

Abstract
Aim
Junctophilins (JPs), a protein family of the junctional membrane complex, maintain the close conjunction between cell surface and intracellular membranes in striate muscle cells mediating the crosstalk between extracellular Ca2+ entry and intracellular Ca2+ release. The small‐conductance Ca2+‐activated K+ channels are activated by the intracellular calcium and play an essential role in the cardiac action potential profile. Molecular mechanisms of regulation of the SK channels are still uncertain. Here, we sought to determine whether there is a functional interaction of junctophilin type 2 (JP2) with the SK channels and whether JP2 gene silencing might modulate the function of SK channels in cardiac myocytes.
Methods
Association of JP2 with SK2 channel in mouse heart tissue as well as HEK293 cells was studied using in vivo and in vitro approaches. siRNA knockdown of JP2 gene was assessed by real‐time PCR. The expression of proteins was analysed by Western blotting. Ca2+‐activated K+ current (I K,Ca) in infected adult mouse cardiac myocytes was recorded using whole‐cell voltage‐clamp technique. The intracellular Ca2+ transient was measured using an IonOptix photometry system.
Results
We showed for the first time that JP2 associates with the SK2 channel in native cardiac tissue. JP2, via the membrane occupation and recognition nexus (MORN motifs) in its N‐terminus, directly interacted with SK2 channels. A colocalization of the SK2 channel with its interaction protein of JP2 was found in the cardiac myocytes. Moreover, we demonstrated that JP2 is necessary for the proper cell surface expression of the SK2 channel in HEK293. Functional experiments indicated that knockdown of JP2 caused a significant decrease in the density of I K,Ca and reduced the amplitude of the Ca2+ transient in infected cardiomyocytes.
Conclusion
The present data provide evidence that the functional interaction between JP2 and SK2 channels is present in the native mouse heart tissue. Junctophilin 2, as junctional membrane complex (JMC) protein, is an important regulator of the cardiac SK channels.
Keywords: cardiac myocyte, Junctophilins, siRNA, SK2 channels
1. INTRODUCTION
Junctophilins are a protein family of the junctional membrane complex (JMC), which provides a bridge for the signal transduction in excitable cells between plasma membrane ion channels and intracellular calcium release channels by anchoring the endo/sarcoplasmic reticulum (ER/SR) to the plasma membrane/T‐tubules.1 Among JPs, JP1 is predominantly expressed in skeletal muscle, JP2 is expressed in cardiac, skeletal and smooth muscles, and JP3 and JP4 are mainly present in nervous system.1, 2 Both JP1 and JP2 may be involved in regulating Ca2+ homoeostasis, which aids to excitation‐contraction coupling (e‐c coupling) mechanism.3, 4, 5 JP2 dysfunction has been linked to defects in integral ion channel expression, abnormal intracellular Ca2+ handing and heart disease.6, 7
Small‐conductance Ca2+‐activated K+ channels or SK channels, one family of Ca2+‐activated K+ channels, are widely expressed in various species, including mouse, rat and humans.8, 9, 10 These channels play fundamental roles in regulating cellular excitability.9, 10, 11 The SK channels consist of three subtypes, SK1, SK2, and SK3, encoded by the genes (KCNN1‐KCNN3).9, 12 In cardiac myocytes, the SK channels contribute to the repolarization of the cardiac action potential (AP).13 Knockout of the SK2 channel mouse displayed AP prolongation and atrial fibrillation (AF).14, 15 Inhibition of SK channels by specific inhibitors prolonged AP duration.16, 17, 18 Increased evidences have indicated that the SK2 channel participates in the remodelling in AF19, 20, 21, 22 and heart failure (HF) due to the increased sensitivity of SK channels to intracellular Ca2+ in failing heart.23, 24, 25 Other studies have documented that the dysfunction of the SK2 channel in atrioventricular nodal cells resulted in shortening of spontaneous APs.26 The inhibition of the SK2 channel decreases the firing rate and prolongs APD in sinoatrial node cells.27 Hence, the understanding of the SK channels in the regulation of the cardiac excitability has increasingly become the focus of new therapeutic strategies for the treatment of cardiac arrhythmia.
Ion channels are membrane protein complexes, which significantly affect the channel localization, trafficking, activation and regulation.28 Recent studies have revealed the physiological significance of JPs coupling ion channels.29, 30, 31 JP1 and JP2 could interact with the dihydropyridine receptor (DHPR) that controls Ca2+ homoeostasis and e‐c coupling.31 JP2 was shown to be interacted with TRPC3, a Ca2+‐permeate channel and be functionally linked to ryanodine receptors (RyRs) Ca2+ release in muscle.30, 32 JPs may be an important regulator of the functional interaction between STIM1‐Orai1 protein system and voltage‐gated Ca2+ entry, as demonstrated for Cav1.2 channels.33, 34 Available results highlight the existence of proper interactions between JPs, Ca2+ channels and RyRs.32 We therefore hypothesized the role of JP2 for targeting a Ca2+‐activated K+ channels (SK2 channel) on the plasma membrane in the heart. We investigated (i) an association of JP2 with SK2 channel in native heart tissue as well as in HEK293 cells and (ii) the effects of the knockdown of JP2 on the expression and function of SK2 channel in the mouse cardiomyocytes.
2. RESULTS
2.1. JP2 associates with SK2 channel in native cardiac tissue
Junctophilins are an essential JMC protein which functions as a bridge between transverse tubules and junctional sarcoplasmic reticulum (jSR), the constituents of “dyad”, in cardiac muscle. To define the functional interactome of SK2 channel in the heart, we documented whether JPs interact with ion channels on the T‐tubule/plasma membrane, like the SK channels. Co‐immunoprecipitation experiments were performed in the native mouse heart tissue (Figure 1A) as well as HEK293 cells transfected with SK2 and JP2 (Figure 1B). Anti‐JP2 antibody which was bound to protein A‐agarose precipitated SK2 protein from mouse heart tissues as detected on immunoblots using anti‐SK2 antibody as shown in Figure 1A (right panel) as protein bands at ≈60 kDa. Data indicated that SK2 selectively bound to endogenous JP2 in the mouse heart. Similarly, in the reverse co‐IP, experiments are shown in Figure 1A. Using anti‐SK2 antibody bound to protein A‐agarose to precipitate the detergent‐solubilized protein from the mouse heart tissue, JP2 protein could be detected on immunoblots with anti‐JP2 antibody as shown in Figure 1A (left panel) as protein bands at ≈97 kDa. No specific immunoreactions were observed in the immunoprecipitates of the controls using an unrelated antibody (Figure 1A, Ctrl‐N in left and right panels). These results revealed that JP2 specifically bound to endogenous SK2 in the heart lysates. In our previous study, we demonstrated co‐immunoprecipitation of RyR2 and the SK2 channel in the heart.35 Interestingly, we observed that JP2 was also immunoprecipitated with an antibody against RyR2 (Figure 1C). Take together; these results indicate the specific interaction between JP2 and SK2 in native cardiac tissue. JP2 may form a complex of SK2 with RyR2. The SK2 may be part of a complex made up by JP2 and RyR2.
Figure 1.
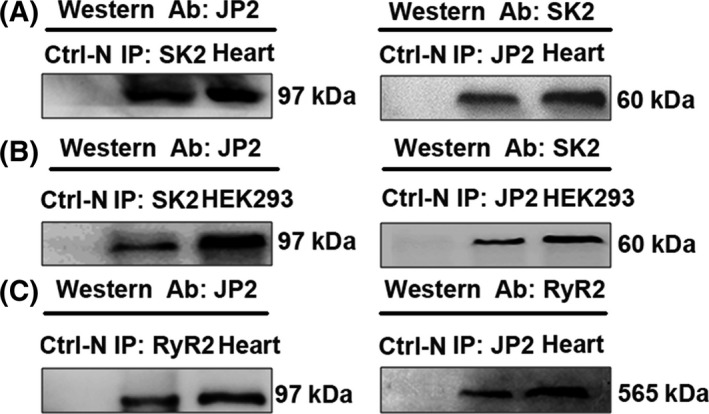
JP2 associates with SK channels in native cardiac tissue and HEK293 cells. A, Solubilized proteins from the mouse heart tissue were immunoprecipitated (IP) with anti‐SK2 (left panel) or anti‐JP2 (right panel) antibodies. Specific anti‐JP2 (left panel) and anti‐SK2 antibodies (right panel) were used to recognize JP2 (MW~97 kDa) and SK2 (~60 kDa) by Western blotting. Ctrl‐N (right and left panels) indicates the negative control using a non‐immobilized gel. B, JP2 co‐IP with SK2 in HEK293 cells transfected with SK2 channel and JP2. Solubilized proteins from transfected HEK293 cells were immunoprecipitated with anti‐SK2 (left panel) or anti‐JP2 (right panel) antibodies. Specific anti‐JP2 (left panel) and anti‐SK2 antibodies (right panel) were individually used to detect JP2 and SK2 channels with Western blot analysis. C, JP2 associates with RyR2 in native heart tissue. Lysates from the mouse heart tissue were immunoprecipitated with anti‐RyR2 (left panel) or anti‐JP2 (right panel) antibodies. Specific anti‐JP2 (left panel) and anti‐RyR2 antibodies (right panel) were individually used to recognize JP2 and RyR2 (~565 kDa) by Western blotting
To confirm whether the interaction between JP2 and SK2 may associate with other specific proteins, we performed a similar set of co‐IP experiments in HEK293 cells transfected with plasmids containing pEGFP‐SK2 and pcDNA‐JP2 (Figure 1B). Solubilized proteins from transfected HEK293 cells were immunoprecipitated with the anti‐SK2 antibody and subsequently immunoblotted with the anti‐JP2 antibody (Figure 1B, left panel). As shown in Figure 1B, JP2 was immunoprecipitated with an antibody against SK2, but was absent in the samples incubated with an unrelated antibody (Figure 1B, Ctrl‐N in left and right panels). The interaction between JP2 and SK2 was also confirmed in immunoprecipitation experiments performed with antibody directed against JP2 followed by immunoblotted with antibody against SK2 (Figure 1B, right panel). These results confirm a direct interaction between JP2 and SK2, indicating that the interaction between two proteins in the HEK293 cells does not associate with other specific proteins.
2.2. Molecular interaction between Junctophilin 2 and SK2 in Vitro
We further attempted to evaluate whether JP2 selectively binds to SK2 in mouse cardiac muscle using GST pulldown experiments. Purified GST and GST‐JP2 fusion proteins were immobilized on glutathione‐Sepharose 4B affinity beads and then incubated for 6 hours at 4°C with 500 μg of proteins solubilized from the heart lysates. As shown in Figure 2B, Western blot analysis with an antibody against SK2 revealed that GST‐JP2 fusion proteins were able to pull down the endogenous SK2, whereas GST alone did not appear to pull down the endogenous SK2. A similar pulldown assays were performed with the cell lysates from HEK293 cells transfected with SK2 as shown in Figure 2C, indicating that GST‐JP2 selectively bound to SK2, but GST alone did not.
Figure 2.
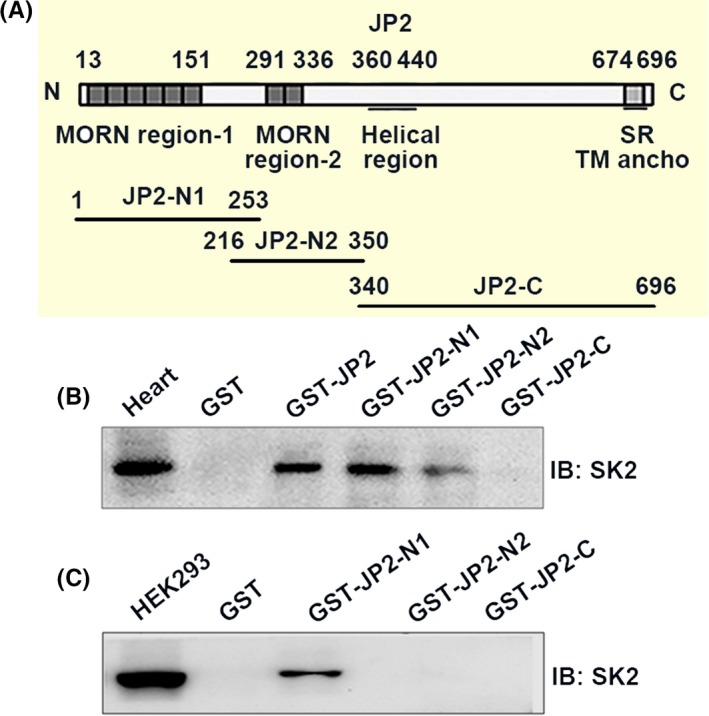
Interaction between JP2 and SK2 in vitro. A, Schematic diagrams of mouse full‐length JP2 and three JP2 constructs. B, GST fusion proteins containing GST‐JP2 and GST‐JP2 constructs (GSTJP2‐N1, GSTJP2‐N2 and GSTJP2‐C) were performed to pull down the mouse heart lysate followed by immunoblotting with an anti‐SK2 antibody. Positive bands displayed interactions of GST‐JP2 with SK2 and GSTJP2‐N1 with SK2. No interaction was observed between SK2 and GSTJP2‐N2, GST‐JP2‐C or GST alone. C, GST fusion proteins were performed to pull down transfected HEK293 cells followed by immunoblotting with an anti‐SK2 antibody. Positive bands showed the binding of GST‐JP2 with SK2 and GST‐JP2‐N1 with SK2. No interaction was observed between SK2 and GST‐JP2‐N2, GST‐JP2‐C or GST alone
Next, we identified the specific residues within JP2 for binding with the cardiac SK2 channel. Previous studies have suggested that JPs contain the MORN motifs within the N‐terminal of JP2 that is highly conserved among species, postulated to play a role in the targeting of ion channels on the surface membrane.1, 2 Thus, we prepared three constructs based on full‐length sequences of JP2 to define the requirements on the MORN motifs of JP2 for the SK2 channel binding (Figure 2A). They are individually named as JP2‐N1 (aa 1‐253 containing the MORN I region of JP2), JP2‐N2 (aa 216‐350 containing the MORE II region) and JP2‐C (aa 340‐696 containing the C‐terminal region of JP2). GST pulldown assays were again performed with three GST fused constructs, GST‐JP2‐N1 (aa 1‐253), GST‐JP‐N2 (aa 216‐350) and GST‐JP2‐C (aa 340‐696). As shown in Figure 2B, GST‐JP2‐N1, but not GST‐JP2‐C and GST alone, was able to pull down the endogenous SK2 protein, as revealed by Western blotting with the anti‐SK2 antibody. GST‐JP2‐N2 shows weak binding with SK2. A similar pulldown assays were performed on the HEK293 cells transfected with the SK2 channel (Figure 2C). When three GST fused constructs were immobilized on glutathione column and then incubated with solubilized proteins prepared from the cell lysates, GST‐JP2‐N1 selectively bound to SK2, as revealed by Western blotting with the anti‐SK2 antibody, whereas GST‐JP‐C and GST alone did not appear to bind SK2 (Figure 2C). Our data indicated that the N1 domain, 1‐253‐amino acid region of JP2, contains the majority of the binding sites for SK2, suggesting that the binding site could comprise the MORN I motifs of JP2.
2.3. Colocalization of JP2 and SK2 in adult mouse cardiomyocytes
To test the physiological association between JP2 and SK2, we assessed whether these proteins are colocalized in the same cellular compartments in isolated mouse cardiac myocytes. Confocal imaging showed that the JP2 protein was distributed in striated patterns (A and B, Figure 3) in single isolated cardiac cells, which have regularly spaced transverse striations corresponding to the positions of the Z‐lines in the ventricular myocytes (B, Figure 3) or Z‐tubules in the atrial myocytes (A, Figure 3).7, 33 In agreement with our previous work,27 positive SK2 staining was predominantly distributed along the Z‐lines or Z‐tubules (A and B, Figure 3) and partial colocalized with JP2, as observed via double staining. The clear colocalization of JP2 and the SK2 channel was found at the plasma membrane of HEK 293 cells cotransfected with the plasmids containing JP2 and SK2 as shown in Figure 3D. These data demonstrated the clear colocalization of JP2 and the SK2 channel in the cardiac myocytes and mammalian cells, supporting the interaction between JP2 protein and the SK2 channel.
Figure 3.
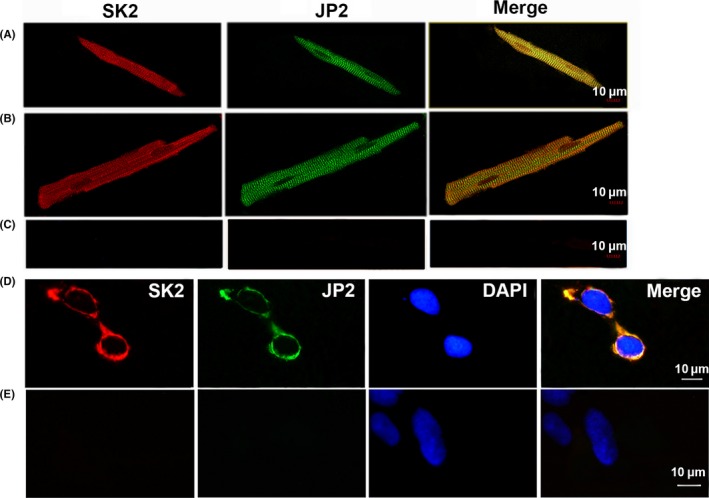
Colocalizations of JP2 and SK2 channels in adult mouse cardiac myocytes and HEK293 cells. Confocal images show double immunostaining with anti‐JP2 antibody (green) and anti‐SK2 antibody (red) in single isolated mouse atria (A) and ventricular (B) myocytes as well as HEK293 cells (D) cotransfected with JP2 and SK2. Negative control experiments (Control) were performed with the secondary antibodies with anti‐rabbit‐IgG TRITC‐conjugated and with anti‐mouse‐IgG FITC‐conjugated in atria myocytes (C) and HEK293 cells (E). Merge images show the colocalization of JP2 and SK2 channels near the Z‐lines in the cardiac cells or on the surface membrane of HEK293 cells. Scale bars, 10 μm
2.4. JP2 increases SK2 channel surface expression in HEK293 Cells
To rule out the physiological significant of the interaction between JP2 and SK2, we investigated whether the targeting of SK2 channels on the membrane requires the JP2 protein base on the HEK293 cells stably expressing SK2 and SK2+JP2 with cell ‐surface biotinylation assay. As shown in Figure 4A and 4C, the total expression of the SK2 protein from HEK293 cells cotransfected with JP2 and SK2 plasmids does not change significantly compared with the cells transfected with the SK2 channel alone (P = .778, n = 5 for each). The surface expression of the SK2 protein from HEK293 cells cotransfected with JP2 and SK2 plasmids significantly increased by 1.02 ± 0.14% (n = 5 for each) compared with the cells transfected with the SK2 channel alone (0.13 ± 0.05%) (P < .05, Figure 4B, C). The results indicated that JP2 regulates the targeting of SK2 channel on the membrane.
Figure 4.
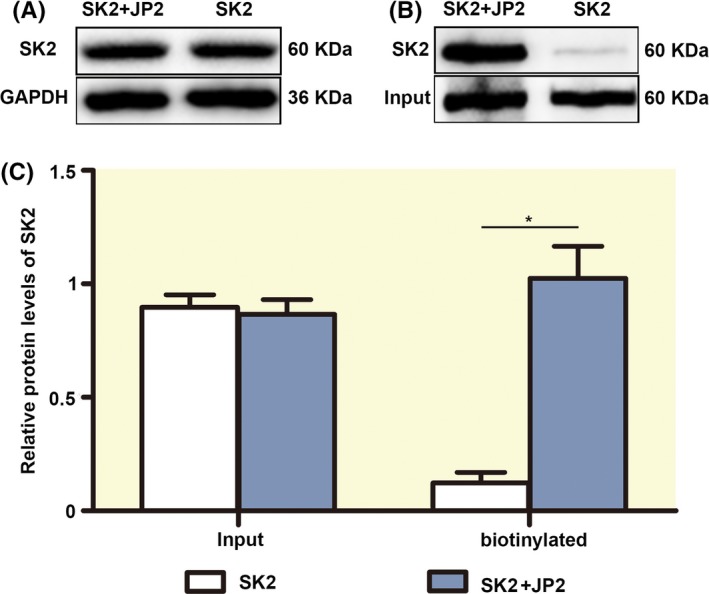
JP2 increases SK2 channel surface expression in HEK293 cells. A, Western blot analysis showing the total expression of the SK2 protein from HEK293 cells cotransfected with JP2 and SK2 plasmids and with the cells transfected with the SK2 channel alone. B, The surface expression of the SK2 protein from HEK293 cells stably expressing SK2 and SK2+JP2 with cell surface biotinylation assay. Signals were corrected to total channel protein input and normalized. C, The bar graphs show no changes in the total expression of the SK2 protein (input), but the surface expression of the SK2 protein (biotinylated protein) from HEK293 cells cotransfected with JP2 and SK2 plasmids significantly increased compared with the cells transfected with the SK2 channel alone (*P < .05, n=5 per group)
2.5. Knockdown of JP2 depresses Ca2+‐activated K+ currents in cardiac myocytes
To expand our understanding of the physiological significance of JP2 binding to the SK2 channel in cardiomyocytes, we examined whether JP2 specifically modulates the SK2 channel function using an RNA interference technique. A small interference RNA oligonucleotide probe against a specific cDNA sequence of JP2 was found to be highly effective in knocking down of JP2 expression in the cardiac myocytes (see Supplementary Information).
Western blotting analysis revealed that the level of JP2 expression in the adult mouse cardiac tissue transduced with Ad‐mediated siRNA specifically targeted against JP2 for 7 days was reduced to 47% and 48%, respectively, of the levels observed in the non‐infected adult cardiac tissue (Ctrl‐NT) and infected scramble siRNA (Ad‐NC) cardiac tissue (P = .0047 and P = .0098, n = 5 per group from 5 hearts, Figure 5C, D). No significant change in expression levels of the SK2 channel was examined in the adult cardiac tissue infected with Ad‐siJP2 compared with the controls (n = 5 per group from 5 hearts, Figure 5C, E).
Figure 5.
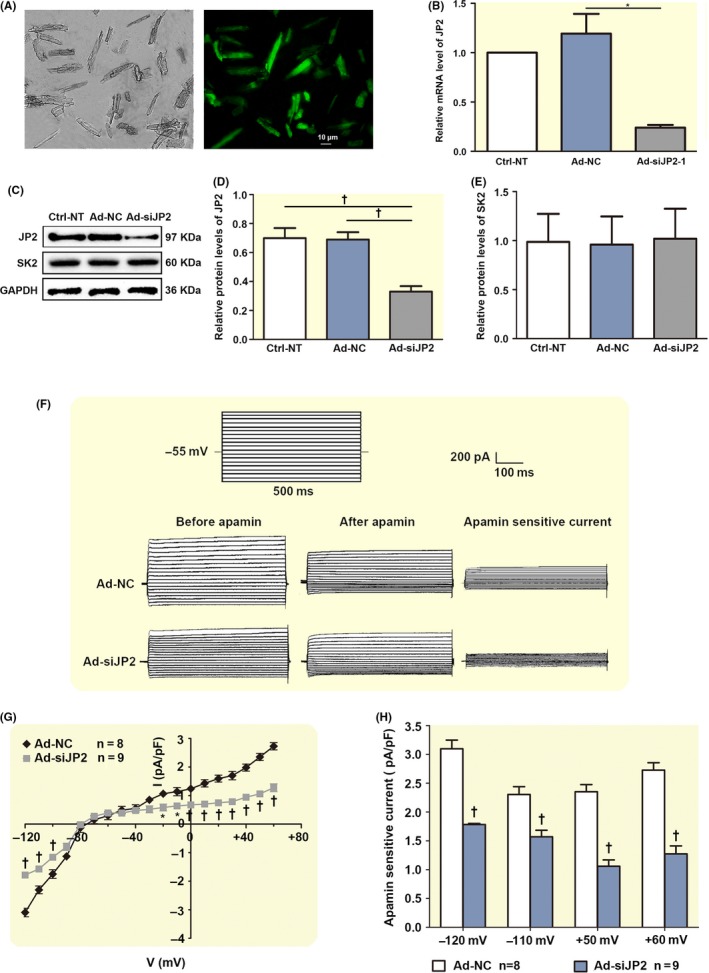
siRNA knockdown of JP2 depresses I K,Ca in the adult mouse cardiac myocytes. A, A photomicrograph of the fluorescent microscopy of EGFP expression in the isolated adult mouse cardiac myocytes after injection. B, A significant suppression of the levels of JP2 mRNA in the adult mouse cardiac myocytes transduced with Ad‐siJP2 vectors was observed using real‐time PCR compared to the scramble siRNA (Ad‐NC) myocardium and non‐transfected (Ctrl‐NT) myocardium (each n = 3). C, Western blot analysis showing the expressions of JP2 and SK2 proteins in adult mouse myocardium with different treatments. D, The bar graphs show significant decreases in the expression of JP2 in adult mouse myocardium transduced by Ad‐mediated siRNA specific to JP2. *P < .05 with respect to Ad‐NT or Ctrl‐NC groups (n = 5 per group). E, The bar graphs show no changes in the expression of SK proteins in siRNA‐infected adult mouse myocardium compared with Ad‐NT or Ctrl‐NC groups (n = 5 per group). F, Representative traces of I K,Ca evoked by depolarization steps from −120 mV to +60 mV from a holding potential of −55 mV in infected adult mouse cardiac myocytes. The protocol is illustrated above the current traces. Apamin‐sensitive currents in the right panels were obtained from the digital subtraction of the total current recorded in the presence and absence of apamin. G, Summary data of the current density‐voltage relationships showing a reduced apamin‐sensitive current densities in the adult mouse cardiac myocytes transduced with siRNA specific to JP2 (‡P < .01, †P < .01, *P < .05 vs control siRNA groups, n = 8 per group). F, The bar graphs show reduced I K,Ca in infected adult mouse cardiac myocytes when the test potentials were −120, −110, +50 and +60 mV, respectively. *P < .05, †P < .01 vs control siRNA groups (n = 8 for each group)
We further investigated whether siRNA knockdown of JP2 may interfere with the SK2 channel function. The Ca2+‐activated K+ current (I K,Ca) in the single adult mouse cardiac myocytes infected with Ad‐siJP2 for 7 days through the caudal vein injection was measured using patch‐clamp technique. Figure 5A shows the GFP expression in single adult mouse cardiac myocytes transfected with Ad‐siJP2 for 7 days under a fluorescent microscope. Figure 5F shows representative whole‐cell I K,Ca traces recorded at a holding potential of −55 mV to various voltage steps from both the control and siRNA JP2‐EGFP‐positive cardiac myocytes. The current density‐voltage relations of I K,Ca were summarized in Figure 5G, which shows a significant decrease in both the inward and outward current densities in the infected cardiac myocytes compared with the control cells (*P < .05, †P < .01, respectively, n = 8 for Ad‐NC; n = 9 for Ad‐siJP2). At −120, −110, +50 and +60 mV, the I K,Ca densities in the cardiac myocytes infected with siRNA against JP2 decreased by about 42.4%, 31.8%, 55% and 53.2%, respectively, compared with the control cells (†P = .01, n = 8 for Ad‐control; n = 9 for Ad‐shRNA) as shown in Figure 5H. Altogether, knockdown of JP2 led to a decrease in the I K,Ca, suggesting that JP2 is required for proper function of SK2 channel in cardiac myocytes. In combination with our immunoprecipitation data showing that JP2 binds to SK2, further supporting a novel role of JP2 in regulating SK2 channel.
2.6. Knockdown of JP2 depresses intracellular Ca2+ transient
Based on the observation of reduced Ca2+‐activated K+ current in isolated adult cardiac myocytes infected with Ad‐siJP2, we investigated whether the alteration of the SK2 channel function in Ad‐siJP2 cardiac myocytes is related to the intracellular Ca2+ abnormalities caused by knockdown of JP2. Typical recordings of the intracellular Ca2+ ([Ca2+]i) transients are shown in Figure 6A. Summarized data are displayed in Figure 6B‐C, illustrating that the resting calcium levels were unchanged in the cardiac myocytes infected with Ad‐siJP2 compared with the control cells (1.17 ± 0.09 vs 1.18 ± 0.09 ratio unites, P = .92) (Figure 5B). siRNA knockdown of JP2 significantly decreased the amplitude of the [Ca2+]i transient (0.249 ± 0.020 vs 0.49 ± 0.0389 ratio units in control cells, P = .004, n = 35 from 8 hearts) (Figure 6B) and increased its time to 50% peak (25.28 ± 4.46 vs 17.54 ± 2.86 ms in control cells, P = .013, n = 35 from 8 hearts). Whereas the time for Ca2+ transient to 50% decay was unchanged in Ad‐siJP2 cardiac myocytes compared with the controls (71.57 ± 6.94 vs 67.55 ± 9.06 ms, P = 0.67, n = 26 from 8 hearts) (Figure 6C).
Figure 6.
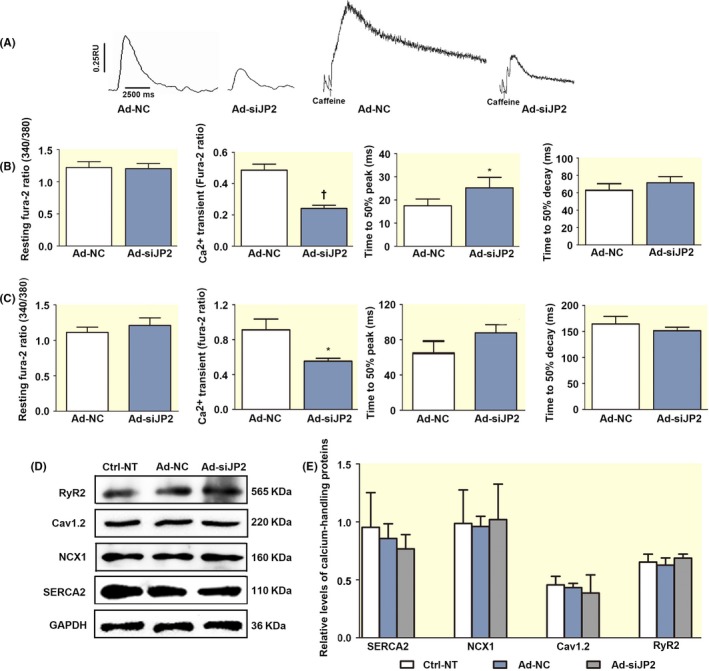
Knockdown of JP2 reduced the amplitude of the Ca2+ transient in the adult mouse cardiac myocytes. A, Samples of the Ca2+ transient of Furo‐2 loaded adult mouse cardiac myocytes treated with siRNA specific to JP2. The intracellular Ca2+ transient induced by field stimulation at 1 Hz. B, The bar graphs show resting Fura‐2 ratio (340/380 nm), the amplitude of the [Ca2+]i transient, time to 50% peak and time to 50% decay in Ad‐siJP2 infected adult mouse cardiac myocytes. Compared with Ad‐NC (siRNA‐negative control) group, siRNA knockdown of JP2 significantly decreased the amplitude of the [Ca2+]i transient, increased its time to 50% peak. C, The bar graphs show examples of Ca2+ transients elicited by application of 10 mmol L−1 caffeine. The adult cardiac myocytes infected with Ad‐shJP2 reduced Ca2+ transient amplitude (Fura‐2 ratio) compared to Ad‐NC without significantly changing in the Ca2+ transients dynamics, such as resting Fura‐2 ratio, time to 50% peak and time to 50% decay. D, Western blot analysis showing the expressions of calcium‐handling proteins in adult mouse heart E, The bar graphs show no changes in the expression of RyR2, SERCA2, Cav1.2 LTCC and NCX1 in siRNA‐infected adult mouse heart compared with Ad‐NT or Ctrl‐NC groups (n = 5 per group)
To study whether reduced Ca2+ transient was due to change in the SR store Ca2+, we measured RyR2‐dependent SR store Ca2+ content by caffeine. Figure 5C shows examples of Ca2+ transients elicited by application of 10 mmol L−1 caffeine. The adult cardiac myocytes infected with Ad‐shJP2 showed a significantly lower caffeine‐evoked Ca2+ release (0.55 ± 0.07, ratio units, N = 25) compared to control cells (0.91 ± 0.12, N = 48; P = .0001) without significantly changing in the Ca2+ transients dynamics (Figure 6C). These results indicated that knockdown of JP2 significantly decreased caffeine‐mediated SR Ca2+ store levels, and this could account for reduced [Ca2+]i transients amplitude. We next explore whether knockdown of JP2 affects the expression of calcium‐handling proteins. As shown in Figure 6D, E, there was no significant alteration in the expression of RyR2, SERCA2, Cav1.2 LTCC and NCX1 in Ad‐siJP2 cardiac cells compared to control cells.
3. DISCUSSION
Junctophilins are key molecules that located in the junctional membrane complex of skeletal and cardiac muscle cells. The role of JP2 as a membrane‐binding protein has been previously established.1, 2, 3 However, only recently has JP2 been identified as an interacting membrane protein with DHPR and TRPC3 in skeletal muscle cells that localizes on the plasma membrane.30, 31 In the present study, we demonstrated that JP2 is the SK2 channels interacting protein in native cardiac tissue using co‐immunoprecipitation and pulldown experiments. The same experiments indicated that JP2 and SK2 were immunoprecipitated and pull down with solubilized proteins from HEK293 cells expressing heterologous proteins, indicating that JP2 and SK2 interact directly. An association of these proteins in native cardiac tissue and their close proximity in the same cellular compartment was confirmed in adult mouse cardiac cells by confocal immunofluorescent imaging, where the majority of both JP2 and SK2 proteins was localized near the Z‐lines in the native cardiac myocytes or at the plasma membrane in the HEK293 cells. And the significance of the interaction between the SK2 channels with JP2 is further extended by our data that JP2, an MJC protein, enhanced the proper cell surface expression of the SK2 channel. The molecular mechanisms of this effect may be mediated by protein‐protein interactions or by the participation of other molecules.
JP2 is predicted to interact with the plasma membrane or T‐tubule membranes via its MORN motif at the N‐terminus, and it is also thought to interact with SR membranes via its transmembrane domain at the C‐terminus.1, 2 These characteristics allow JP2 to crosslink the T‐tubule membrane to the SR membrane, leading to a direct interaction with ion channels on the plasma membrane. In the present study, we demonstrated that JP2 is the SK2 channels interacting protein in native cardiac tissue. Our study provides new evidence regarding important molecular determinants involving the MORN motifs of JP2. The MORN motifs (amino acids 1‐253) within the N‐terminus of JP2 are responsible for a physical and functional interaction with SK2 channels. Recent reports showed that mutation of two residues (N101R and Y141H) within the MORN motifs of JP2 was found in patients with hypertrophic cardiomyopathy,34, 36, 37 indicating potential physiological and pathophysiological significant of JPs MORN domain.
The SK channels are voltage‐independent and have a high gate affinity for the intracellular Ca2+ signalling.10, 38 We previously showed that RyR2 alters SK2 channel activity in cardiac myocytes due to changed in the intracellular Ca2+ levels.35 Our present results reveal that interfering RNA silencing of endogenous JP2 caused a significantly reduced density of I k,Ca. and reduced amplitude of the Ca2+ transient in the mice cardiomyocytes. It is possible that the interaction between JP2 and SK2 channels establishes a crosstalk between the SK channels on the surface membrane and the intracellular Ca2+ signalling. We provided data of the electrophysiological and intracellular Ca2+ to support the decrease in the SK2 channel current may be related to a reduction in intracellular calcium levels. Silencing of JP2 may cause defective coupling between the SK2 channel activation and the intracellular Ca2+ signalling, and consequently to dysfunction of SK2 channels. Indeed, a decrease in intracellular Ca2+ is thought to result in a prolonging of AP, leading to the development of atrial fibrillation and arrhythmias.14, 39
Ca2+ is essential in cardiac electrical activity and acts as the direct activator of the myofilaments which cause contraction. JP2 plays a notable role in the maintaining Ca2+ flux,2, 7, 40 and its functional coupling with Ca2+ is further explained in our study. We demonstrated that JP2 expression knockdown caused a decrease in the peak of the [Ca2+]i transients and the slow time to the Ca2+ transient peak. Knockdown of JP2 effects on the [Ca2+]i transient and its dynamics may predominantly reflect a slowed down of RyR2 release rate and /or the SR Ca2+ load. There was no significant alteration in the expression of RyR2, SERCA2, Cav1.2 LTCC and NCX1 in Ad‐siJP2 cardiac cells compared to control cells. One possibility is that the alteration of RyR2 Ca2+ release may be due to increased distance of RyR2 from the flux of Ca2+ entering through L‐type Ca2+ channels, or otherwise disrupted cellular ultrastructure.7, 41 In the presence of 10 mmol L−1 caffeine, the reduced Ca2+ levels we observed indicated that knockdown of JP2 reduced the SR Ca2+ stores, consistent with reduced the peak of the [Ca2+]i transients. This closely follows Golini's work31 in which the reduced [Ca2+]i transients seen in skeletal muscle caused by JP1 and JP2 knockdown as well as cardiac hypertrophy and failure.6, 42, 43
Ion‐channel targeting is a dynamic and complex process, which is associated with a number of interacting proteins.44 Recent studies focused on the role of cytoskeletal proteins in SK2 channel targeting and traffic in cardiac myocytes have revealed a new study field for SK channel interaction proteins in physiology and disease.40, 45, 46 JPs have a key role in anchoring the SR to the T‐tubule/sarcolemma membrane in striated muscles, which imply that JP2 may act as the anchor for the SK2 channel at the membrane. Thus, the decrease in I K,Ca may be not simply due to reduced intracellular Ca2+ levels in the cardiac cells. Additional studies are necessary to determine the roles of JPs in the localization and integration of SK channels. It will be important in future work to determine the regulation of SK2 channel targeting by JPs using JP2 knockdown mice (JP2 knockout mouse does not survive). A better understanding of the coupling mechanisms of JP2 with ion channels and JP2 binding partners may represent a new therapeutic strategy for certain heart disturbances.
4. MATERIALS AND METHODS
4.1. Animals
Three‐month‐old adult C57BL/6 mice of both sexes used in these studies were obtained from Beijing Weitong Lihua Experimental Animal Technology, Limited (No. SCXK Jing 2012‐0001). Animal care and procedures were approved by the Committee on the Ethics of Animal Experiments of the University of Zhengzhou (No. SYXK‐2010‐0001). Animal use was in accordance with National Institutes of Health and institutional guidelines. All mice were group housed and kept on a 12‐hour light/dark cycle with food and water available ad libitum.
4.2. Plasmids and antibodies
Full‐length mouse JP2 cDNA (accession number, NM_001205076.1) was inserted at the BamHI/XhoI sites of the pGEX‐4T‐1 vector (Shanghai Genechem Biotechnology, Shanghai, China) to generate pGEX‐4T‐1‐JP2. Three constructs contained cDNA fragments (JP21‐253, amino acid residues 1‐253; JP2216‐350, residues 216‐350; JP2340‐696, residues 340‐696) were subcloned into the glutathione S‐transferase (GST) fusion vector pGEX‐4T‐1 with the BamHI/XhoI sites to generate pGEX‐4T‐1‐JP21‐253, pGEX‐4T‐1‐JP2216‐350 and pGEX‐4T‐1‐JP2340‐696. Full‐length mouse SK2 cDNA (accession number AY258141) was subcloned into pIRES‐EGFP (Clontech Laboratories, Palo Alto, CA, USA) to obtain pSK2‐IRES‐EGFP. Full‐length cDNA of JP2 was subcloned into pcDNA3.1 vector (Shanghai Genechem Biotechnology, Shanghai, China) to obtain pCDNA3.1‐JP2. A pSK2‐IRES2 + JP2 plasmid, containing full‐length cDNA of SK2 and JP2, was generated by subcloning the full‐length cDNA of JP2 into pSK2‐IRES2. All plasmids were verified by sequencing.
The following commercial antibodies were used for immunoblotting and/or immunostaining: anti‐JP2 antibody (Abcam, Cambridge, MA, USA), anti‐SK2 antibody (Alomone labs, Jerusalem, Israel), anti‐SERCA2 antibody (Alomone labs), anti‐NCX1 antibody (Alomone labs), anti‐Cav1.2 LTCC antibody (Alomone labs) and anti‐RyR2 antibody (Thermo, Rockford, IL, USA). Secondary antibodies used for immunoblotting were HRP‐conjugated goat anti‐rabbit (Bio‐Rad, Hercules, CA, USA) and HRP‐conjugated goat anti‐mouse (Bio‐Rad). Secondary antibody used for immunostaining was TRITC‐conjugated goat anti‐rabbit IgG (Jackson ImmunoResearch, West Grove, PA, USA) and FITC‐conjugated goat anti‐mouse IgG (Jackson Immuno Research).
4.3. HEK293 cell culture and transfection
HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 1% penicillin‐streptomycin (Invitrogen) at 37°C in an incubator with a mixture of air and 5% CO2. Cultured cells were split 24 h prior to transfection and plated on 100‐mm plates to obtain ≧90% confluence at time of transfection. Lipofectamine 2000 reagent (Invitrogen) was used to transfect HEK293 cells with pSK2‐IRES‐EGFP, pSK2‐IRES‐EGFP+JP2 and pSK2‐IRES‐EGFP+JP2 plasmids according to the manufacturer's instructions. To establish the cells stably expressing SK2 and SK2+JP2, they were cultured in the presence of 100 μg/mL Geneticin (G418; Sigma, St. Louis, MO, USA) for 5 weeks of continuous passage and then maintained in the medium supplemented with 50 μg/mL G418. Untransfected cells were used as a negative control. After transfection for 48 hour, the cells were harvested and the lysates isolated from transfected cells were used for further experiments.
4.4. Co‐immunoprecipitation
Reciprocal co‐immunoprecipitation (co‐IP) was performed as previously reported.35 In brief, 300 μg of solubilized proteins isolated from the adult mouse heart or from HEK293 cells was incubated with anti‐JP2, anti‐SK2 or anti‐RyR2 antibodies overnight at 4°C, and control groups (Ctrl‐N) were incubated with IgG antibodies of non‐mouse and non‐rabbit sources, followed by an additional incubation with protein G Sepharose (Santa Cruz Biotechnology, La Jolla, CA, USA) for 6 hour at 4°C. Protein‐bead complexes were washed three times with a high‐salt buffer (50 mmol L−1 Tris pH 8.0, 1 mol L−1 NaF, 500 mmol L−1 NaCl, 0.5% NP‐40 and complete protease inhibitor mixture), then a low‐salt buffer (50 mmol L−1 Tris pH 8.0, 1 mol L−1 NaF, 250 mmol L−1 NaCl, 0.5% NP‐40, and complete protease inhibitor mixture) and finally a salt‐free buffer (50 mmol L−1 Tris pH 8.0, 1 mol L−1 NaF, 0.5% NP‐40, and complete protease inhibitor mixture) to remove non‐specific binding. The immune complexes were assessed with immunoblot analysis with anti‐JP2, anti‐SK2 or anti‐RyR2 antibodies.
4.5. Western blot analysis
Mouse heart or HEK293 cell lysates were prepared in RIPA buffer (50 mmol L−1 Hepes, pH 7.5, 50 mmol L−1 NaCl, 50 mmol L−1 NaF, 10 mmol L−1 sodium pyrophosphate, 5 mmol L−1 EDTA, 1 mmol L−1 NaVO4, 0.25% sodium deoxycholate, 1% NP‐40) and protease inhibitor cocktail (Sigma‐Aldrich, St. Louis, MO, USA). Protein samples were separated by SDS‐PAGE and transferred onto a polyvinylidene difluoride membrane (Roche, Basel, Switzerland). The membrane was hybridized overnight at 4°C with the primary antibody against JP2 (dilution 1:300) or SK2 (dilution 1:300) or RyR2 (dilution 1:600). After extensive washings and incubation for 2 hour at room temperature with secondary antibodies, immunoreactivity was analysed with a standard ECL protocol (Pierce, MA, USA).
4.6. GST pulldown assays
GST, GST‐JP2 fusion protein or GST constructs of GST‐JP2 (GST‐JP21‐253, GST‐JP2216‐350 and GST‐JP2340‐696) were transformed into Escherichia coli BL21 cells (Novagen, Darmstadt, Germany). The recombinant GST proteins were expressed and purified using a Pierce GST pulldown kit (Pierce) according to the manufacturer's instruction. 500 μg of GST, GST‐JP2 or GST‐JP2 constructs premixed with glutathione‐Sepharose 4B was incubated with the mouse heart lysates or with the HEK293 cell lysates overnight at 4°C. Bound GST fusion proteins were washed five times with the pulldown buffer (Pierce GST pulldown kit). The pulldown complexes with GST, GST‐JP2 or GST‐JP2 constructs were eluted from the glutathione‐agarose by 10 mmol L−1 reduced glutathione in TBS (pH 8.0). The bound proteins were separated by SDS‐PAGE, visualized by Coomassie Blue staining and then subjected to Western blot analysis with appropriate antibody.
4.7. Single cardiac myocyte isolation
The adult mouse was anesthetized with sodium pentobarbital i.p. (80 mg/kg). The heart was rapidly removed, cannulated in ice‐cold Ca2+‐free modified Tyrode's solution containing 140 mmol L−1 NaCl, 5.4 mmol L−1 KCl, 1 mmol L−1 MgCl2, 10 mmol L−1 N‐2‐hydroxyethylpiperazine‐N‐2‐ethanesulfonic acid (HEPES) and 10 mmol L−1 glucose at pH 7.4 and mounted onto a Langendorff perfusion system. The hearts were then perfused with a fresh enzyme solution containing collagenase type II (Sigma‐Aldrich) and protease (type XIV, Sigma‐Aldrich) for 35‐40 min and a high‐K+ solution (120 mmol L−1 potassium glutamate, 20 mmol L−1 KCl, 1 mmol L−1 MgCl2, 0.3 mmol L−1 EGTA, 10 mmol L−1 glucose and 10 mmol L−1 HEPES, adjusted to pH 7.4 with KOH) for 5 min. The hearts were removed from the Langendorff apparatus. Single atrial and ventricular cells were isolated and kept in the high‐K+ solution for the immunofluorescence labelling. The entire perfusion procedure was performed at 37°C and 100% O2. 35
4.8. Immunocytochemistry
Freshly isolated adult mouse cardiac cells or cultured HEK 293 cells were fixed on glass coverslips with 4% paraformaldehyde and permeabilized with 0.4% Triton X‐100 at room temperature. The cells were then incubated overnight at 4°C with anti‐SK2 (dilution 1:100) and or anti‐JP2 antibodies (dilution 1:100). Following three washes in PBS, the cells were incubated with FITC‐conjugated goat anti‐mouse antibody (dilution 1:250) and/or TRITC‐conjugated goat anti‐rabbit antibody (dilution 1:250) for 3 hour at room temperature.26 Negative control experiments (Control) were performed with the secondary antibodies with anti‐rabbit‐IgG TRITC‐conjugated and with anti‐mouse‐IgG FITC‐conjugated in cardiac myocytes and HEK293 cells. Confocal images were captured with an Olympus FV1000 confocal laser scanning microscopy (Olympus Corporation, Tokyo, Japan).
4.9. Biotinylation of surface proteins
The stably transfected HEK293 cells were washed with ice‐cold PBS and then treated with sulfo‐NHS‐SS‐Biotin (0.5 mg/mL; Pierce, Thermo Scientific, Waltham, MA, USA) at 4°C for 30 minutes.47 The cells were lysed in an extraction buffer (10 mmol L−1 Tris‐HCl pH 7.5, 10 mmol L−1 EDTA, 1% Triton X‐100 and 1% SDS) and a 1% mammalian protease inhibitor cocktail (Sigma‐Aldrich, St. Louis, USA). After centrifugation (10 000 g for 20 minutes at 4°C), the samples were normalized for protein concentration and incubated with immobilized NeutrAvidin Gel (agarose beads; Pierce, Thermo Scientific) overnight at 4°C. The beads were washed 3 times with a washing buffer including protease inhibitors and eluted using a sample buffer containing 50 mmol L−1 DTT and a trace amount of bromophenol blue. The eluates were resolved by SDS‐PAGE gel and immunoblotted with the SK2 antibody.
4.10. Construction and production of small interfering RNA
Two optimal 19‐mer target sequences (GCCACAATGTGCTGGTCAA, location 1528, GTATGGTGATCTTGCTGAA, location 2554) were selected by scanning mouse‐specific JP2 cDNA sequences (accession number NM_001205076.1).48 Another siRNA sequence (TTCTCCGAACGTGTCACGT) that had no homology to any known mammalian gene was used as a siRNA‐negative control. Two pairs of oligonucleotides encoding shRNAs and a negative control shRNA were designed and chemically synthesized by Shanghai Genechem Biotechnology, Shanghai, China. Virus particles were packaged by transfection each short hairpin RNA‐expressing vector into the HEK293‐based packaging cells using the ADMAX™ (Microbix Biosystems, Toronto, Canada) according to the manufacturer's protocol. The viral titre was stored at −70°C before use.
A total dose of 1 × 109 PFU adenovirus with control siRNA and JP2 siRNA were delivered into mice by tail vein injection, respectively, and the experiments were carried out 7 days after injection.49
4.11. Whole‐cell patch‐clamp recordings
Whole‐cell Ca2+‐activated K+ current (I K,Ca) was recorded using voltage‐clamp protocol as previously described.35 Whole‐cell recordings of I K,Ca in the infected adult cardiac myocytes were performed at room temperature (20‐22°C). Patch pipettes had resistances of 3‐4 MΩ and were filled with an intracellular solution containing 144 mmol L−1 potassium gluconate, 1.15 mmol L−1 MgCl2, 5 mmol L−1 EGTA, 10 mmol L−1 HEPES (adjusted to pH 7.25 using KOH). 500 nmol L−1 of a free [Ca2+] was applied by a calcium titration programme used to calculate the free [Ca2+], bound and dissociated. The external solution contained 140 mmol L−1 N‐methyl‐d‐glucamine (NMG), 4 mmol L−1 KCl, 1 mmol L−1 MgCl2, 5 mmol L−1 glucose and 10 mmol L−1 HEPES (adjusted to pH 7.4 using HCl). I K,Ca was elicited by a voltage ramp from −120 to +60 mV. After stable traces were obtained, apamin (500 pmol/L; Santa Cruz Biotechnology) was applied to the bath, and the apamin‐sensitive current was obtained by digital subtraction. The currents were normalized to cell capacity to obtain the current density (pA/pF). The series resistance compensation of ≥90% was obtained. Whole‐cell patch‐clamp currents were acquired with an EPC800 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany), filtered at 10 kHz, digitized at sampling frequency of 50 kHz. Data were acquired and analysed with pCLAMP10 software (Axon Instruments Inc., Foster, CA, USA).
4.12. Ca2+ transient measurements
The intracellular Ca2+ ([Ca2+]i) transient was measured as previous described.50 The [Ca2+]i transient was recorded from infected adult cardiac myocytes loaded with 2 μmol L−1 Fura‐2‐AM in the dark at room temperature for 30 minutes. After the loading of Fura‐2, the cells were washed twice with the superfusion buffer containing 1.8 mmol L−1 CaCl2 and kept in the same buffer in the dark at room temperature for 30 minutes. To evoke [Ca2+]i transients, the cells were stimulated at 1 Hz by field stimulation applied by two parallel platinum electrodes. For the determination of the SR calcium content, the cells were treated with 10 mM caffeine. Cytosolic free Ca2+ was measured as the 340 to 380 nm (340/380) ratio of Fura‐2 fluorescence excited at 510 nm using an IonOptix photometry system (PMT‐300; IonOptix, Westwood, MA, USA). Data were acquired and analysed with Signal Averager software IonWizard 6.6 (IonOptix).
4.13. Statistical analysis
The data are expressed as the means ± SEM. Differences between the groups were assessed using one‐way ANOVA or Student's t test, as appropriate. A P value of <.05 was considered statistically significant.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Supporting information
{kind=link}
ACKNOWLEDGEMENTS
We thank Dr. Chiamvimonvat N for technical advice and helpful suggestions on the manuscript. This work was supported by grants from the National Natural Science Foundation of China (grant No. 81570311 and 81270248).
Fan HK, Luo TX, Zhao WD, et al. Functional interaction of Junctophilin 2 with small‐ conductance Ca2+‐activated potassium channel subtype 2(SK2) in mouse cardiac myocytes. Acta Physiol. 2018;222:e12986 10.1111/apha.12986
REFERENCES
- 1. Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11‐22. [DOI] [PubMed] [Google Scholar]
- 2. Garbino A, Van Oort RJ, Disit SK, Landstrom AP, Ackerman MJ, Wehrens HT. Molecular evolution of the junctophilin gene family. Physiol Genomics. 2009;37:175‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cuttell L, Vaughan A, Silva E, et al. Undertaker, a Drosophila junctophilin, links Draper‐mediated phagocytosis and calcium homeostasis. Cell. 2008;135:524‐534. [DOI] [PubMed] [Google Scholar]
- 4. Garbino A, Wehrens XH. Emerging role of junctophilin‐2 as a regulator of calcium handling in the heart. Acta Pharmacol Sin. 2010;31:1019‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beavers DL, Landstrom AP, Chiang DY, Wehrens XH. Emerging roles of junctophilin‐2 in the heart and implications for cardiac diseases. Cardiovasc Res. 2014;103:198‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Landstrom AP, Kellen CA, Dixit SS, et al. Junctophilin‐2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium‐handling. Circ Heart Fail. 2011;4:214‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Oort RJ, Garbino A, Wang W, et al. Disrupted junctional membrane complexes and hyperactive ryanodine receptors following acute junctophilin knockdown in mice. Circulation. 2011;1239:979‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bond CT, Maylie J, Adelman JP. Small conductance Ca2+‐activated K+ channels. Ann N Y Acad Sci. 1999;868:370‐378. [DOI] [PubMed] [Google Scholar]
- 9. Köhler M, Hirschberg B, Bond CT, et al. Small‐conductance, calcium‐activated potassium channels from mammalian brain. Science. 1996;273:1709‐1714. [DOI] [PubMed] [Google Scholar]
- 10. Stocker M. Ca (2+)‐activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci. 2004;5:758‐770. [DOI] [PubMed] [Google Scholar]
- 11. Nanou E, Alpert MH, Alford S, El Manira A. Differential regulation of synaptic transmission by pre‐ and postsynaptic SK channels in the spinal locomotor network. J Neurophysiol. 2013;109:3051‐3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adelman JP, Maylie J, Sah P. Small‐conductance Ca2+‐activated K+ channels: form and function. Annu Rev Physiol. 2012;74:245‐269. [DOI] [PubMed] [Google Scholar]
- 13. Xu Y, Tuteja D, Zhang Z, et al. Molecular identification and functional roles of a Ca2+‐activated K+ channel in human and mouse hearts. J Biol Chem. 2003;278:49085‐49094. [DOI] [PubMed] [Google Scholar]
- 14. Li N, Timofeyev V, Tuteja D, et al. Ablation of a Ca2+‐activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol. 2009;587:1087‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nattel S. Calcium‐activated potassium current: a novel ion channel candidate in atrial fibrillation. J Physiol. 2009;587:1385‐1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosa JC, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH. Bis‐quinolinium cyclophanes: 6,10‐diaza‐3(1,3),8(1,4)‐dibenzena‐1,5(1,4)‐ diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non‐peptidic blocker of the apamin‐sensitive Ca(2+)‐activated K+ channel. J Med Chem. 1998;41:2‐5. [DOI] [PubMed] [Google Scholar]
- 17. Skibsbye L, Poulet C, Diness JG, et al. Small‐conductance calcium‐activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc Res. 2014;103:156‐167. [DOI] [PubMed] [Google Scholar]
- 18. Hsueh CH, Chang PC, Hsieh YC, Reher T, Chen PS, Lin SF. Proarrhythmic effect of blocking the small conductance calcium activated potassium channel in isolated canine left atrium. Heart Rhythm. 2013;10:891‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ozgen N, Dun W, Sosunov EA, et al. Early electrical remodeling in rabbit pulmonary vein results from trafficking of intracellular SK2 channels to membrane sites. Cardiovasc Res. 2007;75:758‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Strobaek D, Hougaard C, Johansen TH, et al. Inhibitory gating modulation of small conductance Ca2+‐activated K+ channels by the synthetic compound(R)‐N‐(benzimidazol‐2‐yl)‐1,2,3,4‐tetrahydro‐1‐naphtylamine(NS8593) reduces after‐hyperpolarizing current in hippocampal CA1 neurons. Mol Pharmacol. 2006;70:1771‐1782. [DOI] [PubMed] [Google Scholar]
- 21. Yu T, Deng C, Wu R, et al. Decreased expression of small‐conductance Ca2+‐activated K+ channels SK1 and SK2 in human chronic atrial fibrillation. Life Sci. 2012;90:219‐227. [DOI] [PubMed] [Google Scholar]
- 22. Qi XY, Diness JG, Brundel BJ, et al. Role of small‐conductance calcium‐activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation. 2014;129:430‐440. [DOI] [PubMed] [Google Scholar]
- 23. Chua SK, Chang PC, Maruyama M, et al. Small‐conductance calcium‐activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res. 2011;108:971‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee YS, Chang PC, Hsueh CH, et al. Apamin‐sensitive calcium‐activated potassium currents in rabbit ventricles with chronic myocardial infarction. J Cardiovasc Electrophysiol. 2013;24:1144‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonilla IM, Long VP III, Vargas‐Pinto P, et al. Calcium‐activated potassium current modulates ventricular repolarization in chronic heart failure. PLoS One. 2014;9:e108824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Q, Timofeyev V, Lu L, et al. Functional roles of a Ca2+‐activated K+ channels in atrioventricular node. Circ Res. 2008;102:465‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang XD, Timofeyev V, Li N, et al. Adelman. J, Lieu, DK, Chiamvimonvat N: critical roles of a small conductance Ca²⁺‐activated K⁺ channel (SK3) in the repolarization process of atrial myocytes. Cardiovasc Res. 2014;101:317‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang XD, Lieu DK, Chiamvimonvat N. Small‐conductance Ca2+ ‐activated K+ channels and cardiac arrhythmias. Heart Rhythm. 2015;12:1845‐1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang W, Landstrom AP, Wang Q, et al. Reduced junctional Na+/Ca2+‐exchanger activity contributes to sarcoplasmic reticulum Ca2+ leak in junctophilin‐2‐deficient mice. Am J Physiol Heart Circ Physiol. 2014;307:H1317‐H1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woo JS, Hwang JH, Ko JK, Kim DH, Ma J, Lee EH. Glutamate at position 227 of junctophilin‐2 is involved in binding to TRPC3. Mol Cell Biochem. 2009;328:25‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Golini L, Chouabe C, Berthier C, et al. Junctophilin 1 and 2 Proteins Interact with the L‐type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J Biol Chem. 2011;286:43717‐43725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee EH, Cherednichenko G, Pessah IN, Allen PD. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J Biol Chem. 2006;281:10042‐10048. [DOI] [PubMed] [Google Scholar]
- 33. Chopra N, Yang T, Asghari P, et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation– contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:7636‐7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woo JS, Cho CH, Lee KJ, Kim DH, Ma J, Lee EH. Hypertrophy in skeletal myotubes induced by junctophilin‐2 mutant, Y141H, involves an increase in store‐operated Ca2+ entry via Orai1. J Biol Chem. 2012;287:14336‐14348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mu YH, Zhao WC, Duan P, et al. RyR2 modulates a Ca2+‐activated K+ current in mouse cardiac myocytes. PLoS ONE. 2014;9:e94905‐e94913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Landstrom AP, Weisleder N, Batalden KB, et al. Mutations in JPH2‐encoded junctophilin‐2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beavers DL, Wang W, Ather S, et al. Mutation E169K in junctophilin‐2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. 2013;62:2010‐2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schumacher MA, Rivard AF, Bächinger HP, Adelman JP. Structure of the gating domain of a Ca2+‐activated K+ channel complexed with Ca2+ /calmodulin. Nature. 2001;410:1120‐1124. [DOI] [PubMed] [Google Scholar]
- 39. Diness JG, Sørensen US, Nissen JD, et al. Inhibition of small‐conductance Ca2+‐activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:380‐390. [DOI] [PubMed] [Google Scholar]
- 40. Nishi M, Mizushima A, Nakagawara K, Takeshima H. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun 2000;273:920‐927. [DOI] [PubMed] [Google Scholar]
- 41. Reynolds O, Quick AP, Wang Q, et al. Junctophilin‐2 gene therapy rescues heart failure by normalizing RyR2‐mediated Ca2+ release. Int J Cardiol. 2016;225:371‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lindner M, Erdmann E, Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol. 1998;30:743‐749. [DOI] [PubMed] [Google Scholar]
- 43. Zhang C, Chen B, Guo A, et al. Microtubule‐mediated defects in junctophilin‐2 trafficking contribute to myocyte transverse‐tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation. 2014;129:1742‐1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delisle BP, Anson BD, Rajamani S, January CT. Biology of cardiac arrhythmias: ion channel protein trafficking. Circ Res. 2004;94:1418‐1428. [DOI] [PubMed] [Google Scholar]
- 45. Rafizadeh S, Zhang Z, Woltz RL, et al. Functional interaction with filamin A and intracellular Ca2+ enhance the surface membrane expression of a small‐conductance Ca2+‐activated K+ (SK2) channel. Proc Natl Acad Sci U S A. 2014;111:9989‐9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu L, Timofeyev V, Li N, et al. α‐Actinin2 cytoskeletal protein is required for the functional membrane localization of a Ca2+‐activated K+ channel (SK2 channel). Proc Natl Acad Sci U S A. 2009;106:18402‐18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Howes MT, Kirkham M, Riches J, et al. Clathrin‐independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J Cell Biol. 2010;190:675‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hirata Y, Brotto M, Weisleder N, et al. Uncoupling store‐operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J . 2006;90:4418‐4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu W, Zhang Y, Wang L, et al. Bone morphogenic protein 4‐smad induced upregulation of platelet derived growth factor AA impairs endothelial function. Arterioscler Thromb Vasc Biol. 2016;36:553‐560. [DOI] [PubMed] [Google Scholar]
- 50. Howarth FC, Shafiullah M, Qureshi MA. Chronic effects of type 2 diabetes mellitus on cardiac muscle contraction in the Goto‐Kakizaki rat. Exp Physiol. 2007;92:1029‐1036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials