath.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Jemal A., Siegel R., Ward E., Murray T., Xu J., Thun M. J. Ca-Cancer J. Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- Sharma S. V., Bell D. W., Settleman J., Haber D. A. Nat. Rev. Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Rimawi M. F., Shetty P. B., Weiss H. L., Schiff R., Osborne C. K., Chamness G. C., Elledge R. M. Cancer. 2010;116:1234–1242. doi: 10.1002/cncr.24816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon D. S., Brandt R., Ciardiello F., Normanno N. Crit. Rev. Oncol. Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- Di Carlo G., Mascolo N., Izzo A. A., Capasso F. Life Sci. 1999;65:337–353. doi: 10.1016/s0024-3205(99)00120-4. [DOI] [PubMed] [Google Scholar]
- Mahapatra D. K., Asati V., Bharti S. K. Eur. J. Med. Chem. 2015;92:839–865. doi: 10.1016/j.ejmech.2015.01.051. [DOI] [PubMed] [Google Scholar]
- Mahapatra D. K., Bharti S. K., Asati V. Eur. J. Med. Chem. 2015;98:69–114. doi: 10.1016/j.ejmech.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Mahapatra D. K., Bharti S. K., Asati V. Eur. J. Med. Chem. 2015;101:496–524. doi: 10.1016/j.ejmech.2015.06.052. [DOI] [PubMed] [Google Scholar]
- Ducki S. IDrugs. 2007;10:42. [PubMed] [Google Scholar]
- Ji T., Lin C., Krill L. S., Eskander R., Guo Y., Zi X., Hoang B. H. Mol. Cancer. 2013;12:55. doi: 10.1186/1476-4598-12-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E.-B., Zhang K., Cheng L. Y., Mack P. Biochem. Biophys. Res. Commun. 1998;245:435–438. doi: 10.1006/bbrc.1998.8452. [DOI] [PubMed] [Google Scholar]
- Heathcote D. A., Patel H., Kroll S. H., Hazel P., Periyasamy M., Alikian M., Kanneganti S. K., Jogalekar A. S., Scheiper B., Barbazanges M. J. Med. Chem. 2010;53:8508–8522. doi: 10.1021/jm100732t. [DOI] [PubMed] [Google Scholar]
- Gao M., Duan L., Luo J., Zhang L., Lu X., Zhang Y., Zhang Z., Tu Z., Xu Y., Ren X. J. Med. Chem. 2013;56:3281–3295. doi: 10.1021/jm301824k. [DOI] [PubMed] [Google Scholar]
- Selleri S., Bruni F., Costagli C., Costanzo A., Guerrini G., Ciciani G., Gratteri P., Besnard F., Costa B., Montali M. J. Med. Chem. 2005;48:6756–6760. doi: 10.1021/jm058002n. [DOI] [PubMed] [Google Scholar]
- Popik P., Kostakis E., Krawczyk M., Nowak G., Szewczyk B., Krieter P., Chen Z., Russek S. J., Gibbs T. T., Farb D. H. J. Pharmacol. Exp. Ther. 2006;319:1244–1252. doi: 10.1124/jpet.106.107201. [DOI] [PubMed] [Google Scholar]
- Bondock S., Fadaly W., Metwally M. A. Eur. J. Med. Chem. 2010;45:3692–3701. doi: 10.1016/j.ejmech.2010.05.018. [DOI] [PubMed] [Google Scholar]
- Behbehani H., Ibrahim H. M., Makhseed S., Mahmoud H. Eur. J. Med. Chem. 2011;46:1813–1820. doi: 10.1016/j.ejmech.2011.02.040. [DOI] [PubMed] [Google Scholar]
- Cherukupalli S., Karpoormath R., Chandrasekaran B., Hampannavar G. A., Thapliyal N., Palakollu V. N. Eur. J. Med. Chem. 2016;126:298–352. doi: 10.1016/j.ejmech.2016.11.019. [DOI] [PubMed] [Google Scholar]
- Kamal A., Tamboli J. R., Nayak V. L., Adil S., Vishnuvardhan M., Ramakrishna S. Bioorg. Med. Chem. Lett. 2013;23:3208–3215. doi: 10.1016/j.bmcl.2013.03.129. [DOI] [PubMed] [Google Scholar]
- Brahmkshatriya P. S., Dobes P., Fanfrlik J., Rezac J., Paruch K., Bronowska A., Lepsík M., Hobza P. Curr. Comput.-Aided Drug Des. 2013;9:118–129. doi: 10.2174/1573409911309010011. [DOI] [PubMed] [Google Scholar]
- McClue S. J., Blake D., Clarke R., Cowan A., Cummings L., Fischer P. M., MacKenzie M., Melville J., Stewart K., Wang S. Int. J. Cancer. 2002;102:463–468. doi: 10.1002/ijc.10738. [DOI] [PubMed] [Google Scholar]
- Kamal A., Tamboli J. R., Ramaiah M. J., Adil S., Koteswara Rao G., Viswanath A., Mallareddy A., Pushpavalli S., Pal-Bhadra M. ChemMedChem. 2012;7:1453–1464. doi: 10.1002/cmdc.201200205. [DOI] [PubMed] [Google Scholar]
- Viegas-Junior C., Danuello A., da Silva Bolzani V., Barreiro E. J., Fraga C. A. M. Curr. Med. Chem. 2007;14:1829–1852. doi: 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- Bansal Y., Silakari O. Eur. J. Med. Chem. 2014;76:31–42. doi: 10.1016/j.ejmech.2014.01.060. [DOI] [PubMed] [Google Scholar]
- Tietze L. F., Bell H. P., Chandrasekhar S. Angew. Chem., Int. Ed. 2003;42:3996–4028. doi: 10.1002/anie.200200553. [DOI] [PubMed] [Google Scholar]
- Muregi F. W., Ishih A. Drug Dev. Res. 2010;71:20–32. doi: 10.1002/ddr.20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepali K., Sharma S., Sharma M., Bedi P., Dhar K. Eur. J. Med. Chem. 2014;77:422–487. doi: 10.1016/j.ejmech.2014.03.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.