

Abstract
Chronic ethanol consumption causes hepatic steatosis and inflammation, which are associated with liver hypoxia. Monocyte chemoattractant protein‐1 (MCP‐1) is a hypoxia response factor that determines recruitment and activation of monocytes to the site of tissue injury. The level of MCP‐1 is elevated in the serum and liver of patients with alcoholic liver disease (ALD); however, the molecular details regarding the regulation of MCP‐1 expression are not yet understood completely. Here, we show the role of liver X receptor α (LXR α) in the regulation of MCP‐1 expression during the development of ethanol‐induced fatty liver injury, using an antagonist, 22‐S‐hydroxycholesterol (22‐S‐HC). First, administration of 22‐S‐HC attenuated the signs of liver injury with decreased levels of MCP‐1 and its receptor CCR2 in ethanol‐fed mice. Second, hypoxic conditions or treatment with the LXR α agonist GW3965 significantly induced the expression of MCP‐1, which was completely blocked by treatment with 22‐S‐HC or infection by shLXR α lentivirus in the primary hepatocytes. Third, over‐expression of LXR α or GW3965 treatment increased MCP‐1 promoter activity by increasing the binding of hypoxia‐inducible factor‐1α to the hypoxia response elements, together with LXR α. Finally, treatment with recombinant MCP‐1 increased the level of expression of LXR α and LXR α‐dependent lipid droplet accumulation in both hepatocytes and Kupffer cells. These data show that LXR α and its ligand‐induced up‐regulation of MCP‐1 and MCP‐1‐induced LXR α‐dependent lipogenesis play a key role in the autocrine and paracrine activation of MCP‐1 in the pathogenesis of alcoholic fatty liver disease, and that this activation may provide a promising new target for ALD therapy.Copyright © 2014 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: LXRα, MCP‐1, HIF‐1, alcoholic liver disease
Introduction
Alcoholic liver disease (ALD) remains the most common cause of liver‐related mortality worldwide 1. The clinical spectrum of ALD includes simple steatosis, alcoholic steatohepatitis (ASH), progressive fibrosis, cirrhosis and hepatocellular carcinoma 1. Ethanol metabolism changes the redox state of the liver, which leads to alterations in the hepatic metabolism of lipids, carbohydrates, proteins and uric acids that subsequently produce a wide spectrum of hepatic injury, the most characteristic being interference in lipid metabolism and sustained inflammation 2. The essential roles of nuclear receptors in the pathogenesis of ethanol‐induced liver injury have been evident, given the diverse roles of nuclear receptors in cellular lipid metabolism and inflammation, and potential functions of nuclear receptor ligands such as fatty acids, cholesterol derivatives and other metabolites that are possibly generated after ethanol ingestion 3. However, the molecular details for the nuclear receptor functions in lipid metabolism and inflammation in the ethanol‐injured liver are not yet completely understood.
Monocyte chemoattractant protein‐1 (MCP‐1) and its receptor, chemokine (C–C motif) receptor 2 (CCR2), are major determinants for recruitment and activation of monocytes/macrophages to the site of tissue injury 4. A series of reports suggested a critical role for MCP‐1 in the pathogenesis of ALD. The level of MCP‐1 was elevated in the serum, liver and mononuclear cells of patients with ALD 5, 6. Recently, Manderkar et al 7 showed that deficiency of MCP‐1 protects mice against alcoholic liver injury by inhibiting the production of proinflammatory cytokines, such as tumour necrosis factor α (TNFα), IL‐1β and IL‐6, and by inducing fatty acid oxidation‐related genes, indicating an important role of MCP‐1 that links functions of proinflammatory cytokines to hepatic lipid metabolism. However, the mechanism by which expression of MCP‐1 is regulated during ethanol consumption has not been understood clearly. Recently, it was reported that chronic ethanol intake caused liver hypoxia and subsequent activation of hypoxia‐inducible factor‐1 (HIF‐1) 8. Interestingly, ethanol consumption up‐regulated MCP‐1, which triggered lipid accumulation via HIF‐1α activation, resulting in hepatic steatosis in ALD mice 8. HIF‐1α is directly involved in the hypoxia‐mediated transcriptional induction of MCP‐1 9. Recently, we reported that HIF‐1α and liver X receptor (LXRα) are activated via positive crosstalk that enhanced both lipogenesis and inflammatory response in macrophages 10. Together, these observations suggest a potential involvement of LXRα and HIF‐1α in the regulation of MCP‐1 in ethanol‐induced liver injury.
LXRα is a nuclear receptor that regulates genes controlling de novo hepatic lipogenesis, such as sterol regulatory element‐binding proteins (SREBP‐1c) and fatty acid synthase (FAS) 11. Expression of LXRα and the downstream lipogenic genes is enhanced in liver biopsies from diverse liver diseases accompanying steatosis, such as non‐alcoholic fatty liver disease (NAFLD) and hepatitis C virus‐associated hepatic steatosis 12. Ethanol consumption increases the rate of hepatic triglyceride synthesis and secretion in humans 13. Ethanol feeding enhances the fatty acid biosynthetic pathway by activation of SREBP‐1, which may contribute to the development of alcoholic fatty liver in mice 14. Ethanol‐induced hepatic steatosis is mediated by carbohydrate response element‐binding protein, which is an important regulator of hepatic lipogenesis and a downstream target of LXR 15. Based on these observations, we hypothesized that LXR may have a central role in ethanol‐induced hepatic steatosis, and that treatment of LXRα antagonists may have a beneficial effect in protecting against alcoholic liver injury.
Oxysterols, oxidized derivatives of cholesterol, including 22(R)‐HC, 24(S)‐HC, 24(S),25‐epoxycholesterol, 20(S)‐HC and 27‐HC, were reported to be activating ligands of LXRs 16. Interestingly, serum levels of agonistic oxysterols, such as 25‐HC and 27‐HC, are significantly increased in NAFLD patients 17. By contrast, 22‐S‐HC is considered to be an antagonistic ligand of LXRα, because it generates steric hindrance that inhibits the formation of the activating configuration of LXRα 16. 22‐S‐HC down‐regulated expression of the FAS gene through an LXR response element (LXRE) located in the promoter and abolished the effect of the synthetic LXRα agonist TO901317 18. In a rat model of high‐fat‐induced fatty liver, administration of 22‐S‐HC attenuated hepatic steatogenesis 19. Here, we report that inhibition of LXRα by 22‐S‐HC dramatically represses hepatic steatosis and the HIF‐1‐mediated activation of MCP‐1 in ethanol‐induced fatty liver injury. Importantly, LXRα and HIF‐1α mediated autocrine and paracrine activation of MCP‐1 in both hepatocytes and Kupffer cells (KCs), which function as a major determinant in the pathogenesis of alcoholic liver injury. Therefore, LXRα and its ligands are promising candidates for ALD therapy.
Materials and methods
Animal experiments
Eight week‐old male C57BL/6 mice were gradually habituated to a liquid Lieber–DeCarli liquid diet (Dyets, Bethlehem, PA, USA) with 5% v/v ethanol over a period of 2 weeks, then maintained on the 5% ethanol diet (36% ethanol‐derived calories) for 5 weeks. Consumption was recorded daily and isocaloric amounts of a nonethanol‐containing diet (in which dextran–maltose replaced calories from ethanol) were dispensed to pair‐fed animals. Body weights were recorded weekly. After 3 weeks of feeding with a 5% ethanol diet, mice from both the ethanol‐fed and pair‐fed groups were administered 22‐S‐HC (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a dose of 10 mg/kg/day in 0.2 ml 0.5% carboxymethyl cellulose solution by oral gavage for 2 weeks. The pair‐fed control group and the ethanol‐fed group consisted of six and eight mice, respectively. Serum was separated from whole blood and frozen at –80 °C. Liver tissue was rapidly excised and portions of the liver were stored for RNA extraction and western blotting, or fixed in 10% formalin for histopathological analysis. All experiments were conducted according to the guidelines of Seoul National University Institutional Animal Care and Use Committee.
For histological examination, a 3 µm section of Paraplast‐embedded tissue was stained routinely with haematoxylin and eosin (H&E). Oil red O staining of the frozen liver section was performed as described previously 20. Immunohistochemistry was examined with anti‐MCP‐1 antibody (Abcam, Cambridge, MA, USA) and anti‐LXRα antibody (Perseus Proteomics, Tokyo, Japan). Hepatic triglyceride was measured using a triglyceride colorimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA) and MCP‐1 protein was quantified using an ELISA kit (R&D Systems, Minneapolis, MN, USA).
Isolation and culture of primary mouse hepatocytes and Kupffer cells (KCs)
Primary hepatocytes and KCs were isolated from 8–10 week‐old male C57BL/6 N mice (Charles River Laboratories, Wilmington, MA, USA). The liver was perfused with Hanks's balanced salts solution (HBSS; Sigma‐Aldrich, St Louis, MO, USA) containing 0.05 mm ethylene glycol tetra‐acetic acid for 3 min and subsequently with HBSS containing 0.05% collagenase type IV (Sigma‐Aldrich). After centrifugation at 500 rpm for 3 min, the cell pellet containing hepatocytes was plated on a collagen‐coated plate with Medium 199 (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS), 23 mm HEPES and 10 nm dexamethasone. For isolation of KCs, non‐parenchymal, cell‐enriched supernatant was centrifuged in 50%/25% Percoll (GE Healthcare, Waukesha, WI, USA). The KCs‐enriched layer was plated and cultured in RPMI 1640 medium (HyClone) with 10% FBS. The cells were grown in an incubator with 5% CO2 and 95% air at 37 °C.
For a co‐culture system, the primary hepatocytes (1.5 × 105) were seeded together with the KCs (7 × 105); 4 h after seeding, the cells were treated with 1 µm GW3965 for 48 h; at the end of treatment, the medium was collected for measurement of MCP‐1 and the cells were stained with Nile red or were harvested for measurement of mRNA.
Cell lines and reagents
Huh‐7 cells were maintained in RPMI containing 10% FBS at 37 °C in a 5% CO2/95% air incubator. The cells were exposed to hypoxia or treated with TO901317 (Alexis Biochemicals, San Diego, CA, USA), GW3965 (Sigma‐Aldrich), 22‐S‐HC or recombinant human MCP‐1 (R&D Systems). Hypoxic conditions were generated by exposing cells to 0.1% O2 or treating cells with desferrioxamine (DFO), a known hypoxia‐mimicking reagent 21.
Plasmids, small interfering RNA (siRNA), and lentiviral‐shLXR α
The reporters, human LXRα promoter–Luc, human SREBP‐1c promoter–Luc, LXRE–Luc and the eukaryotic expression vector pCMV–Myc–LXRα, were as described previously 20, 21. A 98 bp HIF‐1 binding sequence from human wild‐type or mutant MCP‐1 promoter region was cloned into the pGL3–basic vector 22. Transient transfection, reporter gene analysis and transfection of siRNA were described previously 19, 20. The siRNA duplexes targeting LXRα and non‐specific siRNA and siGFP are described in Table S1 (see supplementary material).
The lentiviral vector containing the sequences of shRNA specifically targeting the mouse LXRα and scrambled control sequence were cloned into the pLKO.1 TRC cloning vector. All of the newly constructed vectors were verified by DNA sequencing. The sequences of shLXRα and shGFP are described in Table S1 (see supplementary material) 23. The recombinant lentiviruses were produced by transient transfection of the lentiviral vector, the lentiviral packaging vector psPAX2 and pMD2.G envelope plasmid into HEK293, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The supernatant containing viruses was collected from the culture on day 3 after transfection by centrifugation at 2500 rpm for 3 min, and the titre was determined.
Western blotting, real‐time PCR (qRT–PCR) and chromatin immunoprecipitation (ChIP)
Western blotting was basically performed as previously described, using specific antibodies against MCP‐1 (Abcam, Cambridge, UK), LXRα (Affinity BioReagents, Golden, CO, USA), HIF‐1α, SREBP‐1c, SCD‐1, FAS, β‐actin (Santa Cruz Biotechnology) or α‐tubulin (Calbiochem, San Diego, CA, USA) 19, 20. qRT–PCR amplifications were performed using specific primers, as shown in Table S2 (see supplementary material) 19. ChIP assays were carried out using specific antibodies against LXRα (Affinity BioReagents), HIF‐1α, CBP and p300 (Santa Cruz Biotechnology) 21. Immunoprecipitated DNA was amplified by PCR with specific primers corresponding to the flanking region of the LXRE on the human LXRα promoter and SREBP‐1 promoter, or the hypoxia response element (HRE), on the human MCP‐1 promoter, as described previously 21, 22, 24.
Nile red staining and FACS analysis
Huh‐7 cells, mouse primary hepatocytes, or KCs were seeded and incubated overnight. In Huh‐7 cells, siRNAs were transfected twice at 48 h intervals with FuGENE siRNA, and then the cells were treated with rhMCP‐1, GW3965, DFO or 22‐S‐HC 20, 21. At the end of treatment, Nile red staining and FACS analysis were performed as previously described 20, 21.
Results
22‐S‐HC, an LXR α antagonist, prevents the development of ethanol‐induced fatty liver injury in mice
To investigate the role of LXRα in ethanol‐induced liver damage, both the ethanol‐fed and pair‐fed groups were administered 22‐S‐HC at 10 mg/kg/day for 2 weeks. Prolonged ethanol feeding resulted in fatty liver injury, as assessed by the serum levels of glutamate pyruvate transaminase (GPT), glutamic oxaloacetic transaminase (GOT) and triglyceride (TG) (Figure 1A). The number and size of hepatic lipid droplets were largely increased after ethanol exposure when examined by H&E and oil red O staining (Figure 1B; see also supplementary material, Figure S1A). Consistently, the hepatic levels of TG and malondialdehyde (MDA) were significantly increased after ethanol feeding (Figure 1A). However, the administration of 22‐S‐HC dramatically lowered all these fatty liver injury variables, as well as the ratio of liver weight:body weight (Figure 1A, B; see also supplementary material, Figure S1A, B). Also, the increased levels of proinflammatory cytokines, such as TNFα, IL‐1β and IL‐6 in the ethanol‐fed mice were significantly decreased by the administration of 22‐S‐HC (see supplementary material, Figure S1C). The expression of LXRα and its lipogenic downstream genes was highly up‐regulated at both the mRNA and protein levels in the livers of ethanol‐fed mice, but was maintained at control levels by administration of 22‐S‐HC (Figure 1C; see also supplementary material, Figure S2). These findings suggest that activation of LXRα is closely associated with ethanol‐induced liver injury.
Figure 1.
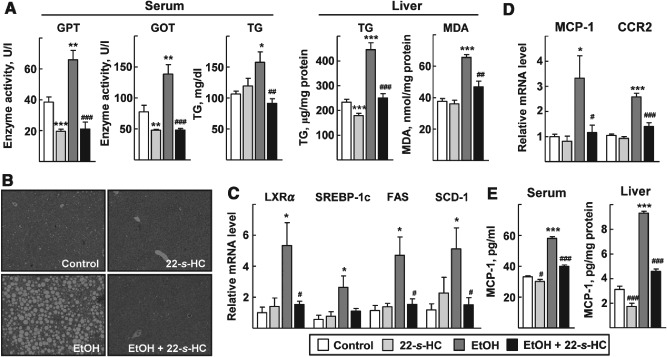
22‐S‐HC protects against ethanol‐induced fatty liver injury in mice. After an ethanol (EtOH)‐containing diet or control diet for 3 weeks, mice were administered vehicle or 22‐S‐HC for 2 weeks. (A) GPT and GOT enzyme activities and the amount of TG and MDA in the serum or liver tissues were quantified. (B) Liver sections were stained with H&E (×200). (C, D) Total RNAs obtained from liver tissues were subjected to qRT–PCR for determination of mRNA levels of the indicated genes. (E) Amounts of MCP‐1 protein in the serum or liver tissues were quantified by ELISA; numbers represent mean ± SD (n = 6 for the pair‐fed control and 22‐S‐HC groups; n = 8 for the ethanol‐fed and ethanol‐with ‐22‐S‐HC group); *p < 0.05, **p < 0.05 and ***p < 0.001 compared with the pair‐fed control group; # p < 0.05, ## p < 0.01 and ### p < 0.001 compared with the ethanol‐fed group; comparisons between groups were performed using the Mann–Whitney U‐test
Because MCP‐1 is one of the major cytokines that plays a role in the pathogenesis of ethanol‐induced liver injury 7, we determined the level of hepatic expression of MCP‐1 and its receptor CCR2 after administration of 22‐S‐HC. The hepatic levels of MCP‐1 and CCR2 were increased significantly in the ethanol‐fed mice; however, the levels were dramatically lowered by 22‐S‐HC (Figure 1D). The amount of MCP‐1 in the serum and liver increased after ethanol feeding, but was decreased by administration of 22‐S‐HC (Figure 1E). These data clearly showed a preventive role for 22‐S‐HC against ethanol‐induced liver injury and suggested involvement of LXRα and MCP‐1 in the protective function of 22‐S‐HC.
LXR α and HIF‐1 induce expression of MCP‐1 synergistically in hepatocytes
Because expression of MCP‐1 is transcriptionally activated by HIF‐1 under hypoxia, and the crosstalk between LXRα and HIF‐1α plays an important role in regulation of lipogenic genes and proinflammatory cytokines, we hypothesized that LXRα may contribute to the HIF‐1‐mediated transcriptional activation of MCP‐1 in the ethanol‐induced fatty liver injury 9, 10. Thus, we examined a potential involvement of LXRα in MCP‐1 expression in primary hepatocytes. First, activation of LXRα or HIF‐1α by treatment with the LXRα agonist GW3965, or a hypoxia‐mimicking agent, DFO, respectively, increased the protein level of MCP‐1 when examined by ELISA and western blotting. Co‐treatment with GW3965 and DFO further increased MCP‐1 levels (Figure 2; see also supplementary material, Figure S3). Expression of LXRα and its target SREBP‐1c protein was increased by either GW3965 or DFO treatment (Figure 2A; see also supplementary material, Figure S3). Similar results were obtained when LXRα and HIF‐1α were activated by another LXRα agonist, TO901317, or actual hypoxic conditions, respectively (see supplementary material, Figure S4). However, these increases in the level of MCP‐1 were largely repressed by 22‐S‐HC treatment (Figure 2A; see also supplementary material, Figure S3A) or lentiviral shLXRα infection (Figure 2B; see also supplementary material, Figure S3B). To address directly the involvement of LXRα in MCP‐1 expression, LXRα was exogenously introduced into the liver cells. Over‐expression of LXRα induced the mRNA level of MCP‐1 and additional treatment with DFO further increased MCP‐1 mRNA, suggesting that LXRα is associated with hypoxia‐induced MCP‐1 expression (Figure 2C). Rapamycin and bortezomib, which are known to inhibit HIF‐1, largely decreased the GW3965‐induced mRNA level of MCP‐1, demonstrating that the induction of MCP‐1 was both LXRα‐ and HIF‐1α‐dependent (Figure 2D) 25, 26.
Figure 2.
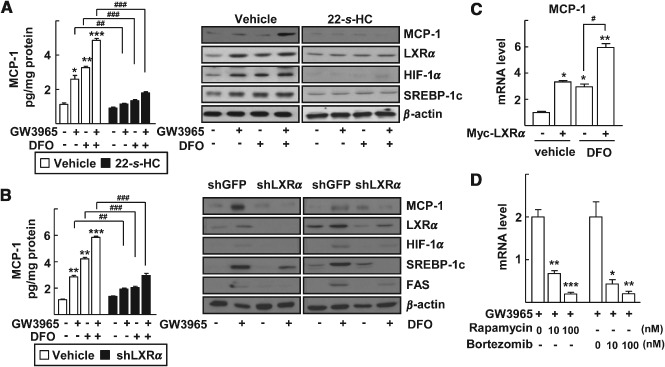
Induction of MCP‐1 by LXRα and HIF‐1α in primary mouse hepatocytes. (A) Primary hepatocytes were treated with the indicated combinations of 1 µm GW3965, 100 µm DFO and 1 µm 22‐S‐HC for 24 h. (B) Primary hepatocytes were infected by lenti‐shGFP or lenti‐shLXRα viruses. After 24 h of viral infection, the cells were treated with the indicated combinations of 1 µm GW3965 and 100 µm DFO for an additional 24 h. MCP‐1 secreted into the culture supernatants was quantified by ELISA (left). The numbers represent mean ± SD (n = 3); *p < 0.05, **p < 0.01 and ***p < 0.001 compared with no treatment; ## p < 0.01 and ### p < 0.001 compared with vehicle or shGFP, as indicated: expressions of protein levels in whole‐cell lysates were analysed by western blotting (right). (C) Huh‐7 cells were transfected with empty vector or Myc‐LXRα; after 24 h, the cells were treated with vehicle or 100 µm DFO; mRNA levels of MCP‐1 were measured by qRT–PCR; numbers represent mean ± SD (n = 3); *p < 0.05 and **p < 0.01 compared with empty vector transfection and vehicle treated control; ## p < 0.01 and ### p < 0.001 compared with the treatment of DFO. (D) Huh‐7 cells were treated with 1 µm GW3965 in the presence or absence of rapamycin or bortezomib for 24 h; mRNA levels of MCP‐1 were measured by qRT–PCR; numbers represent mean ± SD (n = 3); *p < 0.05, **p < 0.01 and ***p < 0.001 compared with GW3965 alone; statistical significance was evaluated by two‐way ANOVA
Next, we considered the molecular mechanism of induction of MCP‐1 by LXRα. DFO treatment increased the mRNA levels of MCP‐1 and vascular endothelial growth factor (VEGF) in the primary hepatocytes, as previously shown in human astrocytes (Figure 3A) 9. Similarly, treatment by GW3965 led to a significant up‐regulation of MCP‐1 mRNA levels and those of LXRα and its target genes (Figure 3A). The promoter of the MCP‐1 gene is known to encode three HREs that are activated by hypoxia 9. When we examined activity of the reporter encoding MCP‐1 promoter, it was dramatically increased by exotic expression of HIF‐1α or LXRα, and by treatment of DFO or GW3965. However, activity of the MCP‐1 promoter reporter containing mutations in the HREs was much lower compared with that of the wild‐type promoter by the same treatment (Figure 3B).
Figure 3.
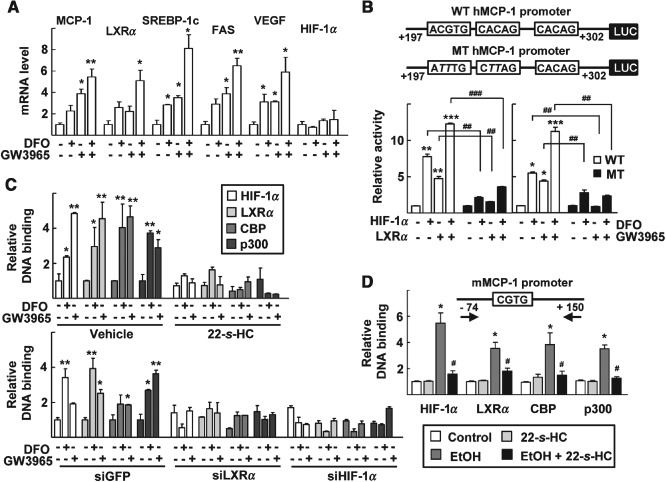
LXRα induces transcription of MCP‐1 through activation of the HRE present in MCP‐1 promoter. (A) Primary hepatocytes were treated with 1 µm GW3965 and/or 100 µm DFO for 24 h; mRNA levels of the indicated genes were analysed by qRT–PCR; numbers represent mean ± SD (n = 3); *p < 0.05 and **p < 0.01 compared with the vehicle‐treated control. (B) Schematic representation of the human MCP‐1 promoter investigated in this study; squares, HRE and its mutant sequence (top); Huh‐7 cells were transiently transfected with the wild‐type (WT; □) or mutant (MT; ▪) human MCP‐1 promoter reporter, together with empty vector or expression vectors for HIF‐1α and LXRα, as indicated (left); or the transfected cells were treated with 100 µm DFO and/or 1 µm GW3965 for 24 h (right); numbers represent mean ± SD (n = 3); *p < 0.05, **p < 0.01 and ***p < 0.001 compared with empty vector transfection or vehicle‐treated control; ## p < 0.01 and ### p < 0.001 compared with treatment with the WT promoter. (C) Huh‐7 cells were treated with 1 µm GW3965 or 100 µm DFO in the absence or presence of 1 µm 22‐S‐HC for 24 h (top): Huh‐7 cells were transfected with siGFP, siLXRα or siHIF‐1α for 24 h and then treated with 1 µm GW3965 or 100 µm DFO for an additional 24 h (bottom); numbers represent mean ± SD (n = 3); *p < 0.05 and **p < 0.01 compared with the vehicle‐treated control; statistical significance was evaluated by two‐way ANOVA. (D) Schematic representation of the mouse‐specific primers used in the ChIP assay (top). As described in Materials and methods, after an ethanol‐containing diet treatment for 3 weeks, mice were administered with vehicle or 22‐S‐HC for an additional 2 weeks. ChIP DNA samples were prepared from the indicated mouse livers to amplify the MCP‐1 promoter regions; numbers represent mean ± SD (n = 3); *p < 0.05 compared with the pair‐fed control group; # p < 0.05 compared with the ethanol‐fed group; comparisons between groups were performed using the Mann–Whitney U‐test; DNA fragments that immunoprecipitated using specific antibodies as indicated were amplified by PCR, using specific primers
ChIP analysis showed that treatment by DFO or GW3965 increased DNA binding of HIF‐1α on the HREs in the MCP‐1 promoter (Figure 3C). Interestingly, LXRα also bound the HREs together with co‐activators CBP and p300. The recruitment of these proteins was efficiently inhibited by treatment with 22‐S‐HC or by silencing of LXRα or HIF‐1α (Figure 3C). Together, these results suggest that LXRα enhances the transcription of MCP‐1, probably by recruiting HIF‐1α together with co‐activators into the promoter of the MCP‐1 gene. In vivo ChIP assays further showed that HIF‐1α, LXRα and their co‐activators bound a HRE in the mouse MCP‐1 promoter in the livers of ethanol‐fed mice, but these bindings were significantly reduced after administration of 22‐S‐HC (Figure 3D).
MCP‐1 induces LXR α‐dependent lipogenesis in hepatocytes
MCP‐1 plays an important role in the induction of hepatic lipogenesis, which contributes to ethanol‐induced fatty liver disease 7, 8. Therefore, we examined whether LXRα is involved in MCP‐1‐induced lipogenesis. Treatment with recombinant MCP‐1 protein induced accumulation of lipid droplets in the cytosol of primary hepatocytes; however, it was completely blocked by treatment with 22‐S‐HC (Figure 4A, B). Silencing of either LXRα or HIF‐1α repressed the MCP‐1‐induced lipogenesis (Figure 4C, left). The protein levels of LXRα, its downstream lipogenic genes and HIF‐1α were increased by MCP‐1 treatment, but were abolished by silencing LXRα (Figure 4C, right). These results indicate that MCP‐1 increases the expression of LXRα, which may mediate MCP‐1‐induced lipogenesis in the liver.
Figure 4.
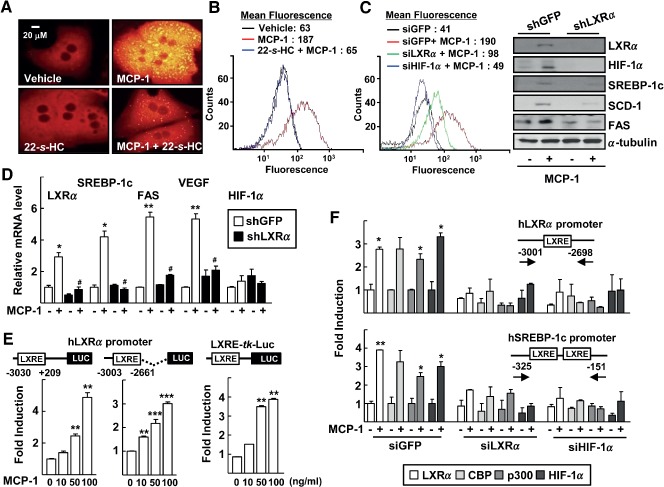
MCP‐1 induces LXRα‐dependent lipid accumulation in primary hepatocytes. (A) Hepatocytes were treated with 50 ng rhMCP‐1 protein and/or 1 µm 22‐S‐HC for 48 h. At the end of treatment, lipid droplets were stained using Nile red and visualized by fluorescence microscopy (×400). (B) Huh‐7 cells were treated with recombinant human MCP‐1 and/or 22‐S‐HC for 48 h and then their neutral lipid content was analysed by flow cytometry after Nile red staining; geometric mean fluorescence intensity values for peaks were indicated (top). (C) Huh‐7 cells were transfected with siGFP, siLXRα or siHIF‐1α and then treated with rhMCP‐1 for 48 h; their neutral lipid content was analysed by flow cytometry after Nile red staining (left); hepatocytes were infected by lenti‐shGFP or lenti‐shLXRα viruses for 24 h and then treated with 50 ng rhMCP‐1 protein for an additional 24 h; whole‐cell lysates were analysed for protein expression by western blotting (right). (D) Hepatocytes were treated with 50 ng rhMCP‐1 protein for 24 h; total RNA was prepared and analysed for expression of the indicated transcripts by qRT–PCR; numbers represent mean ± SD (n = 3); *p < 0.05 and **p < 0.01 compared with vehicle‐treated control; # p < 0.05 compared with the MCP‐1 treatment with shGFP transfection. (E) Huh‐7 cells were transiently transfected with the indicated human LXRα promoter–Luc (left and centre) or LXRE‐tk‐Luc (right); transfected cells were treated with rhMCP‐1 for 24 h; numbers represent mean ± SD (n = 3); **p < 0.01 and ***p < 0.01 compared with the vehicle‐treated control; statistical significance was evaluated by two‐way ANOVA. (F) Huh‐7 cells were transfected with siLXRα or siHIF‐1α for 24 h and then treated with rhMCP‐1 for an additional 24 h. DNA fragments that immunoprecipitated using the indicated specific antibodies were amplified by PCR, using specific primers for LXRα promoter (top) or the SREBP‐1 promoter (bottom); numbers represent mean ± SD (n = 3); *p < 0.05 and **p < 0.01 compared with vehicle‐treated control; statistical significance was evaluated by two‐way ANOVA
Next, we examined the mechanism by which MCP‐1 induces activation of LXRα. As shown in Figure 4D, MCP‐1 treatment increased levels of LXRα at the mRNA level. Consistent with this finding, the 3 kb LXRα upstream promoter (–3030 to +209) was activated by recombinant MCP‐1 treatment in a dose‐dependent manner. The LXRα promoter reporters encoding the LXRE (–3003 to –2661) or the reporter encoding a consensus LXRE were activated by MCP‐1, suggesting that MCP‐1 activates the transcription of LXRα through the LXRE site present on the LXRα promoter (Figure 4E). A ChIP assay showed that both LXRα and HIF‐1α bound to the LXRE present on the human LXRα promoter or human SREBP‐1c promoter in the presence of MCP‐1. This DNA binding was inhibited by siLXRα or siHIF‐1α, indicating that crosstalk between LXRα and HIF‐1α plays a critical role in MCP‐1‐induced LXRα gene expression and other LXRα‐dependent lipogenic gene expression (Figure 4F).
Regulation of MCP‐1 expression and lipid droplet accumulation in KCs
Although hepatocytes secrete MCP‐1, KCs are considered to be the main source of MCP‐1, which induces autocrine and paracrine activation of inflammatory signalling in the liver 4. In the primary cultured KCs, treatment by GW3965 or DFO increased mRNA expression not only of MCP‐1 but also CCR2 (Figure 5A). Like effects seen in hepatocytes, treatment by MCP‐1 induced mRNA expression of genes in the lipogenic pathway, such as LXRα and FAS, and proinflammatory cytokines including TNFα, IL‐1β and MCP‐1 itself in the KCs (Figure 5B). Co‐treatment with 22‐S‐HC significantly decreased the expression of these genes (Figure 5B). Consistently, treatment by TO901317 or DFO induced lipid droplet accumulation in the KCs (Figure 5C). Also, treatment by MCP‐1 largely induced lipid droplet accumulation in the cells, which was dramatically decreased by co‐treatment with 22‐S‐HC (Figure 5C). When the primary hepatocytes were co‐cultured with KCs, the level of secreted MCP‐1 protein after GW3965 treatment was much higher compared with hepatocyte or KC cultures (Figure 5D). Consistently, the mRNA level of MCP‐1 was significantly increased in the co‐culture system (see supplementary material, Figure S5A). Accumulation of lipid droplets was dramatically increased when hepatocytes were co‐cultured with KCs (see supplementary material, Figure S5B). Together, these results suggest that the crosstalk between LXRα and HIF‐1α plays key roles in both hepatocytes and the KCs, which mediates autocrine and paracrine activation of the MCP‐1‐induced lipogenesis and inflammation network in ethanol‐induced liver injury (Figure 5E).
Figure 5.
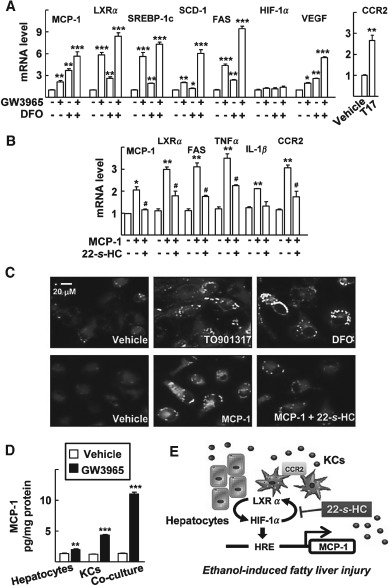
MCP‐1 induces lipogenesis in KCs via activation of LXRα and HIF‐1α. (A) KCs were treated with 1 µm GW3965 and/or 100 µm DFO for 24 h; mRNA levels of the indicated genes were analysed by qRT–PCR. (B) KCs were treated with 50 ng recombinant human MCP‐1 protein and/or 1 µm 22‐S‐HC for 48 h; numbers represent mean ± SD (n = 3); *p < 0.05, **p < 0.01 and ***p < 0.01 compared with vehicle‐treated control; # p < 0.05 compared with MCP‐1 treatment; statistical significance was evaluated by two‐way ANOVA. (C) KCs were treated with vehicle (DMSO), 100 µm DFO or 1 µm TO901317 for 48 h, or with vehicle (H2O), 50 ng recombinant human MCP‐1 protein and/or 1 µm 22‐S‐HC for 48 h; following treatment, lipid droplets were stained using Nile red and visualized by fluorescence microscopy (×400). (D) The primary hepatocytes were cultured with or without KCs for 4 h, and with vehicle or GW3965 1 µm for 48 h; MCP‐1 protein secreted into the culture supernatants was quantified by ELISA; numbers represent mean ± SD (n = 3); **p < 0.01 and ***p < 0.001 compared with vehicle treatment; statistical significance was evaluated by two‐way ANOVA. (E) Schematic model for the activation of MCP‐1 by LXRα and HIF‐1α, which mediates ethanol‐induced fatty liver injury
Discussion
Preferential elevation of MCP‐1 in the serum and the liver of ALD patients and its importance in the modulation of proinflammatory cytokines such as TNFα, IL‐1β and IL‐6 support the pathological importance of MCP‐1 in the development of ALD 5, 7. However, the mechanism by which expression of MCP‐1 is regulated during consumption of ethanol has not yet been clearly understood. Here, our observation that LXRα potentiated the activation of HIF‐1α, especially through the upstream promoter of MCP‐1, provides a potential mechanism for the ethanol‐induced induction of MCP‐1. The activation of LXRα during ethanol exposure may induce the positive activation circuit of HIF‐1α and LXRα on the HRE of the MCP‐1 promoter, which was similarly seen in the generation of foam cells in atherosclerotic lesions 10. Similarly, it was reported that activation of LXRα potentiates LPS responses, such as induction of TLR4 and secretion of TNFα and MCP‐1 in human macrophages 27. These observations indicate that LXRα has primary functions in the pathological roles of MCP‐1 by inducing its expression, especially in the hypoxic conditions generated during alcoholic fatty liver injury.
MCP‐1 exerts its action via CCR2 or independent of CCR2, which is expressed in monocyte/macrophages but not in liver cells 7. We observed that the ligand‐induced activation of LXRα increased MCP‐1 expression in both hepatocytes and KCs, indicating that both autocrine and paracrine activation loops of MCP‐1 could be activated by LXRα (Figure 5E). MCP‐1 may function via a CCR2‐dependent mechanism in KCs, because treatment with recombinant MCP‐1 induced CCR2 expression in KCs (Figure 5A). By contrast, MCP‐1 acts in a CCR2‐independent manner in hepatocytes, because CCR2 was absent in these cells. To understand the autoactivation of MCP‐1, the mechanism by which MCP‐1 induces activation of LXRα should be clarified. Because MCP‐1 induces activation of mitogen‐activated protein kinases (MAPKs), which may phosphorylate LXRα, the MCP‐1‐induced activation of MAPKs might contribute to the activation of LXRα 28, 29. On the other hand, lipid metabolites that are generated during the action of MCP‐1 might function as activating ligands for LXRα. For instance, the amount of 27‐HC was increased in the liver of NAFLD 17. Moreover, the mechanism by which expression of CCR2 is altered during ethanol consumption should be further investigated. Interestingly, CCR2 is a hypoxia‐modulated cytokine receptor and the CCR2 promoter contains a putative HRE 30. Therefore, positive crosstalk between HIF‐1α and LXRα might contribute to ethanol‐induced CCR2 activation in the liver.
Among the nuclear receptors, mostly peroxisome proliferator‐activated receptors (PPARs) have been examined for their roles in ALD 3. For example, hepatocyte damage after ethanol‐feeding was significant in PPARα‐null mice, but not in wild‐type mice 31. Treatment with PPAR agonists, including pioglitazone, reduced the toxic effects of ethanol and reversed hepatic fat accumulation 32, 33. Recently, oestrogen‐related receptor γ was shown to be an important downstream mediator of cannabinoid 1 receptor that activated expression of CYP2E1, resulting in increases in the level of reactive oxygen species and liver injury by ethanol 34. Because crosstalk between nuclear receptors provides divergent and convergent control in lipogenesis and inflammation, the potential crosstalk between LXRα and PPARs or ERRγ may contribute to the pathogenesis of alcoholic liver disease. Indeed, PPARγ is a downstream target of LXRα, which has an LXRE motif in its upstream promoter region 35. Moreover, the involvement of LXRα in oestrogen‐related receptor γ‐mediated CYP2E1 regulation could be possible, because the administration of 22‐S‐HC in ethanol‐fed mice decreased the mRNA levels of ERRγ, cannabinoid 1 receptor and CYP2E1 (see supplementary material, Figure S6). Therefore, further understanding of the nuclear receptor networks in lipogenesis and inflammation may provide useful information by which to understand the pathogenesis of ALD, and for the development of new pathophysiology‐based therapies against ALD.
Dysregulation of cholesterol metabolism is closely associated with human metabolic diseases such as NAFLD and insulin resistance. NAFLD patients showed significantly elevated levels of serum oxysterols, such as 4β‐HC, 25‐HC and 27‐HC, which are agonistic ligands of LXRα 16. In animal models of NAFLD, administration of cholic acid or bile duct ligation had a beneficial effect on fatty liver, and was accompanied by decreased hepatic levels of oxysterols 36. Synthesis of these oxysterols is catalysed by several cytochrome P450 enzymes, such as sterol 27‐hydroxylase (CYP27A1) and sterol 24‐hydroxylase (CYP46A1) 37. Interestingly, expression of sterol 27‐hydroxylase is LXR‐dependent 38. These findings indicate that oxysterols may regulate their own levels by activating nuclear receptors, including LXRα. Cholic acid and cholesterol sulphate, the endogenous ligands of farnesoid X receptor (FXR) and retinoic acid receptor‐related orphan receptor‐α (RORα), respectively, inhibit the activation of LXRα, resulting in attenuation of hepatic steatosis 24, 36. Therefore, a balance between different cholesterol metabolites and their receptors seems to be an important determinant that decides the progression of metabolic diseases. However, to our knowledge, changes in the levels of fatty acids, cholesterol and their metabolites during chronic ethanol ingestion have not been examined. Therefore, further studies are necessary to understand cholesterol metabolism, including various metabolites and their potential as ligands of nuclear receptors in ALD patients.
Current clinical management for severe forms of alcoholic steatohepatitis focuses on alcohol abstinence and specific pharmacological treatments, including corticosteroids, pentoxifylline, anti‐TNF agents and antioxidant substances such as N‐acetylcysteine 1. However, benefits from these treatments remain controversial and appropriate guidelines have not been recommended for intermediate severity or low‐risk ALD. To overcome these issues, complete understanding of the pathophysiology of ALD and identification of new therapeutic targets are required. Here, our study shows the critical roles of the LXRα‐mediated autocrine/paracrine activation of MCP‐1 in a mouse model of alcoholic steatohepatitis. Together with more clinical and experimental research that elucidates LXR as a potential therapeutic target, development of LXRα antagonists may provide novel therapeutics that block the development and progression of ALD.
Author contributions
TN, BL, SK and ML designed the study and interpreted the results; TN, YH, NK, HP and YK performed the experiments; and TN, ML wrote the manuscript.
Supplementary material on the internet.
The following supplementary material may be found in the online version of this article:
Figure S1. 22‐S‐HC, antagonist of LXRα, inhibits ethanol‐induced fatty liver
Figure S2. Expression of LXRα from ethanol‐fed mice
Figure S3. Activation of LXRα and HIF‐1 induces MCP‐1 expression
Figure S4. LXRα and HIF‐1α synergistically induce expression of MCP‐1
Figure S5. MCP‐1 mRNA levels and neutral lipids in the co‐culture system of the primary hepatocytes with KCs
Figure S6. Antagonist of LXRα inhibits ERRγ‐mediated induction of CYP2E1
Table S1. Sequences used for RNA interference
Table S2. Primer sequences used for qRT–PCR and ChIP
Supporting information
22‐S‐HC, antagonist of LXRα inhibits ethanol‐induced fatty liver. After an ethanol‐containing diet treatment for 3 weeks, mice were administered vehicle or 22‐S‐HC for 2 weeks. (A) Liver sections were fixed in formalin and stained with oil red O (×100). (B) Liver weight:body weight ratio was measured after 5 weeks of ethanol feeding. (C) Total RNA from liver tissue was subjected to qRT–PCR for the determination of mRNA levels of TNFα, IL‐1β and IL‐6; numbers represent mean ± SD (n = 6 for the pair‐fed control and 22‐S‐HC groups; n = 8 for the ethanol‐fed and ethanol with 22‐S‐HC groups); *p < 0.05 and **p < 0.01 compared with the pair‐fed control group; # p < 0.05 and ## p < 0.01 compared with the ethanol‐fed group; comparisons between groups were performed using the Mann–Whitney U‐test.
Expression of LXRα from ethanol‐fed mice. (A) After an ethanol‐containing diet treatment for 3 weeks, mice were administered vehicle or 22‐S‐HC for 2 weeks; immunohistochemical staining for LXRα in liver (×100). (B) Whole‐cell lysates were obtained from the livers of the indicated mice and they were analysed for protein levels by western blotting (top); the density of each protein band was determined using an image‐analysis system and normalized to that of the corresponding α‐tubulin (bottom); y axis represents the relative protein level for comparison with that of pair‐fed control group; **p < 0.005 compared with the pair‐fed control group (n = 6); ## p < 0.01 compared with the ethanol‐fed group (n = 8); comparisons between groups were performed using the Mann–Whitney U‐test
Activation of LXRα and HIF‐1 induces MCP‐1 expression. The band intensities of western blots shown in Figure 2A, B, and two other blots with the same experiments were measured and normalized with that of β‐actin; numbers represent mean ± SD (n = 3); *p < 0.05, **p < 0.01 and ***p < 0.001 compared with vehicle treatment; # p < 0.05, ## p < 0.01 and ### p < 0.001 compared with vehicle or shGFP, as indicated; statistical significance was evaluated by two‐way ANOVA
LXRα and HIF‐1α synergistically induce expression of MCP‐1. Huh‐7 cells were treated with 1 µm TO901317 or exposed with 0.1% O2 for 24 h. MCP‐1 protein in culture supernatants was quantified using an ELISA kit; numbers represent mean ± SD (n = 3); **p < 0.01 and ***p < 0.001 compared with vehicle‐treated control; statistical significance was evaluated by two‐way ANOVA
MCP‐1 mRNA levels and neutral lipids in the co‐culture system of the primary hepatocytes with KCs. Primary hepatocytes were seeded with or without KCs for 4 h and treated with vehicle or GW3965 1 µm for 48 h. (A) mRNA levels of MCP‐1 were measured by qRT–PCR. The numbers represent mean ± SD (n = 3); **p < 0.01 and ***p < 0.001 compared with vehicle treatment; # p < 0.01 and ## p < 0.001 compared with the indicated culture; statistical significance was evaluated by two‐way ANOVA. (B) The cells were stained using Nile red and visualized by fluorescence microscopy (×320)
Antagonist of LXRα inhibits ERRγ‐mediated induction of CYP2E1. After an ethanol‐containing diet treatment for 3 weeks, mice were administered vehicle or 22‐S‐HC for 2 weeks. Total RNA from liver tissue was subjected to qRT–PCR for determination of mRNA levels of ERRγ, CB1R and CYP2E1; numbers represent mean ± SD (n = 6 for the pair‐fed control and the 22‐S‐HC groups; n = 8 for the ethanol‐fed and the ethanol with 22‐S‐HC groups); **p < 0.01 compared with pair‐fed control group; # p < 0.05 and ## p < 0.01 compared with ethanol‐fed group; comparisons between groups were performed using the Mann–Whitney U‐test
Sequences used for RNA interference
Primer sequences used for qRT–PCR and ChIP
Acknowledgements
This study was supported by grants from the National Research Foundation of Korea (NRF; Grant Nos 2013R1A1A2063195, 2014R1A2A1A10052265, 2012M3A9B6055338, and 2007‐0056817).
No conflicts of interest were declared.
References
- 1. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 2012; 57: 399–420. [DOI] [PubMed] [Google Scholar]
- 2. Zakhari S, Li TK. Determinants of alcohol use and abuse: impact of quantity and frequency patterns on liver disease. Hepatology 2007; 46: 2032–2039. [DOI] [PubMed] [Google Scholar]
- 3. Gyamfi MA, Wan YJ. Pathogenesis of alcoholic liver disease: the role of nuclear receptors. Exp Biol Med 2010; 235: 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deshmane SL, Kremlev S, Amini S, et al. Monocyte chemoattractant protein‐1 (MCP‐1): an overview. J Interferon Cytokine Res 2009; 29: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fisher NC, Neil DA, Williams A, et al. Serum concentrations and peripheral secretion of the β‐chemokines monocyte chemoattractant protein 1 and macrophage inflammatory protein 1α in alcoholic liver disease. Gut 1999; 45: 416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186: 82–89. [DOI] [PubMed] [Google Scholar]
- 7. Mandrekar P, Ambade A, Lim A, et al. An essential role for monocyte chemoattractant protein‐1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology 2011; 54: 2185–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nath B, Levin I, Csak T, et al. Hepatocyte‐specific hypoxia‐inducible factor‐1α is a determinant of lipid accumulation and liver injury in alcohol‐induced steatosis in mice. Hepatology 2011; 53: 1526–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mojsilovic‐Petrovic J, Callaghan D, Cui H, et al. Hypoxia‐inducible factor‐1 (HIF‐1) is involved in the regulation of hypoxia‐stimulated expression of monocyte chemoattractant protein‐1 (MCP‐1/CCL2) and MCP‐5 (Ccl12) in astrocytes. J Neuroinflamm 2007; 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Na TY, Lee HJ, Oh HJ, et al. Positive cross‐talk between hypoxia inducible factor‐1α and liver X receptor α induces formation of triglyceride‐loaded foam cells. Arterioscler Thromb Vasc Biol 2011; 31: 2949–2956. [DOI] [PubMed] [Google Scholar]
- 11. Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol‐activated receptors LXR and FXR. Nat Rev Mol Cell Biol 2012; 13: 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lima‐Cabello E, García‐Mediavilla MV, Miquilena‐Colina ME, et al. Enhanced expression of pro‐inflammatory mediators and liver X‐receptor‐regulated lipogenic genes in non‐alcoholic fatty liver disease and hepatitis C. Clin Sci 2011; 120: 239–250. [DOI] [PubMed] [Google Scholar]
- 13. Crouse JR, Grundy SM. Effects of alcohol on plasma lipoproteins and cholesterol and triglyceride metabolism in man. J Lipid Res 1984; 25: 486–496. [PubMed] [Google Scholar]
- 14. You M, Fischer M, Deeg MA, et al. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element‐binding protein (SREBP). J Biol Chem 2002; 277: 29342–29347. [DOI] [PubMed] [Google Scholar]
- 15. Liangpunsakul S, Ross RA, Crabb DW. Activation of carbohydrate response element‐binding protein by ethanol. J Investig Med 2013; 61: 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Svensson S, Ostberg T, Jacobsson M, et al. Crystal structure of the heterodimeric complex of LXRα and RXRβ ligand‐binding domains in a fully agonistic conformation. EMBO J 2003; 22: 4625–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ikegami T, Hyogo H, Honda A, et al. Increased serum liver X receptor ligand oxysterols in patients with non‐alcoholic fatty liver disease. J Gastroenterol 2012; 47: 1257–1266. [DOI] [PubMed] [Google Scholar]
- 18. Kase ET, Andersen B, Nebb HI, et al. 22‐Hydroxycholesterols regulate lipid metabolism differently than T0901317 in human myotubes. Biochim Biophys Acta 2006; 1761: 1515–1522. [DOI] [PubMed] [Google Scholar]
- 19. Tranheim Kase E, Nikolić N, Pettersen Hessvik N, et al. Dietary supplementation with 22‐S‐hydroxycholesterol to rats reduces body weight gain and the accumulation of liver triacylglycerol. Lipids 2012; 47: 483–493. [DOI] [PubMed] [Google Scholar]
- 20. Na TY, Shin YK, Roh KJ, et al. Liver X receptor mediates hepatitis B virus X protein‐induced lipogenesis in hepatitis B virus‐associated hepatocellular carcinoma. Hepatology 2009; 49: 1122–1131. [DOI] [PubMed] [Google Scholar]
- 21. Na TY, Lee HJ, Oh HJ, et al. Positive cross‐talk between hypoxia inducible factor‐1α and liver X receptor α induces formation of triglyceride‐loaded foam cells. Arterioscler Thromb Vasc Biol 2011; 31: 2949–2956. [DOI] [PubMed] [Google Scholar]
- 22. Mojsilovic‐Petrovic J, Callaghan D, Cui H, et al. Hypoxia‐inducible factor‐1 (HIF‐1) is involved in the regulation of hypoxia‐stimulated expression of monocyte chemoattractant protein‐1 (MCP‐1/CCL2) and MCP‐5 (Ccl12) in astrocytes. J Neuroinflamm 2007; 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rippmann JF, Schoelch C, Nolte T, et al. Improved lipid profile through liver‐specific knockdown of liver X receptor‐α in KKAy diabetic mice. J Lipid Res 2009; 50: 22–31. [DOI] [PubMed] [Google Scholar]
- 24. Kim EJ, Yoon YS, Hong S, et al. Retinoic acid receptor‐related orphan receptor α‐induced activation of adenosine monophosphate‐activated protein kinase results in attenuation of hepatic steatosis. Hepatology 2012; 55: 1379–1388. [DOI] [PubMed] [Google Scholar]
- 25. Land SC, Tee AR. Hypoxia‐inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007; 282: 20534–20543. [DOI] [PubMed] [Google Scholar]
- 26. Befani CD, Vlachostergios PJ, Hatzidaki E, et al. Bortezomib represses HIF‐1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J Mol Med (Berl) 2012; 90: 45–54. [DOI] [PubMed] [Google Scholar]
- 27. Fontaine C, Rigamonti E, Nohara A, et al. Liver X receptor activation potentiates the lipopolysaccharide response in human macrophages. Circ Res 2007; 101: 40–49. [DOI] [PubMed] [Google Scholar]
- 28. Werle M, Schmal U, Hanna K, et al. MCP‐1 induces activation of MAP‐kinases ERK, JNK and p38 MAPK in human endothelial cells. Cardiovasc Res 2002; 56: 284–292. [DOI] [PubMed] [Google Scholar]
- 29. Chen M, Bradley MN, Beaven SW, et al. Phosphorylation of the liver X receptors. FEBS Lett 2006; 580: 4835–4841. [DOI] [PubMed] [Google Scholar]
- 30. Ricciardi A, Elia AR, Cappello P, et al. Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res 2008; 6: 175–185. [DOI] [PubMed] [Google Scholar]
- 31. Nakajima T, Kamijo Y, Tanaka N, et al. Peroxisome proliferator‐activated receptor‐α protects against alcohol‐induced liver damage. Hepatology 2004; 40: 972–980. [DOI] [PubMed] [Google Scholar]
- 32. Kong L, Ren W, Li W, et al. Activation of peroxisome proliferator activated receptor‐α ameliorates ethanol induced steatohepatitis in mice. Lipids Health Dis 2011; 10: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Enomoto N, Takei Y, Hirose M, et al. Prevention of ethanol‐induced liver injury in rats by an agonist of peroxisome proliferator‐activated receptor‐γ, pioglitazone. J Pharmacol Exp Ther 2003; 306: 846–854. [DOI] [PubMed] [Google Scholar]
- 34. Kim DK, Kim YH, Jang HH, et al. Estrogen‐related receptor‐γ controls hepatic CB1 receptor‐mediated CYP2E1 expression and oxidative liver injury by alcohol. Gut 2013; 62: 1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seo JB, Moon HM, Kim WS, et al. Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator‐activated receptor‐γ expression. Mol Cell Biol 2004; 24: 3430–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gabbi C, Bertolotti M, Anzivino C, et al. Effects of bile duct ligation and cholic acid treatment on fatty liver in two rat models of non‐alcoholic fatty liver disease. Dig Liver Dis 2012; 44: 1018–1026. [DOI] [PubMed] [Google Scholar]
- 37. Russell DW. Oxysterol biosynthetic enzymes. Biochim Biophys Acta 2000; 1529: 126–135. [DOI] [PubMed] [Google Scholar]
- 38. Gilardi F, Viviani B, Galmozzi A, et al. Expression of sterol 27‐hydroxylase in glial cells and its regulation by liver X receptor signaling. Neuroscience 2009; 164: 530–540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
22‐S‐HC, antagonist of LXRα inhibits ethanol‐induced fatty liver. After an ethanol‐containing diet treatment for 3 weeks, mice were administered vehicle or 22‐S‐HC for 2 weeks. (A) Liver sections were fixed in formalin and stained with oil red O (×100). (B) Liver weight:body weight ratio was measured after 5 weeks of ethanol feeding. (C) Total RNA from liver tissue was subjected to qRT–PCR for the determination of mRNA levels of TNFα, IL‐1β and IL‐6; numbers represent mean ± SD (n = 6 for the pair‐fed control and 22‐S‐HC groups; n = 8 for the ethanol‐fed and ethanol with 22‐S‐HC groups); *p < 0.05 and **p < 0.01 compared with the pair‐fed control group; # p < 0.05 and ## p < 0.01 compared with the ethanol‐fed group; comparisons between groups were performed using the Mann–Whitney U‐test.
Expression of LXRα from ethanol‐fed mice. (A) After an ethanol‐containing diet treatment for 3 weeks, mice were administered vehicle or 22‐S‐HC for 2 weeks; immunohistochemical staining for LXRα in liver (×100). (B) Whole‐cell lysates were obtained from the livers of the indicated mice and they were analysed for protein levels by western blotting (top); the density of each protein band was determined using an image‐analysis system and normalized to that of the corresponding α‐tubulin (bottom); y axis represents the relative protein level for comparison with that of pair‐fed control group; **p < 0.005 compared with the pair‐fed control group (n = 6); ## p < 0.01 compared with the ethanol‐fed group (n = 8); comparisons between groups were performed using the Mann–Whitney U‐test
Activation of LXRα and HIF‐1 induces MCP‐1 expression. The band intensities of western blots shown in Figure 2A, B, and two other blots with the same experiments were measured and normalized with that of β‐actin; numbers represent mean ± SD (n = 3); *p < 0.05, **p < 0.01 and ***p < 0.001 compared with vehicle treatment; # p < 0.05, ## p < 0.01 and ### p < 0.001 compared with vehicle or shGFP, as indicated; statistical significance was evaluated by two‐way ANOVA
LXRα and HIF‐1α synergistically induce expression of MCP‐1. Huh‐7 cells were treated with 1 µm TO901317 or exposed with 0.1% O2 for 24 h. MCP‐1 protein in culture supernatants was quantified using an ELISA kit; numbers represent mean ± SD (n = 3); **p < 0.01 and ***p < 0.001 compared with vehicle‐treated control; statistical significance was evaluated by two‐way ANOVA
MCP‐1 mRNA levels and neutral lipids in the co‐culture system of the primary hepatocytes with KCs. Primary hepatocytes were seeded with or without KCs for 4 h and treated with vehicle or GW3965 1 µm for 48 h. (A) mRNA levels of MCP‐1 were measured by qRT–PCR. The numbers represent mean ± SD (n = 3); **p < 0.01 and ***p < 0.001 compared with vehicle treatment; # p < 0.01 and ## p < 0.001 compared with the indicated culture; statistical significance was evaluated by two‐way ANOVA. (B) The cells were stained using Nile red and visualized by fluorescence microscopy (×320)
Antagonist of LXRα inhibits ERRγ‐mediated induction of CYP2E1. After an ethanol‐containing diet treatment for 3 weeks, mice were administered vehicle or 22‐S‐HC for 2 weeks. Total RNA from liver tissue was subjected to qRT–PCR for determination of mRNA levels of ERRγ, CB1R and CYP2E1; numbers represent mean ± SD (n = 6 for the pair‐fed control and the 22‐S‐HC groups; n = 8 for the ethanol‐fed and the ethanol with 22‐S‐HC groups); **p < 0.01 compared with pair‐fed control group; # p < 0.05 and ## p < 0.01 compared with ethanol‐fed group; comparisons between groups were performed using the Mann–Whitney U‐test
Sequences used for RNA interference
Primer sequences used for qRT–PCR and ChIP