

 A series of new triazolo linked 4β-amidopodophyllotoxin conjugates (9a–l) were synthesized using click chemistry and evaluated for their antitumor activity.
A series of new triazolo linked 4β-amidopodophyllotoxin conjugates (9a–l) were synthesized using click chemistry and evaluated for their antitumor activity.
Abstract
A series of new triazolo linked 4β-amidopodophyllotoxin conjugates (9a–l) were synthesized using click chemistry and evaluated for their antitumor activity against four human cancer cell lines. Among them, two compounds (9c and 9j) showed significant anticancer activity with IC50 values of 0.9 and 0.07 μM, respectively. Biological studies are conducted into the cell-cycle distribution of these conjugates inducing G2/M-phase arrest, apart from an increase in the levels of caspase-3 proteins, followed by apoptotic cell death. A tubulin polymerization assay analysis showed that these compounds effectively inhibit microtubule assembly in HeLa cells and, moreover, Hoechst 33258 and Immunohistochemistry staining suggest that these compounds induce cell death by apoptosis. The docking studies showed that compounds 9c and 9j interact and bind efficiently with the tubulin protein at the colchicine site.
Introduction
Nature has always provided us with a broad range of bioactive products for the treatment of life threatening human ailments such as neurological and immune system disorders, infections and cancer.1 Amongst these, podophyllotoxin (PPT, 1) is one of the most abundant naturally occurring cyclolignans, and it is isolated from Podophyllum peltatum L and Podophyllum hexandrum2 (Fig. 1) and has been extensively used in clinical practice against a variety of malignancies. In contrast to their parent compound, podophyllotoxin, the semi-synthetic derivatives etoposide (2) and teniposide (3) differ substantially in their mechanism of action.3,4 These podophyllotoxins (2 and 3) are DNA topoisomerase II inhibitors since they induce cell death and enhance the topoisomerase II-mediated DNA cleavage via the stabilization of the transient DNA topoisomerase II cleavage complex. In such a complex, DNA is cleaved on both strands and covalently linked to the enzyme. The topoisomerase II inhibition prevents DNA from dissociating5 while podophyllotoxin inhibits the assembly in the microtubulin.6,7 Etoposide has been reported to be toxic and presents several limitations such as moderate potency, poor water solubility, development of drug resistance, metabolic inactivation and other toxic effects.8,9
Fig. 1. Naturally occurring and semisynthetic podophyllotoxin.

These observations have led to several studies on the modification of the structure of etoposide, including etopophos (4) where the aspect of bioavailability is addressed. The most important modification was the substituent in the 4β-position leading to the potent inhibition of topoisomerase II. The replacement of the C-4 sugar unit of etoposide with heterocycles was studied10 and led to the composite pharmacophore model proposed by MacDonald and co-workers,11 which designated the C-4 molecular area of podophyllotoxin (1) as a potential variable region for future investigations. The comparative molecular field analysis (CoMFA) models generated by Lee and co-workers12,13 further demonstrated that bulky substituents at C-4 might be favorable for DNA topo-II inhibition. These postulates are compatible with the excellent activity profiles of NK 611 (5), TOP-53 (6), and GL-331.14 In addition, both GL-331 and TOP-53 showed enhanced DNA topo-II inhibition, as well as antitumor potential, and interestingly the drug-resistance profiles of these were significantly different from those of 1. This recommends the key role of substitution at C4, with respect to the activity profiles in these classes of compounds, and the feasibility of optimizing through rational modification at this position.15
1,2,3-Triazoles are five-membered nitrogen containing heterocyclic compounds; these triazole derivatives, due to their wide spectrum of biological activities, anti-HIV, antifungal, antitubercular and antitumor being prominent among them,16–19 have attracted the interest of medicinal chemists in recent times. They have also been used as building blocks for the synthesis of important bioactive conjugates. Considering the biological implication of triazoles, researchers have used this moiety in the development of various biologically potent motifs. The classic synthesis of the 5-membered triazole ring is accomplished through a synthetic approach known as “click chemistry”.
In our ongoing effort to develop more potent anticancer agents, we have been involved in the development of new synthetic strategies20 for podophyllotoxin-based compounds and construction of a new class of podophyllotoxin congeners as potential anticancer agents.21,22 We decided to exploit the click chemistry tool to construct C-4 1,2,3-triazole linked congeners of podophyllotoxin. There are a considerable number of successful applications of catalytic cycloaddition reactions to the design of biologically active compounds. The [2+3] cycloaddition reaction of azides with acetylenes was utilized to construct 1,2,3-triazole based conjugates. Based on these facts, in this study, we report our investigation on the synthesis of several 4β-aminopodophyllotoxins linked with 1,2,3-triazole via an amide spacer as potential anticancer agents.
Results and discussions
Chemistry
In Scheme 1, 4β-[(amido-4-substituted)-1,2,3-triazol-1-yl]podophyllotoxin hybrids (9a–l) were synthesized by reducing 4β-azido-podophyllotoxin235 with Pd–C to corresponding 4β-aminopodophyllotoxin 6. 4β-Amidopodophyllotoxin 8 was obtained by the coupling of 2-azidoacetic acid 7 and 4β-aminopodophyllotoxin 6 using EDCI/HOBt. The triazolo-conjugates of 4β-amidopodophyllotoxin (9a–l) were obtained in excellent yields by reacting one equivalent of substituted aryl acetylene, 5–10 mol% of sodium ascorbate and 2 mol% of CuSO4·5H2O in t-BuOH : H2O (1 : 2). The list of substitutions and their yields are found in Table 1.
Scheme 1. 4β-[(Amido-4-substituted)-1,2,3-triazol-1-yl] podophyllotoxin hybrids (9a–l).

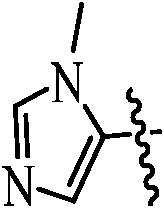
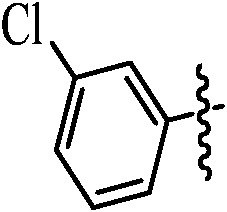
Table 1. Podophyllotoxin 4β-amido [(4-substituted)-1,2,3-triazol-1-yl] derivatives (9a–l).
| Entry | Ar | Yield a (%) | Entry | Ar | Yield a (%) |
| 9a |
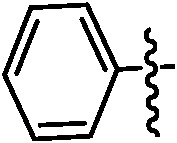
|
91 | 9h |
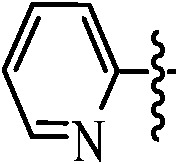
|
89 |
| 9b |
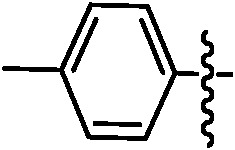
|
89 | 9i |
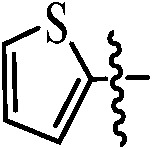
|
91 |
| 9c |
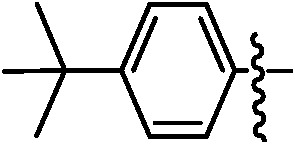
|
92 | 9j |
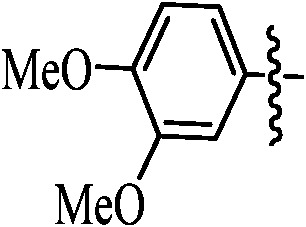
|
85 |
| 9d |
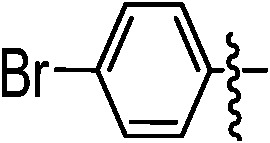
|
89 | 9k |
|
87 |
| 9e |
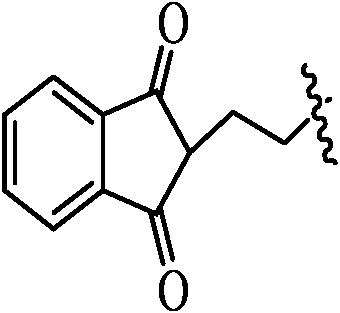
|
86 | 9l |
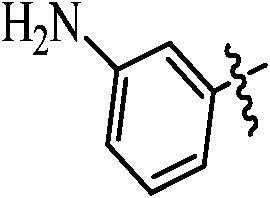
|
88 |
| 9f |
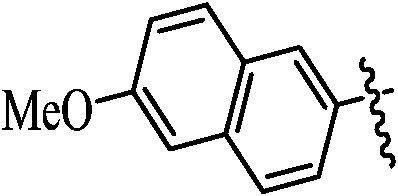
|
85 | |||
| 9g |
|
89 |
aIsolated yields after precipitation in diethyl ether.
All the products were characterized by 1H NMR, 13C NMR and IR spectroscopy and ESI-MS. In 1H NMR spectroscopy, the cyclization of azides to triazoles was confirmed by the resonance of H-5 in the triazole ring in the aromatic region and that of C4α-H around δ 6.0. 13C NMR spectroscopy also supported this structure, which showed all the carbon signals corresponding to triazole derivatives. ESI-MS of all the compounds showed an M + Na or M + K adduct ion as the parent molecular ion.
Biological evaluation
Cytotoxic activity
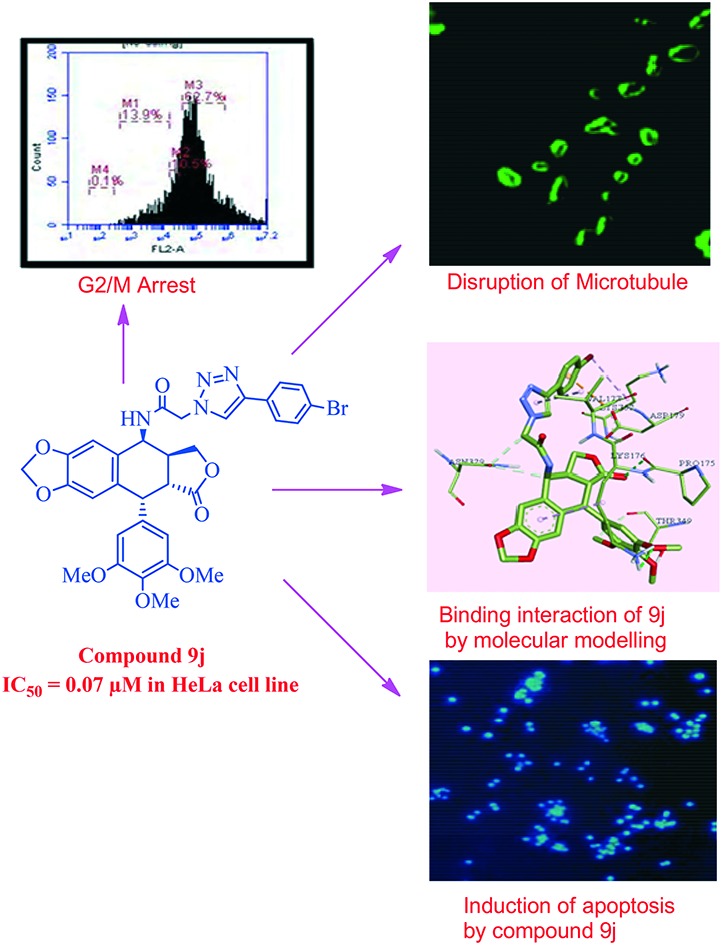
These synthesized compounds were evaluated for their cytotoxic activity against four human cancer cell lines, namely MCF-7 (breast cancer), B16 (oral cancer), HT 29 (colon cancer) and HeLa (cervical cancer), by employing MTT assay and using podophyllotoxin and etoposide as the reference drugs. The structure activity relationship (Fig. 2) was examined using the cytotoxicity values from Table 2 of the HeLa (cervical cancer) cell line. The synthesized scaffold consists of 4 rings, for ease we have named them A, B, C and D, and whilst rings A–C were kept constant, changes were made at the D ring. For compounds 9a, 9b and 9c, the ring was not substituted with a weak electron donating group like a methyl or tertiary butyl group. It was observed that the cytotoxicity was greater for compound 9c compared to 9b and least for compound 9a, in which the substituents were absent. The cytotoxicity was enhanced by 15 folds for compound 9j (0.07 μM) compared to podophyllotoxin (2.036 μM) when an electron withdrawing atom like bromine was introduced at the fourth position of the ring. Moreover when a hetero aromatic ring, such as pyridine and thiophene, was introduced in place of the benzene ring in compounds like 9h and 9i, the cytotoxicity increased by 3 folds. Whereas in compounds 9e and 9k, where the ring was replaced by a pyrazole or an indinone ring, no significant change was observed in comparison to podophyllotoxin. An observation regarding the substituents on ring D suggests that the potency is maximum when either a bromine or a tertiary butyl group is present on ring D, and the potency decreases with other donating and withdrawing groups.
Fig. 2. The structure activity relationship of triazolo linked podophyllotoxin conjugates.

Table 2. The cytotoxic activity (IC50) a of the podophyllotoxin 4β-amido [(4-substituted)-1,2,3-triazol-1-yl] podophyllotoxin derivatives (9a–l).
| Compound | MCF-7 b | B16 c | HT29 d | HeLa e |
| 9a | 8.1 | 21.5 | 20.1 | 19.6 |
| 9b | 17.1 | 23.5 | 22.5 | 16.8 |
| 9c | 11.3 | 10.6 | 18.6 | 0.9 |
| 9d | 15.8 | 22.4 | 13.9 | 17.9 |
| 9e | 22.4 | 10.8 | 10.6 | 2.3 |
| 9f | 21.9 | 22.5 | 15.6 | 25.4 |
| 9g | 38.2 | 23.6 | 17.5 | 28.4 |
| 9h | 14.5 | 19.5 | 18.4 | 23.6 |
| 9i | 8.9 | 18.2 | 19.5 | 6.5 |
| 9j | 0.9 | 10.1 | 0.1 | 0.07 |
| 9k | 7.2 | 14.7 | 18.5 | 2.6 |
| 9l | 6.9 | 14.3 | 13.6 | 15.4 |
| Podophyllotoxin | 3.75 | 2.12 | 1.26 | 2.36 |
| Etoposide | 1.36 | 1.85 | 1.98 | 2.24 |
a50% Inhibitory concentration after 48 h of drug treatment.
bHuman breast cancer.
cMacrophage cancer.
dHuman colon cancer.
eHuman cervical cancer.
Effect on cell cycle arrest
In general, the G2/M cell cycle arrest is strongly associated with the inhibition of tubulin polymerization, and it is well established that podophyllotoxin derivatives cause G2/M arrest in cell cycles. The effect of compound 9c and 9j on cell cycle progression was investigated in HeLa cells by flow cytometry.24 In this study HeLa cells were treated with the conjugates 9c and 9j at 0.5 μM for 48 h. As shown in Fig. 3 and Table 3, the percentages of cells in the G2/M phase were 63.3% and 62.7% when the cells were treated with compounds 9c and 9j for 48 h at a concentration of 0.5 μM, respectively. In the control (untreated cells) 19.3% of accumulation in the G2/M phase was observed. It was clearly demonstrated that compounds 9c and 9j caused a G2/M arrest, and this was consistent with the nature of tubulin-binding agents.
Fig. 3. The effects of the compound control, 9c and 9j on the DNA content/cell following the treatment of HeLa cells at 0.5 μM for 48 h. The cell cycle distribution was analysed by the standard propidium iodide procedure as described in the experimental section.

Table 3. The distribution of A549 cells in various phases of the cell cycle.
| Compound | % of cells in sub-G1 phase | % of cells in G1 phase | % of cells in S phase | % of cells in G2/M phase |
| Control | 0.9 | 62.5 | 11.3 | 19.3 |
| 9c | 0.2 | 12.5 | 10.1 | 63.3 |
| 9j | 0.1 | 13.9 | 10.5 | 62.7 |
Immunohistochemistry studies on tubulin
In order to substantiate the observed effects of these compounds on the inhibition of tubulin polymerization to functional microtubules, immunohistochemical25 studies have been carried out to examine the in situ effects of compounds 9c and 9j on the cellular microtubules in HeLa cancer cells. Therefore, the HeLa cells were treated with 9c and 9j at 0.5 μM concentration for 48 h. In this study, the untreated human cervical cancer cells displayed a normal distribution of microtubules (Fig. 4). However, the cells treated with compounds 9c and 9j showed a disrupted microtubule organization, as shown in Fig. 4, which indicates the inhibition of tubulin polymerization.
Fig. 4. IHC analyses of the compounds in the microtubule network: the HeLa cells were treated with compounds 9c and 9j at 0.5 μM concentration for 48 h, followed by staining with an α-tubulin antibody. Microtubule organization was clearly observed by the green colored tubulin network like structures in the control cells and was found to be disrupted in cells treated with compounds 9c and 9j.

Effect of compounds on tubulin polymerization
One of the possibilities was that these conjugates may exhibit cytotoxic activity as well as a G2/M cell cycle arrest by the inhibition of tubulin polymerization,26 since this has been observed in many cases of antimitotic agents. Hence it was considered of interest to investigate the tubulin polymerization aspect. As tubulin subunits heterodimerize and self-assemble to form microtubules in a time dependent manner, we have investigated the progression of tubulin polymerization by monitoring the increase in fluorescence emission at 420 nm (excitation wavelength is 360 nm) in a 384 well plate for 1 h at 37 °C with and without the conjugates 9c and 9j at 3 μM concentration. The conjugates 9c and 9j inhibited tubulin polymerization by 46.40%, and 48.50%, respectively, compared to control podophyllotoxin, which was used as a positive control and showed 50.19% inhibition (Fig. 5).
Fig. 5. The effect on tubulin polymerization: a tubulin polymerization assay was carried out in a reaction mixture that contained PEM buffer and GTP (1 mM) in the presence or absence of the test compounds (9c and 9j) at 3 μM concentration. The reaction was initiated by the addition of GTP to all the wells. Tubulin polymerization was monitored by the increase in fluorescence at 420 nm (excitation wavelength is 360 nm), which was measured for 1 h at 1 min intervals in a multimode plate reader (Tecan) at 37 °C.

Caspase-3 activation
It is known that the cell cycle arrest at the G2/M phase is shown to induce cellular apoptosis, hence it was considered of interest to examine whether the cytotoxicity of 9c and 9j is by virtue of apoptotic cell death.27 In the cysteine aspartase group, caspases in particular play a crucial role in the induction of apoptosis, and amongst them caspase-3 happens to be one of the effector caspases. This prompted the treatment of HeLa cells with compounds 9c and 9j to examine the activation of caspase-3. The results indicate that there is a nearly 4 to 6 fold induction in the caspase-3 activity in cells treated with 0.5 μM concentration of these compounds (Fig. 6). However induction was observed with triazolo linked podophyllotoxin, thus suggesting the activation of caspase-3 by 9c and 9j and indicating that they have the ability to induce apoptosis in HeLa cells.
Fig. 6. The effect of podophyllotoxin conjugates on the caspase 3 protein activity. The HeLa cells were treated with the indicated compounds at 0.5 μM for 48 h and then subjected to fluorimetry, as explained in detail in the experimental section.

Hoechst staining for apoptosis
It is well known that apoptosis has been one of the major pathways that leads to the process of cell death. Chromatin condensation and fragmented nuclei are known as the classic characteristics of apoptosis.28 Thus we investigate the apoptotic inducing effect of 9c and 9j by the Hoechst staining (H33258) method in the HeLa cancer cell line. The HeLa cancer cells were treated with 9c and 9j at 0.5 μM concentrations for 48 h. The manual field quantification of the apoptotic cells based on cytoplasmic condensation, the presence of apoptotic bodies, nuclear fragmentation and the relative fluorescence of the test compounds revealed that there was significant increase in the apoptotic cells, as seen in (Fig. 7).
Fig. 7. Hoechst staining in the HeLa cervical cancer cell line; control cells, and 0.5 μM 9c and 9j.

Molecular modelling study
In silico docking studies were performed to gather information about the binding profile of the two most potent molecules of the series, viz. compounds 9c and 9j, in the colchicine binding site of the tubulin dimer. The three-dimensional crystal structure of tubulin was taken from the RCSB Protein Data Bank (PDB ID ; 3E22) for the docking studies, and Auto dock 4.2 was used for the docking study.29 As a part of the protein preparation, all natural ligands and water molecules were removed from the protein. All polar hydrogen bonds were added to the protein. Throughout the docking study, the protein was kept rigid and the ligand was kept flexible. Kollman and Gasteiger charges were added wherever necessary. A grid box was generated with a box size of 25 Å in each dimension, with a grid spacing of 1.0 Å. The center of the box was chosen as the center of the tubulin active site, which had a large enough space to accommodate all the possible ligand conformations within the box. The maximum number of binding modes saved was set to 10. The conformation with the lowest binding energy was used and assumed to be the best docked. The docking results were viewed in Discovery Studio 2016.30 The study indicated that both these compounds bind at the interface of the α/β chains of the tubulin dimer.
Compound 9c exhibited a greater number of interactions than 9f with the amino acid residues at the binding site. Classical hydrogen bonding was seen between various groups on the compound and the amino acid residues of the target protein. The trimethoxy substituents on the phenyl ring formed strong hydrogen bonds with Tyr210, and the carbonyl oxygen binds with Ile332 and Gly350. Non-conventional hydrogen bonds are observed between 9c and some amino acid residues such as Ser77, Gln11, Asn249, Lys352, Ser178, Val177, Phe351, Gly350, Thr349, Leu248, Pro222, Tyr224, Gly225, Tyr357, Ala247, Val353, and Tyr223, as shown in Fig. 8a. Apart from these, π-bonds were observed between the triazole ring and Thr349; the phenyl ring substituted on the triazole moiety formed bonds with Asp179. Furthermore Π-alkyl bonds were observed between the trimethyl tertiary carbon and Val353.
Fig. 8. a: The interactions of compound 9c, the carbon atoms are shown in green, oxygen atoms in red and nitrogen atoms in violet. b: The interactions of compound 9j, the carbon atoms are shown in green, oxygen atoms in red and nitrogen atoms in violet.

Compound 9j also exhibited varying interactions with the residues in the binding pocket. Ala333 formed a hydrogen bond with the bromine substituted phenyl ring. The oxygen of the amide is seen bound to Val177 via a strong hydrogen bond. Pro175 is seen to form hydrogen bonds with the nitrogen of the amide. Various other amino acid residues are involved in the non-conventional hydrogen bond interactions with the compound including Thr349, Lys336, Tyr224, Asn249, Val353, Ser178, Tyr210, Asn329, Lys176 and Pro175, as shown in Fig. 8b. Other aryl and alkyl π-stacking interactions were observed between the triazole ring and Val177, phenyl ring bearing trimethoxy substituents with Pro348 and Pro175, phenyl ring bearing bromine substituent with Ala333 and oxygen of the furan ring with Asp179.
Conclusion
In conclusion, the most active hybrids 9c and 9j induce apoptotic cell death by the inhibition of tubulin polymerization leading to cell cycle arrest at the G2/M phase of the cell cycle followed by up regulation of the caspase-3 activity. The Hoechst 33258 staining and immunohistochemistry also suggests that 9c and 9j induce cell death by apoptosis. The tubulin polymerization assay showed that these compounds effectively inhibit the microtubule assembly at both the molecular and cellular level in HeLa cells. The molecular modeling experiment displayed the binding mode for the interaction of compounds 9c and 9j with the colchicine binding site, and based on these results it is evident that these new podo hybrids, particularly 9c and 9j, have the potential to be developed as a new class of anticancer agents by further structural modifications. Overall, the current study demonstrates the synthesis of 1,2,3-triazolo linked podophyllotoxin conjugates as promising cytotoxic agents that cause G2/M arrest and induce apoptosis. It is interesting to observe that in our previous study, 4β-[4′-(1-(aryl)ureido)benzamide]podophyllotoxins31 were shown to demonstrate significant G1 cell cycle arrest in comparison to the triazole linked podophyllotoxin conjugates of the present investigation that induce apoptosis by targeting tubulin. Moreover these compounds inhibit microtubule assembly in HeLa cells. Therefore they deserve further detailed investigations for exploitation as newer tubulin binding agents that will lead to apoptotic cell death.
Conflict of interest
The authors declare no competing interests.
Supplementary Material
Acknowledgments
The authors S. R. V. and M. V. P. S. V. are thankful to CSIR and DST, India, respectively, for the award of research fellowships. We also thank the International Scientific Partnership program ISPP at the King Saud University for funding this research work through ISPP#0054.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00273d
References
- Slevin L. M. L. Cancer. 1991;67:319–329. doi: 10.1002/1097-0142(19910101)67:1+<319::aid-cncr2820671319>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Hande K. R. Eur. J. Cancer. 1998;34:1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- Caner B. A. and Longo D. L., Cancer therapy and biotherapy, Lippincott-Raven Publishers, New York, 1996. [Google Scholar]
- Yuan P., Xu B. H., Wang J. Y., Ma F., Fan Y., Li Q., Zhang P. J. Clin. Med. 2012;125:79–139. [Google Scholar]
- Burden D. A., Osheroff N. Biochim. Biophys. Acta, Gen. Subj. 1998;1400:139–154. doi: 10.1016/s0167-4781(98)00132-8. [DOI] [PubMed] [Google Scholar]
- Desben S., Giorgi-Renault S. Curr. Med. Chem. 2002;2:71–90. doi: 10.2174/1568011023354353. [DOI] [PubMed] [Google Scholar]
- Leroy D., Kajava A. V., Frei C., Gasser S. M. Biochemistry. 2001;40:1624–1634. doi: 10.1021/bi0019141. [DOI] [PubMed] [Google Scholar]
- Kobayashi K., Ratain M. J. Cancer Chemother. Pharmacol. 1994;34:S64–S68. doi: 10.1007/BF00684866. [DOI] [PubMed] [Google Scholar]
- Chang J. Y., Han F. S., Liu S. Y., Wang Z. Q., Lee K. H., Cheng Y. C. Cancer Res. 1991;51:195–199. [PubMed] [Google Scholar]
- Moraes R. M., Dayan F. E., Canel C. Stud. Nat. Prod. Chem. 2002;26:149. [Google Scholar]
- Mac Donald T. L., Lehnert E. K., Loper J. T., Chow K. C. and Ross W. E., DNA Topoisomerases in cancer, Oxford University Press, New York, 1991, pp. 119–214.
- Cho S. J., Tropsha A., Suffness M., Cheng Y. C., Lee K. H. J. Med. Chem. 1996;39:1383–1395. doi: 10.1021/jm9503052. [DOI] [PubMed] [Google Scholar]
- Xiao Z., Xiao Y. D., Feng J., Golbraikh A., Tropsha A., Lee K. H. J. Med. Chem. 2002;45:2294–2309. doi: 10.1021/jm0105427. [DOI] [PubMed] [Google Scholar]
- Terada T., Fujimoto K., Nomura M., Yamashita J., Wierzba K., Yamazaki R., Shibata J., Sugimoto Y., Yamada Y., Kobunai T., Takeda S., Minami Y., Yoshida K., Yamaguchi H. J. Med. Chem. 1993;36:1689–1699. doi: 10.1021/jm00064a002. [DOI] [PubMed] [Google Scholar]
- (a) Kamal A., Kumar B. A., Arifuddin M., Dastidar S. G. Bioorg. Med. Chem. 2003;11:5135–5142. doi: 10.1016/j.bmc.2003.08.019. [DOI] [PubMed] [Google Scholar]; (b) Kamal A., Gayatri N. L., Reddy D. R., Reddy P. S. M. M., Arifuddin M., Dastidar S. G., Kondapi A. K., Rajkumar M. Bioorg. Med. Chem. 2005;13:6218–6225. doi: 10.1016/j.bmc.2005.06.032. [DOI] [PubMed] [Google Scholar]; (c) Kamal A., Laxman E., Khanna G. B. R., Reddy P. S. M. M., Rehana T., Arifuddin M., Neelima K., Kondapi A. K., Dastidar S. G. Bioorg. Med. Chem. 2004;12:4197–4201. doi: 10.1016/j.bmc.2004.05.026. [DOI] [PubMed] [Google Scholar]; (d) Kamal A., Kumar B. A., Arifuddin M., Dastidar S. G. Lett. Drug Des. Discovery. 2006;3:205–209. [Google Scholar]; (e) Kamal A. and Kumar B. A., Patent WO136018, 2008.; (f) Kamal A., Kumar B. A. and Arifuddin M., Patent WO07339, 2004.; (g) Kumar B. A., Ph.D. Thesis, Osmania University, 2006. [Google Scholar]
- Kamal A., Rao A. V. S., Vishnuvardhan M. V. P. S., Reddy T. S., Swapna K., Bagul C., Reddy N. V. S., Srinivasulu V. Org. Biomol. Chem. 2015;13:4879–4895. doi: 10.1039/c5ob00232j. [DOI] [PubMed] [Google Scholar]
- Reddy T. S., Kulhari H., Reddy V. G., Rao A. V. S., Bansal V., Kamal A., Shukla R. Org. Biomol. Chem. 2015;13:10136–10149. doi: 10.1039/c5ob00842e. [DOI] [PubMed] [Google Scholar]
- Thomas K. D., Adhikari A. V., Chowdhury I. H., Sumesh E., Pal N. K. Eur. J. Med. Chem. 2011;46:2503–2512. doi: 10.1016/j.ejmech.2011.03.039. [DOI] [PubMed] [Google Scholar]
- He R., Chen Y., Chen Y., Ougolkov A. V., Zhang J. S., Savoy D. N., Billadeau D. D., Kozikowski A. P. J. Med. Chem. 2010;53:1347–1356. doi: 10.1021/jm901667k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Kamal A., Laxman N., Ramesh G. Bioorg. Med. Chem. Lett. 2000;10:2059–2062. doi: 10.1016/s0960-894x(00)00407-8. [DOI] [PubMed] [Google Scholar]; (b) Kamal A., Kumar B. A., Arifuddin M. Tetrahedron Lett. 2004;44:8457–8459. [Google Scholar]
- (a) Kamal A., Kumar B. A., Arifuddin M., Dastidar S. G. Bioorg. Med. Chem. 2003;11:5135–5142. doi: 10.1016/j.bmc.2003.08.019. [DOI] [PubMed] [Google Scholar]; (b) Kamal A., Suresh P., Mallareddy A., Kumar B. A., Reddy P. V., Raju P., Tamboli J. R., Shaik T. B., Jain N., Kalivendi S. V. Bioorg. Med. Chem. 2011;19:2349–2358. doi: 10.1016/j.bmc.2011.02.020. [DOI] [PubMed] [Google Scholar]; (c) Kamal A., Kumar B. A., Suresh P., Juvekar A., Zingde S. Bioorg. Med. Chem. 2011;19:299–2979. doi: 10.1016/j.bmc.2011.03.030. [DOI] [PubMed] [Google Scholar]; (d) Kamal A., Suresh P., Ramaiah M. J., Mallareddy A., Kumar B. A., Raju P., Gopal J. V., Pushpavalli S. N. C. V. L., Lavanya A., Sarma P., Bhadra M. P. Bioorg. Med. Chem. 2011;19:4589–4600. doi: 10.1016/j.bmc.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Kamal A., Azeeza S., Bharathi E. V., Malik M. S., Shetti R. V. C. R. N. C. Mini-Rev. Med. Chem. 2010;10:405–435. doi: 10.2174/138955710791330918. [DOI] [PubMed] [Google Scholar]
- (a) Kamal A., Gayatri N. L., Rao N. V. Bioorg. Med. Chem. Lett. 1998;8:3097. doi: 10.1016/s0960-894x(98)00570-8. [DOI] [PubMed] [Google Scholar]; (b) Kamal A., Laxminarayana B., Gayatri N. L. Tetrahedron Lett. 1997;38:6871. [Google Scholar]; (c) Kamal A., Kumar B. A., Arifuddin M., Dastidar S. G. Bioorg. Med. Chem. 2003;11:5135. doi: 10.1016/j.bmc.2003.08.019. [DOI] [PubMed] [Google Scholar]
- Reddy M. A., Jain N., Yada D., Kishore C., Reddy V. J., Reddy P. S., Anthony A., Kalivendi S. V., Sreedhar B. J. Med. Chem. 2011;54:6751–6760. doi: 10.1021/jm200639r. [DOI] [PubMed] [Google Scholar]
- Jain N., Yada D., Shaik T. B., Vasantha G., Reddy P. S., Kalivendi S. V., Sreedhar B. ChemMedChem. 2011;6:859–868. doi: 10.1002/cmdc.201100019. [DOI] [PubMed] [Google Scholar]
- Cormier A., Marchand M., Ravelli R. B., Knossow M., Gigant B. EMBO Rep. 2008;9:1101–1106. doi: 10.1038/embor.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne L. J., Gude C., Rodriguez H., Steele R. E., Bhatnager A. J. Med. Chem. 1991;34:725–736. doi: 10.1021/jm00106a038. [DOI] [PubMed] [Google Scholar]
- Shankar R., Chakravarti B., Singh U. S., Ansari M. I., Deshpande S., Dwivedi S. K. D., Bid H. K., Konwar R., Kharkwal G., Chandra V., Dwivedi A., Hajela K. Bioorg. Med. Chem. 2009;17:3847–3856. doi: 10.1016/j.bmc.2009.04.032. [DOI] [PubMed] [Google Scholar]
- Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. J. Comput. Chem. 2009;16:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassault Systèmes BIOVIA, DS Visualizer Client, v16.1.0.15350, Dassault Systèmes, San Diego, 2016.
- Kamal A., Suresh P., Janaki Ramaiah M., Srinivasa Reddy T., Ravi Kumar K., Narasimha Rao B., Imthiajali S., Lakshminarayan Reddy T., Pushpavalli S. N. C. V. L., Shankaraiah N., Bhadra M. P. Bioorg. Med. Chem. 2013;21:5198–5208. doi: 10.1016/j.bmc.2013.06.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.