

Abstract
Purpose
Targeted proteomics using MRM with stable‐isotope‐labeled internal‐standard (SIS) peptides is the current method of choice for protein quantitation in complex biological matrices. Better quantitation can be achieved with the internal standard‐addition method, where successive increments of synthesized natural form (NAT) of the endogenous analyte are added to each sample, a response curve is generated, and the endogenous concentration is determined at the x‐intercept. Internal NAT‐addition, however, requires multiple analyses of each sample, resulting in increased sample consumption and analysis time.
Experimental design
To compare the following three methods, an MRM assay for 34 high‐to‐moderate abundance human plasma proteins is used: classical internal SIS‐addition, internal NAT‐addition, and external NAT‐addition—generated in buffer using NAT and SIS peptides. Using endogenous‐free chicken plasma, the accuracy is also evaluated.
Results
The internal NAT‐addition outperforms the other two in precision and accuracy. However, the curves derived by internal vs. external NAT‐addition differ by only ≈3.8% in slope, providing comparable accuracies and precision with good CV values.
Conclusions and clinical relevance
While the internal NAT‐addition method may be “ideal”, this new external NAT‐addition can be used to determine the concentration of high‐to‐moderate abundance endogenous plasma proteins, providing a robust and cost‐effective alternative for clinical analyses or other high‐throughput applications.
Keywords: ExSTA, external standard addition, Multiple Reaction Monitoring (MRM), quantitative proteomics, standard addition, standard curve
1. Introduction
During the past 25 years, proteomics has moved from a qualitative science to a quantitative one. Initially used for protein identification and modification site determination, it is now being advocated as an alternative to ELISA for clinical use.1, 2, 3, 4, 5 Although relative quantitation techniques are still in use and are still valuable for biomarker discovery, for biomarker verification, and clinical analysis, what is required are techniques that allow determination of the absolute amount of material present, so that the amount of protein in a patient sample analyzed in various clinical laboratories can be compared with normal levels. Although these quantitation techniques are sometimes called “absolute”, the concentrations in the biological sample are, in fact, determined by comparison to known amounts of standard materials.6, 7, 8, 9 Exactly how this comparison is performed is the subject of this current paper.
MRM‐based targeted proteomics is now widely accepted as a flexible, precise, and sensitive method for absolute protein quantitation in complex biological samples,10, 11, 12, 13, 14, 15, 16 particularly when performed as part of a bottom‐up approach using stable‐isotope labeled analogues (SIS peptides) of the target peptides as internal standards.17, 18 When SIS peptides are used in conjunction with standard operating procedures (SOPs), reproducible results can be generated within and between laboratories, as the use of SIS peptides helps to compensate for instrumental drift and/or differences in matrix effects between samples,19, 20, 21 as well as facilitating interference testing.10, 22, 23 Ideally, for the highest quantitative precision, the SIS peptide concentrations should be balanced to reflect the expected concentrations in the particular matrix being studied (plasma, urine, cerebrospinal fluid (CSF), etc.).24, 25, 26, 27 The analyte intensities are used in the downstream data analysis to determine the concentrations of the surrogate peptides and thus the proteins of interest. Quantitation is typically done through linear regression analysis of the relative ratios of the internal standard (SIS) peptides to the endogenous (END) peptides, i.e. SIS/END, using for a dilution series of the SIS peptide in a standard sample digest, prepared from a control or pooled sample.23, 25, 28 The actual concentrations of the target analytes are determined by comparison with a calibration curve, where the relative responses of the END analyte and the spiked‐in internal isotopically labeled standard (Figure 1A) are plotted as a function of concentration. These calibration curves also provide information on the linear dynamic range of the assay and the uncertainties (CVs) in the measurements.
Figure 1.
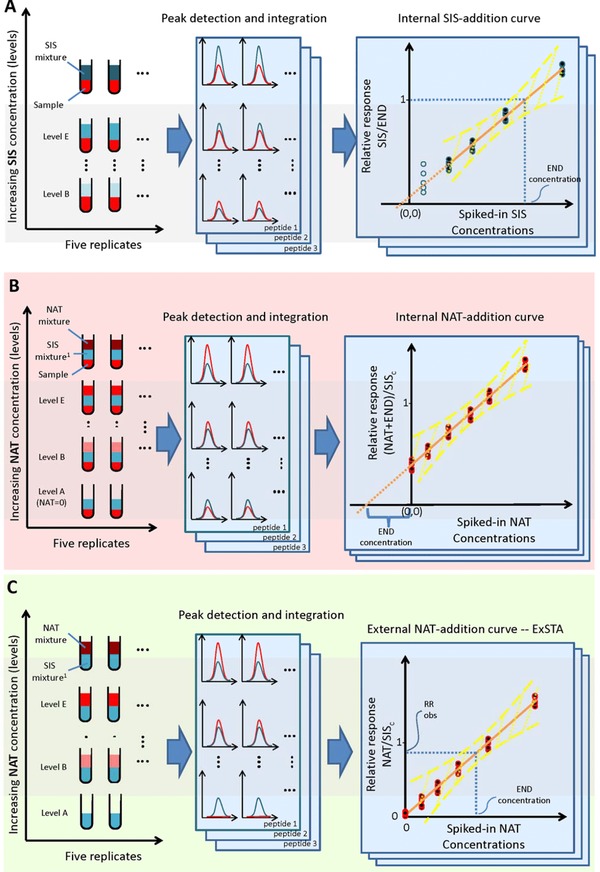
The three methods for generating standard curves. The method shown in panel A—the internal SIS‐addition method—illustrates the common method for generating the standard curve in sample, which uses a SIS peptide mixture spiked in at several concentration levels. The calibration curve is generated by plotting the SIS/END peak area ratios as a function of SIS peptide concentration. The method shown in panel B shows the internal NAT‐addition method where a concentration‐balanced SIS peptide mixture (SISc) is spiked into the sample at a single concentration level and the NAT is added at several concentration levels. The standard curve is generated by plotting the relative response (i.e., R(NAT + END)/RSISc), as a function of the spiked‐in NAT peptide concentration. The concentration of the END peptide is estimated by extrapolating the standard curve and determining the x‐intercept, of which the END concentration is the absolute value. Panel C shows the external NAT‐addition method—ExSTA. In this method, the SISc peptide mixture is spiked into buffer at a single concentration level, and varying levels of NAT are spiked into the mixture. The peak areas of the NAT and SISc peptides are used (the END peptide is not present). After generating the calibration curve in buffer, the END peptide concentration in a sample is estimated from a single point measurement of a sample, to which only a single concentration level of SISc peptide has been added. The yellow lines in all three panels represent the confidence interval associated with the standard curve.
Clinical Relevance
Many clinical cohort studies require rapid, deep, and multiplexed analytical methods to match the extreme complexity of the collected samples and to allow high‐throughput biomarker monitoring at low sample consumption. Targeted proteomics has been widely used to address these challenges when quantifying protein abundances in complex biological matrices. The “gold standard” quantitation method is the internal standard‐addition method, where increasing amounts of synthesized natural form (NAT) of the proteotypic END peptides are added to each sample along with isotopically labeled internal standard (SIS) at a fixed concentration for normalization, response curves are generated, and the END concentrations are determined by extrapolation. This approach, however, requires multiple analyses of each sample, resulting in increased sample consumption and analysis time. Our study of 34 plasma proteins showed that an “external” standard‐addition—generated in buffer using NAT and SIS—can be used to determine the concentration of high‐to‐moderate abundance plasma proteins. While the internal standard‐addition is the still best method, the standard curves derived with our external NAT‐addition method differed by only ≈3.8% in slope, resulting in comparable accuracy and precision values with good CVs, and provided a robust, precise, cost effective, and low sample‐consuming alternative suitable for clinical applications.
A less rigorous variant of this absolute quantitation method is where a single known concentration of SIS peptide is spiked into the sample, and the concentration in the sample is determined from the ratio of the area of the END peak to the area of the labeled peak. This approximation is possible because of the wide linear dynamic range of a triple quadrupole instrument—up to 5–6 orders of magnitude. This “single point” method makes the assumption that the calibration curve is linear within the region spanned by the SIS and the END peaks. Thus, the single‐point method should be used with extreme caution and only over a dynamic range of few orders of magnitude in concentration, where the instrument response is known to be linear from previous experiments.
In addition, while isotopically‐labeled internal standards are considered to be chemically identical to the target analyte, and have identical retention times and fragmentation patterns, there have been occasions where SIS peptides do not entirely compensate for differences in matrix effects. These appear as “outliers” in previous studies,23, 29 and may be caused by a particular sample having a slightly different matrix composition (and potential interferences) than the reference sample used to generate the calibration curve.
An alternative to quantification using calibration curves based on SIS peptides is to use the standard addition approach. In the standard addition method (a well‐established technique in classical analytical chemistry), a standard curve is generated by adding a series of different concentrations of the (unlabeled) target analyte to each sample. Thus, the standard curve is created in exactly the same matrix in which the analysis is performed. Specifically, successive additions of the target analyte are made into each sample, followed by measuring the increasing responses resulting from the increased amount of the analyte in the sample. A straight line fitting the data points is determined by linear regression, and the END concentration can be determined by the extrapolation of the regression line to the x‐intercept. The y‐axis corresponds to the response, and the x‐axis corresponds to the amount of analyte added (Figure 1B).
Although the term “standard addition” is sometimes used incorrectly in the literature, the key feature of the standard addition method (which differentiates it from other methods involving the addition of standards to a sample) is that varying concentrations of the target analyte—in its unlabeled form—are added to each sample. One of its obvious drawbacks of the internal standard addition method (called the internal NAT‐addition method in this paper) is that it requires multiple analyses of each sample, which makes it impractical for LC/MS‐based analyses of large numbers of research or clinical samples due to time and/or sample limitations. Thus, while standard addition is used in many branches of analytical chemistry (including colorimetry), and it is used fairly routinely in MS‐based toxicology, it is not normally used in pharmacology or in clinical chemistry. It does, however, have clear advantages when matrices are complex or variable, or in the presence of strong matrix effect.
The internal NAT‐addition method has been used more often for MALDI‐based analyses—both of drugs and peptides—because of the speed and low sample requirements of this technique,30, 31, 32, 33 sometimes in combination with a labeled internal standard to compensate for instrument drift, as we did in our iMALDI study on angiotensin I.34 Internal NAT addition typically reduces the %CV and/or leads to a lower detection limit. Anderson et al. used a labeled internal standard in combination with standard addition, to determine interferences and matrix effects in Stable Isotope Standards and Capture by AntiPeptide Antibodies (SISCAPA)‐MALDI.35
Because the internal NAT‐addition method requires multiple analyses of the same sample, there have been relatively few applications of this method with LC/ESI–MS, even though this method has the advantage of compensating for matrix effects. Not surprisingly, most of these applications have been for cases where the biological matrix is particularly challenging (such as the analysis of tissues) or extremely variable (such as in postmortem or decomposed samples).36, 37 The internal NAT‐addition method has, however, been used occasionally to provide reference values for LC–MRM‐based proteomic assays.38 Internal NAT addition has also been used for LC/MS‐based quantitation of small molecules in various tissues39, 40, 41, 42 and in postmortem blood.37 A recent multiplexed MRM study used standard addition,43 for the quantitative analysis of 115 veterinary drugs in a variety of food matrices.
Here we compare the conventional method of adding a concentration‐balanced SIS peptide mixture to each sample at multiple concentration levels (Figure 1A), with the internal standard addition method (Figure 1B). Specifically, quantitation in the internal NAT‐addition method is done by linear regression using a dilution series of the synthesized natural unlabeled form of the surrogate peptide (NAT) which is added to a sample where the END peptide is already present. The responses are normalized to the signal from the constant amount of SIS peptide which has been added to the sample.
In addition to these two methods, a third method is also evaluated. This new method, which we call the external NAT‐addition method—external standard addition (ExSTA), is based on a standard curve derived from unlabeled standards spiked into buffer (Table 1 and Figure 1C), again using a constant concentration of SIS peptide for normalization. Although using the same background matrix as the sample is preferable, in real applications it is sometimes impractical, not affordable, or even impossible to obtain sufficient material to generate an internal SIS or NAT calibration curve, as this would require a minimum of 18 injections (3 repeats of 6 levels). The question in such situations is whether an external NAT‐addition method could be considered as an alternative.
Table 1.
Characteristics of the three quantification methods described: internal SIS addition, internal NAT addition, and external NAT addition.
| Internal SIS addition | Internal NAT addition | External NAT addition—ExSTA | |
|---|---|---|---|
| Principle | Standard curve based on SIS peptides added to each sample | Standard curve based on NAT and SIS peptides added to each sample | Standard curve based on NAT and SIS peptides added to buffer |
| Uses synthetic heavy labeled peptide (SIS) | Yes—for calibration curve | Yes—for normalization only | Yes—for normalization only |
| Uses synthetic light peptide (NAT) | No | Yes—for calibration curve | Yes—for calibration curve |
| SIS dilution series is required |
|
|
|
| Standard solutions |
|
|
|
| Generate curve in actual sample matrix |
|
|
|
| Suitable for small amount of sample |
|
|
Yes. |
| Number of analysis required per sample (for a 6 levels curve with 3 repeats/injections) | 18 measurements per sample | 18 measurements per sample |
|
| Requires concentration‐balancing of SIS to END in the reference sample | Recommended | Not required | Not required |
2. Experimental Section
The three methods compared here are shown in Figure 1. For the internal SIS‐addition method (Figure 1A), we spiked varying amounts of a mixture of 34 SIS peptides into a standard plasma sample to give a >100‐fold range of concentrations (as in44, 45) and measured the SIS/END response ratios for the END and heavy‐labeled peptides.
For the internal NAT addition method (Figure 1B), we spiked 34 synthetic heavy‐labeled SIS peptides representing the target proteins into a human plasma sample. The fixed concentration of each SIS peptide was concentration‐balanced to be close to the END concentration of that peptide. We added the synthetic natural (unlabeled) forms (NAT) of these 34 peptides at 5 concentration levels, spanning a 100‐fold concentration range, with level 3 being close to the END peptide concentrations. The (NAT + END)/SIS response ratios for the synthetic (NAT) plus END peptide were calculated as:
For the external NAT‐addition experiments (Figure 1C), we spiked a mixture of 34 SIS peptides into 0.1% FA in water at the same concentrations as would be expected in a plasma sample. We then spiked‐in varying amounts of a concentration‐balanced mixture of the 34 synthetic NAT peptides, to span a 100‐fold concentration range. The response ratios for the synthetic unlabeled peptide; i.e. NAT, and the corresponding SIS peptide were measured as:
Standard curves were generated for all three methods and the concentrations were determined using Qualis‐SIS, as described previously.46
For all calibration curves and standard curves, we used a weighted least‐squares fitting approach to determine the slope and y‐intercept of a straight line. The best‐fit curves were obtained in an iterative four‐step procedure (see Figure S1, Supporting Information). First, the best least‐squares linear fit was calculated without weighting, followed by three similar fittings using weighting. The weights were generated for each step by using the standard curve from the previous iteration to calculate the reciprocal of the squared estimated value, i.e. (1/y i’)2 where y i’ is the estimated value for y at concentration level i. Using this strategy, we reached a stable standard curve after a total of four iterations, after which there were no further changes in the slope and y‐intercept of the curve.
2.1. Internal SIS Addition (Varying Amounts of SIS and no NAT, in Plasma)
In the conventional internal SIS‐addition method, we used our previously described approach44, 45 to determine the concentration of the END peptide. Briefly, we spiked in a series of increasing SIS concentrations across six levels covering a 1000‐fold concentration range. These data are used to generate a calibration curve based on a linear regression analysis of the peak area ratios in the sample. The ratio of SIS peak area to the END peak area is considered as the independent variable and the SIS concentration at each level as the dependent variable. We used the curve to evaluate the dynamic range and to determine the concentration of the END peptide (Figure 1A).
2.2. Internal NAT Addition Method (Constant SIS and Varying Amounts of NAT, in Plasma)
We determined the END peptide concentrations in the sample by using standard addition curves based on the relative responses of the heavy and light peptide forms in the sample (Figure 1B). This method uses linear regression of the relative response (RR) as a function of the spiked‐in NAT concentration. The RR here is the ratio of the total response area for the light form of the peptide in the sample (i.e., NAT + END) to the response area of the SIS peptide. The END peptide concentration is then determined by the x‐intercept of the regression line (see Figure 1B).47 This approach uses a single fixed SIS concentration and series of increasing NAT concentrations across five levels covering a 100‐fold concentration range. The SIS mixture is balanced to the expected END concentrations in the sample.
2.3. External NAT‐Addition Method–ExSTA (Constant SIS and Varying Amounts of NAT, in Buffer)
For this method (Figure 1C), we generated a standard addition curve in buffer using the same concentration‐balanced SIS mixture and NAT amounts as in the internal NAT‐addition method, i.e., a fixed SIS concentration and series of increasing NAT concentrations across five levels covering a 100‐fold concentration range. Using linear regression, we established the relationship between the peak area ratios, i.e., the RR at the different concentrations as the dependent variable and the NAT concentration as the independent variable. We used the regression equation from this standard curve to calculate the concentration of the END peptide in the sample by determining the RR of the peak area ratio when only the SIS peptide is spiked‐in the sample (i.e., the RR for level A where there is no added NAT and where the peak signal in the light channel represents only the END peptide).
2.4. Chemicals and Reagents
For the internal standard experiments, we used human plasma obtained from BioreclamationIVT (catalogue number HMPLEDTA2, lot number BRH796723). This plasma contained K2EDTA as an anticoagulant, and was obtained from healthy race‐ and gender‐matched consenting donors, aged 18–50. The target panel used was composed of 34 peptides corresponding to high‐to‐moderate abundance plasma proteins, i.e. 20 fmol/μL to 200 pmol/μL (see Table 2). All of these proteins are part of the PeptiQuant Workflow Performance Kit (MRM Proteomics, catalogue number WFPK‐A6495‐1) used to monitor LC/MRM–MS performance.48 For each protein, one proteotypic surrogate peptide was chosen. These peptides were selected from our previous panels and experiments according to our standard selection criteria discussed previously,49 and had been empirically optimized and used in various previous studies and projects.16, 45, 50
Table 2.
Proteotypic peptides used and their calculated concentrations with the three methods.
| Protein | Peptide | Concentration (fmol/μL) Internal SIS addition | Concentration (fmol/μL) Internal NAT addition | Concentration (fmol/μL) External NAT addition—ExSTA |
|---|---|---|---|---|
| Afamin | DADPDTFFAK | 171.46 | 82.36 | 90.89 |
| Alpha‐1‐antichymotrypsin | EIGELYLPK | 1854.86 | 1021.24 | 1050.086 |
| Alpha‐1B‐glycoprotein | LETPDFQLFK | 4563.92 | 1363.72 | 1473.71 |
| Alpha‐2‐antiplasmin | LGNQEPGGQTALK | 128.25 | 123.84 | 116.19 |
| Angiotensinogen | ALQDQLVLVAAK | 113.32 | 78.58 | 84.03 |
| Antithrombin‐III | DDLYVSDAFHK | 189.75 | 1422.88 | 1525.57 |
| Apolipoprotein A‐I | ATEHLSTLSEK | 20383.66 | 22835.78 | 24620.63 |
| Apolipoprotein A‐II | SPELQAEAK | 1864.66 | 1524.92 | 1531.51 |
| Apolipoprotein A‐IV | SLAPYAQDTQEK | 233.73 | 163.64 | 174.64 |
| Apolipoprotein B‐100 | FPEVDVLTK | 209.71 | 93.00 | 98.74 |
| Apolipoprotein C‐I | TPDVSSALDK | 19.62 | 20.59 | 25.29 |
| Apolipoprotein E | LGPLVEQGR | 597.86 | 973.18 | 1017.22 |
| Beta‐2‐glycoprotein 1 | ATVVYQGER | 2060.65 | 1786.28 | 1808.34 |
| Ceruloplasmin | EYTDASFTNR | 144.39 | 231.14 | 240.94 |
| Clusterin | ELDESLQVAER | 2806.77 | 952.05 | 937.53 |
| Coagulation factor XII | VVGGLVALR | 209.20 | 181.04 | 190.37 |
| Complement C3 | TGLQEVEVK | 721.00 | 495.48 | 523.08 |
| Complement C4‐B | ITQVLHFTK | 1085.12 | 861.22 | 877.38 |
| Complement component C9 | TEHYEEQIEAFK | 33.30 | 24.35 | 28.13 |
| Complement factor B | EELLPAQDIK | 265.90 | 147.60 | 155.71 |
| Complement factor H | SPDVINGSPISQK | 757.37 | 555.38 | 587.82 |
| Fibrinogen alpha chain | GSESGIFTNTK | 8374.34 | 5455.66 | 5705.70 |
| Fibrinogen beta chain | QGFGNVATNTDGK | 991.74 | 422.34 | 463.29 |
| Gelsolin | TGAQELLR | 96.06 | 122.04 | 114.08 |
| Haptoglobin | VGYVSGWGR | 22742.89 | 17451.28 | 17693.54 |
| Hemopexin | NFPSPVDAAFR | 4479.79 | 1965.02 | 2083.75 |
| Heparin cofactor 2 | TLEAQLTPR | 827.12 | 4171.64 | 4408.87 |
| Inter‐alpha‐trypsin inhibitor heavy chain H1 | AAISGENAGLVR | 371.73 | 413.48 | 417.24 |
| Kininogen‐1 | TVGSDTFYSFK | 281.40 | 195.06 | 210.49 |
| Plasminogen | LFLEPTR | 36.96 | 58.93 | 61.45 |
| Retinol‐binding protein 4 | YWGVASFLQK | 587.93 | 595.58 | 670.90 |
| Serum albumin | LVNEVTEFAK | 460184.3 | 207775.7 | 203717.0 |
| Transthyretin | AADDTWEPFASGK | 3049.86 | 1467.84 | 1516.26 |
| Vitronectin | FEDGVLDPDYPR | 261.70 | 185.44 | 179.54 |
The internal standard peptides are C‐terminal isotopically labeled tryptic peptides. These were synthesized in‐house using a standard Fmoc procedure on an Overture peptide synthesizer (Protein Technologies; Woburn, MA, USA). Purification was performed by HPLC and confirmed by matrix‐assisted laser desorption/ionization time of flight mass spectrometry (MALDI–TOF‐MS) analysis, while characterization was performed by CZE and amino acid analysis (AAA). The AAA and CZE analyses enabled us to determine the SIS peptide concentrations.
The corresponding natural forms of the 34 peptides were purchased from SynPeptide Co. Ltd. with purity higher than 90% for all peptides. Characterization of these 34 peptides was done by CZE and AAA, which enabled us to accurately determine the NAT peptide concentrations. Other chemicals (e.g., ammonium bicarbonate, and dithiothreitol) and solvents (e.g., acetonitrile and water) were LC–MS grade and were purchased from Sigma–Aldrich (St. Louis, MO, USA).
2.5. Sample Preparation
We followed a slightly modified version of the standard sample preparation of the PeptiQuant Workflow Performance Kit.45 For the current study, 60 μL of 10× diluted human plasma was added to 355 μL of 25 mM ammonium bicarbonate and 54 μL of 10% (w/v) sodium deoxycholate for protein denaturation. Disulfide bonds were reduced with a 52.4 μL addition of 50 mM tris(2‐carboxyethyl)phosphine for a final concentration of 5 mM. The sample was incubated in dry air at 60 °C for 30 min, followed by alkylation of free sulfhydryl groups by adding 58 μL of 100 mM iodoacetamide to give a final concentration of 10 mM, and incubating at 37 °C for 30 min in the dark in a dry air incubator. To prevent alkylation of other residues, the remaining iodoacetamide was quenched by adding 58 μL of 100 mM DTT and incubated at 37 °C in a dry air incubator for 30 min. Proteolytic digestion was then initiated by adding an aliquot of TPCK (l‐(tosylamido‐2‐phenyl) ethyl chloromethyl ketone)‐treated bovine trypsin (Worthington; Lakewood, NJ, USA) at a 10:1 substrate/enzyme ratio. Digestion was allowed to proceed over night for 16 h at 37 °C. Upon digestion completion, acidified concentration‐balanced SIS or SIS‐NAT peptide mixture was added for the standard curve method (Figure 1A) and the standard addition method (Figure 1B) respectively. The six SIS mixtures were prepared in 0.1% FA solution to have six concentration levels of the SIS peptides spanning a 1000‐fold concentration range, with a dilution series from the highest concentration of 1:2:5:2:5:10 (labeled F to A, from the highest to the lowest concentration), with level D being balanced to be as close as possible to the END concentration. This dilution series was designed to cover a relatively wide concentration range (1000), with the highest concentration designed to be 10 times as high as the average END peptide concentration, and the lowest concentration to be 100 times lower than the average END peptide concentration. For the standard addition method, the SIS‐NAT mixtures were prepared in 0.1% FA solution to have six concentration levels of the NAT peptides, with a dilution series from the highest concentration of 1:2:5:2:5 for levels F to B, and fixed concentrations of SIS peptides across all levels. The Level A sample was prepared in the same way but without adding any NAT and Level D contained equal amounts of SIS and NAT.
The six standard samples were prepared by combining aliquots of the plasma tryptic digest (200 μL, containing 1.8 μL of original undiluted plasma) with the SIS peptide mixture or the SIS‐NAT mixture (98 μL) and then the adding 202 μL of 1% FA to each standard. After centrifugation at 12 000 × g for 10 min, 300 μL of the supernatant was removed from the acid insoluble sodium deoxycholate, and then desalted and concentrated by SPE with an Oasis HLB cartridge using traditional vacuum manifold processing. The extractions were performed with vacuum bleed valve set to –25 kPa and a <1 mL/min flow rate. The eluted samples (one per concentration level) were then frozen, lyophilized to dryness overnight, and rehydrated in 0.1% FA to give a final concentration of 1 μg/μL (assuming an initial plasma protein concentration of 70 mg/mL) prior to injection. For the preparation of the buffer samples, exactly the same SIS‐NAT mixture was used, except that the standards are added to 0.1% FA instead of to the plasma digest.
2.6. LC/MRM–MS Analysis
LC/MRM‐MS experiments were performed in quintuplicate, by reversed‐phase UHPLC on a 1260 Infinity LC system using a Zorbax Eclipse Plus C18 Rapid Resolution HD column (150 × 2.1 mm, 1.8 μM particles; Agilent Technologies; Palo Alto, CA, USA). The column was maintained at 50 °C and the autosampler was kept at 4 °C. Mobile phase compositions of 0.1% FA in water for solution A and 100% ACN in 0.1% FA for B at a flow rate of 0.4 mL/min were used. The gradient was 0:2.7, 2:9.9, 15:17.1, 22:26.1, 25:40.5, 27:81, 29:81, and 30:2.7 (time, %B). The LC system was interfaced to a 6495 triple quadrupole mass spectrometer (both from Agilent Technologies) via a standard‐flow ESI source—see Table S1, Supporting Information for the list of the general acquisition parameters. Specific peptide parameters, such as collision energy and retention time, had been previously optimized and were not changed.45
2.7. Precision, Repeatability, and Correlation
The precision was evaluated as recommended by the FDA guidelines51, 52 at each concentration level by determining the CV of quintuplicate measurements using the following equation:
The coefficients of determination (R 2) for each standard curve were used to evaluate the intra‐batch repeatability and was determined using the residual and total sum of squares as in following equation:
where y i are the measured values, f i is the estimated values by the standard curve, and is the mean of all measurements.
To evaluate the closeness or discrepancy between the protein concentrations determined using two different methods (method 1 and method 2), we determined both the Pearson and Spearman's rank correlation coefficients using the following equations:
where and the concentration values determined by method 1 and method 2, and and are the mean concentration values determined by method 1 and method 2 respectively. Spearman correlation coefficient was determined by the following equation:
Where cov is the covariance matrix, rankx1 is the rank vector of the concentration values determined by method 1, similarly is the rank vector of the concentration values determined by method 2, and are the standard deviations of the rank vector of method 1 and 2. respectively, is the difference between the ranks of each two corresponding determined concentrations, i.e. , and n is the number of observations, i.e. the number determined protein concentrations by both methods.
2.8. Accuracy
In order to evaluate the accuracies of the three methods, the same experiment was repeated using chicken plasma instead of human plasma as the matrix. Chicken plasma was selected because, with the exception of one peptide—YWGVASFLQK from retinol‐binding protein 4 (a highly conserved protein between human and chicken)—all of the peptides used in our method were not present in the chicken proteome. We, nonetheless, kept the method unchanged and included retinol‐binding protein 4 in the LC–MRM/MS quantification, but it was excluded in the evaluation of the accuracy. The accuracies of the three types of calibration were determined based on the other 33 peptides, keeping the MRM method unchanged. For this test, we spiked a known amount of the natural form of each peptide into a chicken plasma sample. After digestion, the standards (SIS only for internal SIS‐addition method, both SIS and NAT for internal and external NAT‐addition methods) were added to generate the standard curves, as was done for the human samples. The accuracies of each method were calculated based on the known spiked‐in amounts and the concentrations determined using each method's calibration curve.
3. Results and Discussion
The three methods were compared on the basis of precision (as determined by the CVs of quintuplicate measurements), repeatability (as determined by the coefficients of determination), and the correlation between the determined concentrations.
3.1. Precision
Table 3 shows the average CVs of the relative response at each concentration level of all peptides. The average CV was 4.44% for the replicates in the internal SIS‐addition method, 3.77% for the replicates in the internal NAT standard addition method, and 3.50% for the replicates in the external NAT‐addition method. The values were calculated considering concentration levels F to B, which range from 10× the expected END concentration to 0.01× the expected END concentration levels. One extreme outlier (DDLYVSDAFHK from antithrombin‐III) in the external NAT‐addition method was removed after it showed a difference of >5 SD at the lower concentration levels and 4 SD at levels F and E. The three methods, therefore, have similar precisions, but both the internal and external NAT‐addition methods led to more precise measurements than the internal SIS‐addition method. Comparing the maximum CVs across all concentration levels, the internal SIS‐addition method had maximum CVs of 16.02 and 16.03% at concentration levels B and D, respectively, while the internal NAT addition method had a maximum CV of 10.69% for level F and maintained CVs of <10% for all other concentration levels. Similarly, the external NAT‐addition method had its maximum CV in level B (12.09%) and maintained <10% maximum CVs across all other concentration levels. The FDA guideline for the validation of bioanalytical chromatographic methods51, 52 sets the acceptable precision limits at 15% of the nominal value. This shows that, even at their worst, the two standard addition methods still met the FDA precision requirements for plasma proteins with high‐to‐moderate abundance.
Table 3.
A comparison of the CVs and the ranges of the measured relative responses (RRs) at each concentration level in the three methods used.
| Average RR at level B (%CV) | Range RR at level B (%CV) | Average RR at level C (%CV) | Range RR at level C (%CV) | Average RR at level D (%CV) | Range RR at level D (% CV) | Average RR at level E (%CV) | Range RR at level E (%CV) | Average RR at level F (%CV) | Range RR at level F (%CV) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Internal SIS addition | 6.03 | 1.04–16.02 | 4.28 | 1.31–11.38 | 4.17 | 0.77–16.03 | 3.82 | 0.91–12.76 | 3.89 | 0.56–7.97 |
| Internal NAT addition | 3.98 | 1.21–9.28 | 4.08 | 0.83–9.06 | 3.38 | 1.14–8.64 | 3.77 | 0.64–8.13 | 3.63 | 1.07–10.69 |
| External NAT addition—ExSTA | 4.39 | 0.83–12.09 | 3.36 | 0.57–8.68 | 3.16 | 0.64–6.64 | 3.38 | 0.81–7.89 | 3.22 | 1.09–7.48 |
The five concentration levels spanned a 100‐fold concentration range, with a dilution series from the highest concentration of 1:2:5:2:5 (labeled F to B, from the highest to the lowest concentration), with level D being balanced to be as close as possible to the END concentration.
3.2. Repeatability
Three curves were generated for each peptide: one curve using the internal SIS‐addition method, one internal NAT addition curve, and one external NAT‐addition curve (Figure 1). For the internal SIS‐addition method (Figure 1A), the coefficient of determination (R 2) of the standard curve was between 0.9638 and 0.9987, with a mean of 0.9910 (Figure 2). The coefficients of determination for the external NAT‐addition curves were between 0.9623 and 0.9985, with a mean of 0.9912. For the internal NAT‐addition curves, the coefficients of determination were between 0.9671 and 0.9990, with a mean of 0.9902 (Figure 2). Only one peptide (DDLYVSDAFHK, from antithrombin‐III) had an R 2 value below 0.90 in buffer (i.e., with the external NAT‐addition method).
Figure 2.
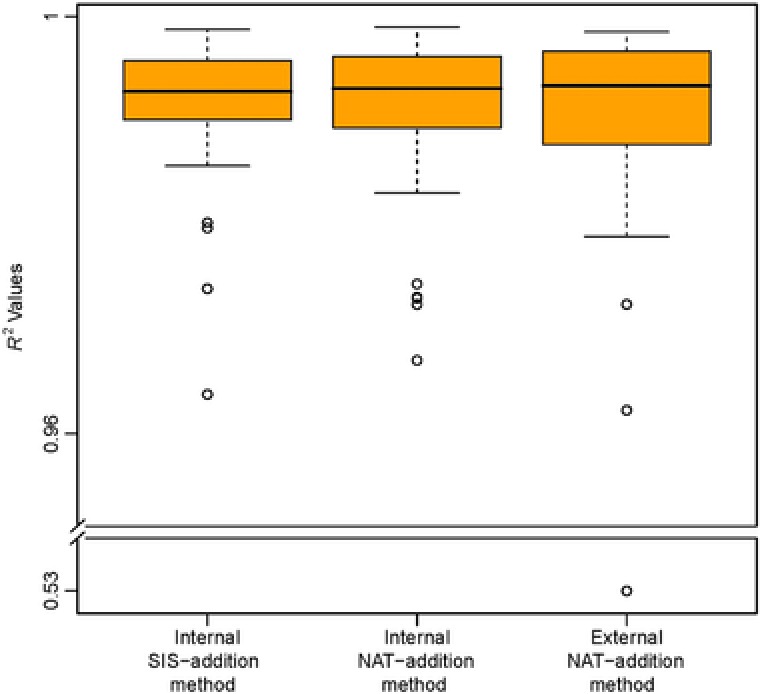
A box‐and‐whisker plot of the R 2 values for the three methods used to generate the standard curves. One outlier in the external NAT‐addition method—ExSTA, originated from antithrombin‐III peptide DDLYVSDAFHK, had an R 2 value of 0.53. The spread of the boxplot corresponds to the variability in the method.
The calculated R 2 values for the internal and external NAT‐addition methods included all of the measured points–i.e., we did not apply any level‐removal strategy or impose any accuracy and precision requirements (see 46 for details). This evaluates the methods for accuracy and precision, and shows any possible deviation from acceptable ranges. In the internal SIS‐addition method, generated by using only the balanced SIS peptide mixture, correspondingly high R 2 values were only observed after applying precision and accuracy filters of 20% each, which removed any lower concentration levels that did not pass our acceptance criteria (see 46 for details). Because all measurements were performed in quintuplicate at each concentration level, the mentioned R 2 values demonstrate the high intra‐laboratory technical repeatability of all three methods.
3.3. Concentration Correlation
The protein concentrations determined by the three methods are shown in Table 2. Figure 3 shows the correlation between the internal SIS addition and the internal NAT‐addition methods. The determined concentrations had a Pearson correlation coefficient of 0.9972 and a Spearman correlation coefficient of 0.9037. The internal SIS‐addition method and the internal NAT‐addition method show an acceptable level of correlation. The concentrations values determined using the internal SIS‐addition method were on average 1.44 times as high as those determined from the internal NAT‐addition method, ranging from a minimum of 0.63 to a maximum of 2.35, with 26 out of the 34 peptides having concentrations higher in the internal SIS‐addition method. This can also be seen in the slope of the regression equation: after removing five outliers—alpha‐1B‐glycoprotein, apolipoprotein A‐I, clusterin, antithrombin‐III, and heparin cofactor 2—the regression equation between the two methods has a slope of 2.22 with a y‐intercept of –284. While this is a good correlation, the high number of outliers is troublesome. Further investigations on the peptide levels of these five proteins will be necessary in order to find the source of the deviation between the two methods. For example, measuring these proteins with alternative peptides and comparing the results with those from the current peptides could help to identify interferences in any of the methods used. It is also important to keep in mind that an error could also originate from incorrect AAA values—any error in the characterization of the SIS peptides would affect both methods similarly, but errors in the characterization of the NAT peptides would affect only the NAT addition method. However, we did not observe any indication of a systematic error in the AAA values of the SIS and NAT peptides used.
Figure 3.
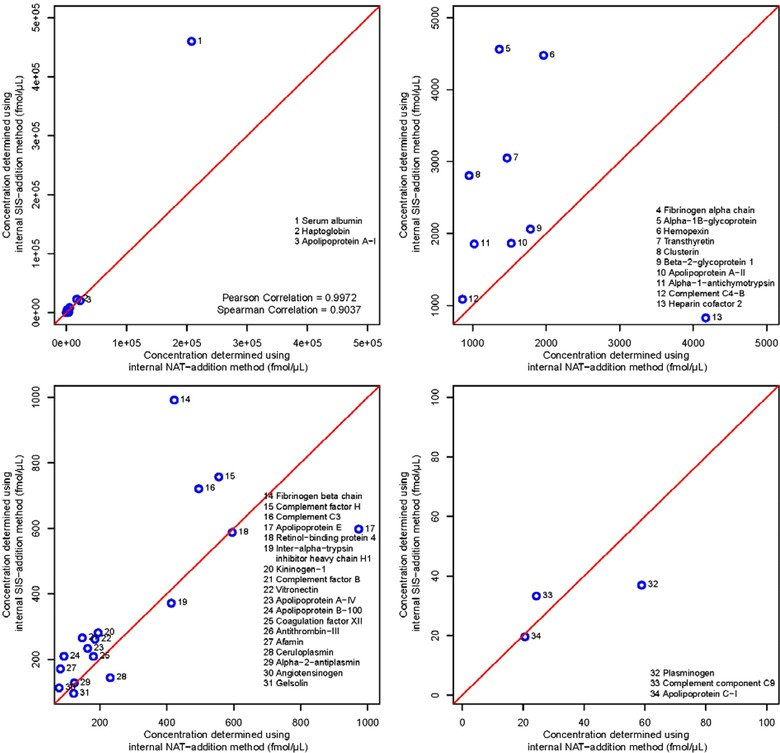
Scatter plots showing the correlation between the concentrations determined by the internal SIS‐addition method and the internal NAT‐addition method. The four plots show four different plotting ranges and the corresponding measured peptide concentration in these ranges, these are 0 to 5e5 fmol/μL for the full range, 1000 to 5000 fmol/μL, 100 to 1000 fmol/μL, and 0 to 100 fmol/μL.
Figure 4 shows the excellent correlation between the concentrations determined from the internal and external NAT‐addition methods with a Pearson correlation coefficient of 0.99994 and a Spearman correlation coefficient of 0.99939. It is evident that these two methods show very similar results, with only the two peptides at the lowest determined concentrations showing a relative difference of >15%, and all differences being <23%. This shows that the external method can be used instead of the internal NAT‐addition method for quantitating peptides with concentrations of ≥60 fmol/μL, with expected deviations of <15% from the concentrations that would have been obtained if the internal NAT addition approach had been used.
Figure 4.
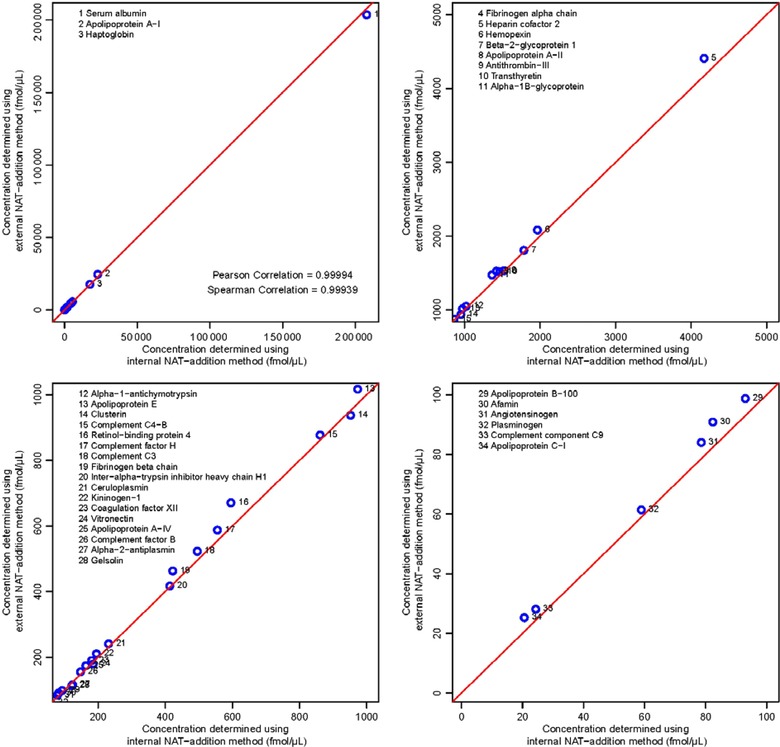
Scatter plots showing the correlation between the concentrations determined by the internal and external NAT‐addition methods, both using the same concentration‐balanced SIS and NAT peptides. The four plots show four different plotting ranges and the corresponding measured peptide concentration in these ranges, these are 0 to 2e5 fmol/μL for the full range, 1000 to 5000 fmol/μL, 100 to 1000 fmol/μL, and 0 to 100 fmol/μL.
3.4. Matrix Effect and Slope Difference in Standard Curves
The slopes of the external NAT‐addition curves in buffer were compared with those of the internal NAT‐addition curves generated in plasma (Figure S2, Supporting Information). There can be no direct comparison with the slope of the calibration curve in the internal SIS‐addition method, because this method is based on addition of increasing SIS concentrations, while the internal and external standard additions involve addition of increasing NAT concentration, while keeping the SIS concentration for each peptide fixed.
The average difference in slope between the internal NAT‐addition method and the external NAT‐addition method was 3.8%, with a range of 0.14–12.45% (Report S1, Supporting Information). This means that the curves generated by both methods using iterative linear regression are very similar. Thus, if the amount of a plasma sample is too small for an internal NAT‐addition curve to be generated, a single‐point measurement might be a method for determining the content of the sample. However, a single‐point measurement approach can also be combined with an external standard curve in case of high‐to‐moderate abundance proteins to obtain information about the figures of merit for measuring a specific peptide from the external NAT‐addition curve. This would add more confidence to the determined concentration value of the protein. In addition, because the sensitivity of an MRM assay for a specific protein is characterized by the slope of the standard curve,53, 54 the small difference in slope between the internal and external NAT‐addition methods indicates that these two methods have very similar nominal sensitivities.
3.5. Accuracy
Using known amounts of NAT peptides spiked into chicken plasma, we evaluated the accuracies of the three methods by the experimentally determined concentrations of each peptide using each of the three calibration methods. For determining the concentrations, we generated one standard curve per peptide per method in chicken plasma (see Report 2, Supporting Information). Table 4 shows the amount of each unlabeled NAT peptide used for this experiment, along with the determined concentrations and the percent recoveries. The spiked‐in peptide amounts varied from 44 fmol/μL to nearly 9000 fmol/μL, simulating the concentration ranges of authentic human plasma (i.e., 20 to ≈5000 fmol/μL) excluding three very high abundant proteins, i.e. serum albumin, apolipoprotein A‐I, and haptoglobin with concentration levels of above 17 (pmol/μL). With exception of two outliers, very good recoveries in all of the three methods were observed, with lower CVs in both of the internal and external NAT‐addition methods compared to the internal SIS‐addition method. The two outliers were heparin cofactor 2 peptide (TLEAQLTPR), which we found to have strong interferences from chicken plasma, as well as retinol‐binding protein 4 peptide (YWGVASFLQK), which is also found in the conserved domain between the human and chicken retinol‐binding protein 4.
Table 4.
Determined accuracies for each of the three methods (internal NAT addition method, external NAT‐addition method, and internal SIS‐addition method) based on using known amount of unlabeled peptides (NAT) spiked into chicken plasma.
| Internal NAT addition | External NAT addition—ExSTA | Internal SIS‐ addition | |||||
|---|---|---|---|---|---|---|---|
| Protein | Added NAT in Chicken plasma (fmol/μL) | Determined NAT in Chicken plasma (fmol/μL) | Percent recovery | Determined NAT in Chicken plasma (fmol/μL) | Percent recovery | Determined NAT in Chicken plasma (fmol/μL) | Percent recovery |
| Afamin | 191.5 | 173.1 | 90.4 | 195.1 | 101.9 | 282.3 | 147.4 |
| Alpha‐1‐antichymotrypsin | 1100.3 | 1014.8 | 92.2 | 1408.8 | 128.0 | 1545.8 | 140.5 |
| Alpha‐1B‐glycoprotein | 667.5 | 574.2 | 86.0 | 1421.0 | 212.9 | NA | NA |
| Alpha‐2‐antiplasmin | 237.0 | 150.9 | 63.7 | 168.4 | 71.0 | 141.4 | 59.7 |
| Angiotensinogen | 148.4 | 124.9 | 84.2 | 179.6 | 121.1 | 97.2 | 65.5 |
| Antithrombin‐III | 172.0 | 145.1 | 84.4 | 174.9 | 101.7 | 16.1 | 9.4 |
| Apolipoprotein A‐I | 7696.0 | 6643.6 | 86.3 | 8362.8 | 108.7 | 6089.1 | 79.1 |
| Apolipoprotein A‐II | 3127.3 | 2343.7 | 74.9 | 2650.4 | 84.8 | 3301.7 | 105.6 |
| Apolipoprotein A‐IV | 245.5 | 195.4 | 79.6 | 220.4 | 89.8 | 241.9 | 98.5 |
| Apolipoprotein B‐100 | 88.8 | 95.2 | 107.2 | 115.0 | 129.5 | 147.2 | 165.8 |
| Apolipoprotein C‐I | 94.8 | 72.3 | 76.3 | 78.5 | 82.8 | 82.8 | 87.3 |
| Apolipoprotein E | 349.6 | 272.6 | 78.0 | 302.6 | 86.6 | 156.4 | 44.7 |
| Beta‐2‐glycoprotein 1 | 1331.3 | 1123.9 | 84.4 | 1265.9 | 95.1 | 1387.3 | 104.2 |
| Ceruloplasmin | 221.3 | 212.4 | 96.0 | 227.0 | 102.6 | 115.3 | 52.1 |
| Clusterin | 525.5 | 522.5 | 99.4 | 575.8 | 109.6 | 702.4 | 133.7 |
| Coagulation factor XII | 115.1 | 93.5 | 81.3 | 106.1 | 92.2 | 96.0 | 83.4 |
| Complement C3 | 395.7 | 310.4 | 78.5 | 335.2 | 84.7 | 355.6 | 89.9 |
| Complement C4‐B | 394.4 | 296.2 | 75.1 | 425.4 | 107.8 | 323.1 | 81.9 |
| Complement component C9 | 223.2 | 168.5 | 75.5 | 243.9 | 109.3 | 185.5 | 83.1 |
| Complement factor B | 209.5 | 193.6 | 92.4 | 225.7 | 107.7 | 224.5 | 107.2 |
| Complement factor H | 219.6 | 148.7 | 67.7 | 167.2 | 76.1 | 177.6 | 80.9 |
| Fibrinogen alpha chain | 7979.7 | 7725.0 | 96.8 | 8364.2 | 104.8 | 9656.8 | 121.0 |
| Fibrinogen beta chain | 2025.8 | 818.2 | 40.4 | 914.7 | 45.2 | 1502.8 | 74.2 |
| Gelsolin | 225.0 | 167.7 | 74.6 | 184.9 | 82.2 | 144.4 | 64.2 |
| Haptoglobin | 8359.3 | 6428.0 | 76.9 | 7168.2 | 85.8 | 9120.5 | 109.1 |
| Hemopexin | 2308.0 | 1635.3 | 70.9 | 2282.3 | 98.9 | 2877.5 | 124.7 |
| Heparin cofactor 2 | 225.0 | 806.4 | 358.4 | 885.2 | 393.4 | 167.5 | 74.4 |
| Inter‐alpha‐trypsin inhibitor heavy chain H1 | 112.6 | 132.8 | 117.9 | 145.5 | 129.2 | 125.3 | 111.3 |
| Kininogen‐1 | 168.0 | 129.6 | 77.1 | 168.1 | 100.0 | 163.1 | 97.1 |
| Plasminogen | 43.8 | 30.0 | 68.5 | 41.0 | 93.5 | 17.8 | 40.6 |
| Retinol‐binding protein 4 | 260.9 | 2638.9 | 1011.4 | 2897.4 | 1110.5 | NA | NA |
| Serum albumin | 8820.0 | 7491.1 | 84.9 | 8788.7 | 99.6 | 11900.1 | 134.9 |
| Transthyretin | 788.1 | 618.3 | 78.5 | 955.1 | 121.2 | 1743.0 | 221.2 |
| Vitronectin | 446.4 | 335.4 | 75.1 | 485.1 | 108.7 | 191.3 | 42.8 |
In the internal NAT‐addition method, the mean accuracy of all of the determined peptide concentrations was 81.7% with a CV of 16.9%; in the external NAT‐addition method, the mean accuracy was 102.3% with a CV of 26.4%; in the internal SIS‐addition method the mean accuracy was 95.5% with a CV of 44%. It is important to note that the variability (represented by %CV here) is approximately 2.5 times higher in internal SIS‐addition compared to the internal NAT‐addition method. Table 4 and Figure 5 show that, while there is a fairly good mean percent accuracy obtained when using the SIS‐addition method, the percent accuracy values have higher variability due to the wider spread of determined concentrations around the true peptide concentrations. Similarly, while the external NAT‐addition shows a better mean accuracy value than the internal NAT‐addition, the percent accuracy values have higher variability in the external NAT‐addition method. In general, however, having tighter accuracy values with lower percent CVs is preferable, as it means that the error is less random and more systematic, which can be corrected for when determining the actual concentration.
Figure 5.
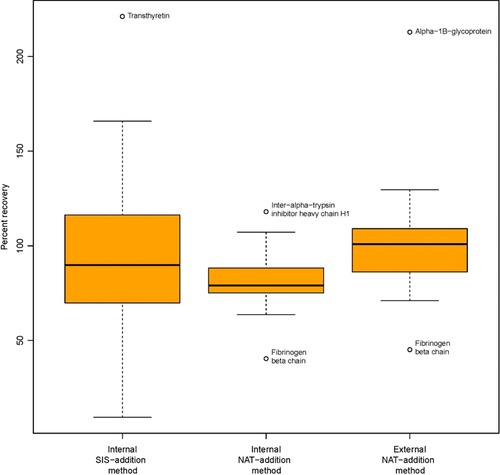
Boxplot of the percent recovery in each of the three methods, internal NAT addition, external NAT addition—ExSTA, and internal SIS‐addition methods. Extreme values appearing as outliers are marked with the name of the corresponding protein.
These accuracy values were obtained after removing the two outliers described above, and are based on all of the other values, including the extreme values appearing above and below the whiskers as shown in Figure 5. When removing these extreme values from all three methods (i.e., transthyretin, inter‐alpha‐trypsin inhibitor heavy chain H1, fibrinogen beta chain, and alpha‐1B‐glycoprotein), we observed mean percent recoveries of 81.9, 98.7, and 91.2% with CVs of 12.6, 14.6, and 40.1%, with the internal NAT addition, external NAT addition—ExSTA, and the internal SIS‐addition methods, respectively.
The FDA guideline for the validation of bioanalytical chromatographic methods51, 52 sets the acceptable accuracy limit at within 15% of the nominal value (except at the LLOQ, where values with <20% accuracy are acceptable). Using the external standard addition method, we obtained a mean difference in concentration of 16.8% when using all peptides, except for the two peptides which showed interferences in chicken plasma. When removing the two extreme values in external standard addition method (i.e., alpha‐1B‐glycoprotein and fibrinogen beta chain, Figure 5), the mean difference dropped to 12.3% with a range between 0.04 and 29.6%. This indicates that accurate concentrations of END peptide surrogates for high‐to‐medium abundance proteins in plasma samples can be obtained by performing single point measurements (i.e., where a single known amount of SIS mixture is spiked into the sample), in conjunction with external standard‐addition curves generated in buffer, using a SIS‐NAT mixture.
4. Conclusions
In a multiplexed targeted LC/MRM–MS experiment, a concentration‐balanced mixture of heavy‐labeled internal standards is usually added to the tryptic sample digest and the responses from both the END and the heavy‐labeled peptides are measured. This is the conventional internal SIS‐addition method. Here we compared this conventional method with two standard‐addition methods, in sample and in buffer. Both standard addition methods showed less variability than the internal SIS‐addition method. Although the quantitated targets in this current study are high‐to‐moderate abundance plasma proteins, we believe that this improvement is due to increases in the S/N ratios, should also improve the quantitation of low‐abundance proteins.
Although the classical internal NAT‐addition method is the best approach for generating a standard curve, it is impractical for routine analysis when the sample volume is limited. It also has a higher cost due to the use of SIS and NAT peptides for each sample at multiple concentration levels. Data acquisition times are also increased, because multiple analyses are required for each sample. The external NAT‐addition technique—ExSTA described in this paper is a robust, fast, and cost‐effective alternative, resulting in an average difference from the slope of the internal NAT‐addition curve of only 3.8%. It needs to be kept in mind that our results were not obtained on random peptides and transitions. In our opinion, the key to the success of the external NAT‐addition method is that the peptides and transitions used have previously been carefully tested and were found to be interference free in a pooled sample of target matrix.
An alternative to generating the external NAT‐addition curve in buffer, external NAT‐addition curves could also be generated in representative matrices, such as BSA in buffer, urine or CSF, or in pooled control samples, if available. In the absence of representative matrix, for instance in cases where pooled control is not available due to limited sample volume (as in tissue biopsies where ≤20 μg of total protein is available for proteomics analysis), external standard addition technique in buffer offers a robust alternative.
Abbreviations
- AAA
amino acid analysis
- END
endogenous
- ExSTA
external standard addition
- NAT
synthesized natural form of the endogenous analyte
- RR
relative response
- SIS
stable‐isotope‐labeled internal‐standard
Conflict of Interest
Dr. Borchers is the Chief Scientific Officer of MRM Proteomics, Inc.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
Y. M. and J. P. contributed equally to this work. We are grateful to Genome Canada and Genome British Columbia for Science and Technology Innovation Centre support and for Genomics Innovation Network support to the University of Victoria ‐ Genome British Columbia Proteomics Centre through the Genome Innovations Network (204PRO for operations; 214PRO for technology development). MRM Proteomics Inc. thanks NRC‐IRAP for project support. CHB is also grateful for support from the Leading Edge Endowment Fund. CHB is also grateful for support from the Segal McGill Family Chair in Molecular Oncology at the Jewish General Hospital (Montreal, Quebec, Canada), and for support from both the Warren Y. Soper Foundation and the Alvin Segal Family Foundation to the Jewish General Hospital (Montreal, Quebec, Canada).
Mohammed Y., Pan J., Zhang S., Han J., Borchers C. H., Prot. Clin. Appl. 2018, 12, 1600180 10.1002/prca.201600180
References
- 1. Boja E. S., Rodriguez H., Korean J. Lab. Med. 2011, 31, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Parker C. E., Pearson T. W., Anderson N. L., Borchers C. H., Analyst 2010, 135, 1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palmblad M., Tiss A., Cramer R., Proteomics Clin. Appl. 2009, 3, 6. [DOI] [PubMed] [Google Scholar]
- 4. Apweiler R., Aslanidis C., Deufel T., Gerstner A., Hansen J., Hochstrasser D., Kellner R., Kubicek M., Lottspeich F., Maser E., Mewes H. W., Meyer H. E., Müllner S., Mutter W., Neumaier M., Nollau P., Nothwang H. G., Ponten F., Radbruch A., Reinert K., Rothe G., Stockinger H., Tarnok A., Taussig M. J., Thiel A., Thiery J., Ueffing M., Valet G., Vandekerckhove J., Verhuven W., Wagener C., Wagner O., Schmitz G., Clin. Chem. Lab. Med. 2009, 47, 724. [DOI] [PubMed] [Google Scholar]
- 5. Percy A. J., Chambers A. G., Yang J., Hardie D. B., Borchers C. H., Biochim. Biophys. Acta 2014, 1844, 917. [DOI] [PubMed] [Google Scholar]
- 6. Gillette M. A., Carr S. A., Nature Methods 2013, 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Di Conza G., Trusso Cafarello S., Loroch S., Mennerich D., Deschoemaeker S., Di Matteo M., Ehling M., Gevaert K., Prenen H., Zahedi R. P., Sickmann A., Kietzmann T., Moretti F., Mazzone M., Cell Rep. 2017, 18, 1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uzozie A. C., Selevsek N., Wahlander A., Nanni P., Grossmann J., Weber A., Buffoli F., Marra G., Mol. Cell. Proteomics 2017, 16, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Faktor J., Sucha R., Paralova V., Liu Y., Bouchal P., Proteomics 2017, 17, 1600323. [DOI] [PubMed] [Google Scholar]
- 10. Picotti P., Aebersold R., Nature Methods 2012, 9, 555. [DOI] [PubMed] [Google Scholar]
- 11. Keshishian H., Addona T., Burgess M., Mani D. R., Shi X., Kuhn E., Sabatine M. S., Gerszten R. E., Carr S. A., Mol. Cell. Proteomics 2009, 8, 2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Selevsek N., Matondo M., Carbayo M. S., Aebersold R., Domon B., Proteomics 2011, 11, 1135. [DOI] [PubMed] [Google Scholar]
- 13. Schiess R., Wollscheid B., Aebersold R., Mol. Oncol. 2009, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fortin T., Salvador A., Charrier J. P., Lenz C., Lacoux X., Morla A., Choquet‐Kastylevsky G., Lemoine J., Mol. Cell. Proteomics 2009, 8, 1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Domanski D., Percy A. J., Yang J., Chambers A. G., Hill J. S., Cohen Freue G. V., Borchers C. H., Proteomics 2012, 12, 1222. [DOI] [PubMed] [Google Scholar]
- 16. Percy A. J., Chambers A. G., Yang J., Borchers C. H., Proteomics 2013, 13, 2202. [DOI] [PubMed] [Google Scholar]
- 17. Altelaar A. F., Frese C. K., Preisinger C., Hennrich M. L., Schram A. W., Timmers H. T., Heck A. J., Mohammed S., J. Proteomics 2013, 88, 14. [DOI] [PubMed] [Google Scholar]
- 18. Rodríguez‐Suárez E., Whetton A. D., Mass Spectrom. Rev. 2013, 32, 1. [DOI] [PubMed] [Google Scholar]
- 19. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A.‐J. L., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega‐Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A., Nat. Biotechnol. 2009, 27, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abbatiello S. E., Mani D. R., Schilling B., Maclean B., Zimmerman L. J., Feng X., Cusack M. P., Sedransk N., Hall S. C., Addona T., Allen S., Dodder N. G., Ghosh M., Held J. M., V. H., Inerowicz H. D., Jackson A., Keshishian H., Kim J. W., Lyssand J. S., Riley C. P., Rudnick P., Sadowski P., Shaddox K., Smith D., Tomazela D., Wahlander A., Waldemarson S., Whitwell C. A., You J., Zhang S., Kinsinger C. R., Mesri M., Rodriguez H., Borchers C. H., Buck C., Fisher S. J., Gibson B. W., Liebler D., Maccoss M., Neubert T. A., Paulovich A., Regnier F., Skates S. J., Tempst P., Wang M., Carr S. A., Mol. Cell. Proteomics 2013, 12, 2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abbatiello S. E., Schilling B., Mani D. R., Zimmerman L. J., Hall S. C., MacLean B., Albertolle M., Allen S., Burgess M. W., Cusack M. P., Ghosh M., Hedrick V., Held J. M., Inerowicz H. D., Jackson A., Keshishian H., Kinsinger C. R., Lyssand J., Makowski L., Mesri M., Rodriguez H., Rudnick P., Sadowski P., Sedransk N., Shaddox K., Skates S. J., Kuhn E., Smith D., Whiteaker J. R., Whitwell C., Zhang S., Borchers C. H., Fisher S. J., Gibson B. W., Liebler D. C., MacCoss M. J., Neubert T. A., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A., Mol. Cell. Proteomics 2015, 14, 2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carr S. A., Abbatiello S. E., Ackermann B. L., Borchers C., Domon B., Deutsch E. W., Grant R. P., Hoofnagle A. N., Uumlttenhain R. H, Koomen J. M., Liebler D. C., Liu T., Maclean B., Mani D. R., Mansfield E., Neubert H., Paulovich A. G., Reiter L., Vitek O., Aebersold R., Anderson L., Bethem R., Blonder J., Boja E., Botelho J., Boyne M., Bradshaw R. A., Burlingame A. L., Chan D., Keshishian H., Kuhn E., Kinsinger C., Lee J., Lee S. W., Moritz R., Oses‐Prieto J., Rifai N., Ritchie J., Rodriguez H., Srinivas P. R., Townsend R. R., Van Eyk J., Whiteley G., Wiita A., Weintraub S., Mol. Cell. Proteomics 2014, 13, 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A., Mol. Cell. Proteomics 2007, 6, 2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H., Mol. Cell. Proteomics 2009, 8, 1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Percy A. J., Chambers A. G., Parker C. E., Borchers C. H., Met. Mol. Biol. 2013, 1000, 167. [DOI] [PubMed] [Google Scholar]
- 26. Percy A. J., Yang J., Chambers A. G., Simon R., Hardie D. B., Borchers C. H., J. Proteome Res. 2014, 13, 3733. [DOI] [PubMed] [Google Scholar]
- 27. Percy A. J., Yang J., Hardie D. B., Chambers A. G., Tamura‐Wells J., Borchers C. H., Methods 2015, 81, 24. [DOI] [PubMed] [Google Scholar]
- 28. Percy A. J., Mohammed Y., Yang J., Borchers C. H., Bioanalysis 2015, 7, 2991. [DOI] [PubMed] [Google Scholar]
- 29. Percy A. J., Chambers A. G., Yang J., Domanski D., Borchers C. H., Anal. Bioanal. Chem. 2012, 404, 1089. [DOI] [PubMed] [Google Scholar]
- 30. Szyszka R., Hanton S. D., Henning D., Owens K. G., J. Am. Soc. Mass Spectrom. 2011, 22, 633. [DOI] [PubMed] [Google Scholar]
- 31. Toghi Eshghi S., Li X., Zhang H., Anal. Chem. 2012, 84, 7626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Notari S., Mancone C., Alonzi T., Tripodi M., Narciso P., Ascenzi P., J. Chromatogr. B 2008, 863, 249. [DOI] [PubMed] [Google Scholar]
- 33. Rubakhin S. S., Sweedler J. V., Anal. Chem. 2008, 80, 7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mason D. R., Reid J. D., Camenzind A. G., Holmes D. T., Borchers C. H., Methods 2012, 56, 213. [DOI] [PubMed] [Google Scholar]
- 35. Anderson N. L., Razavi M., Pearson T. W., Kruppa G., Paape R., Suckau D., J. Proteome Res. 2012, 11, 1868. [DOI] [PubMed] [Google Scholar]
- 36. Peters F. T., Remane D., Anal. Bioanal. Chem. 2012, 403, 2155. [DOI] [PubMed] [Google Scholar]
- 37. Gergov M., Nenonen T., Ojanperä I., Ketola R. A., J. Anal. Toxicol. 2015, 39, 359. [DOI] [PubMed] [Google Scholar]
- 38. Kim J.‐S., Fillmore T. L., Liu T., Robinson E., Hossain M., Champion B. L., Moore R. J., Camp D. G., Smith R. D., Qian W.‐J., Mol. Cell. Proteomics 2011, 10, M110.007302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuepper U., Musshoff F., Madea B., Forensic Sci. Int. 2011, 207, 84. [DOI] [PubMed] [Google Scholar]
- 40. Ahn E. J., Kim H., Chung B. C., Moon M. H., J. Sep. Sci. 2007, 30, 2598. [DOI] [PubMed] [Google Scholar]
- 41. Jiménez‐Díaz I., Vela‐Soria F., Zafra‐Gómez A., Navalón A., Ballesteros O., Navea N., Fernández M. F., Olea N., Vílchez J. L., Talanta 2011, 84, 702. [DOI] [PubMed] [Google Scholar]
- 42. Vela‐Soria F., Jiménez‐Díaz I., Rodríguez‐Gómez R., Zafra‐Gómez A., Ballesteros O., Navalón A., Vílchez J. L., Fernández M. F., Olea N., Talanta 2011, 85, 1848. [DOI] [PubMed] [Google Scholar]
- 43. Dasenaki M. E., N. S. Thomaidis, Anal. Chim. Acta 2015, 880, 103. [DOI] [PubMed] [Google Scholar]
- 44. Percy A. J., Chambers A. G., Yang J., Jackson A. M., Domanski D., Burkhart J., Sickmann A., Borchers C. H., J. Proteomics 2013, 95, 66. [DOI] [PubMed] [Google Scholar]
- 45. Percy A. J., Chambers A. G., Smith D. S., Borchers C. H., J. Proteome Res. 2013, 12, 222. [DOI] [PubMed] [Google Scholar]
- 46. Mohammed Y., Percy A. J., Chambers A. G., Borchers C. H., J. Proteome Res. 2015, 14, 1137. [DOI] [PubMed] [Google Scholar]
- 47. Kelly W. R., Pratt K., Guthrie W. F., Martin K. R., Anal. Bioanal. Chem. 2011, 400, 1805. [DOI] [PubMed] [Google Scholar]
- 48. Percy A. J., Tamura‐Wells J., Albar J. P., Aloria K., Amirkhani A., Araujo G. D. T., Arizmendi J. M., Blanco F. J., Canals F., Cho J.‐Y., Colomé‐Calls N., Corrales F. J., Domont G., Espadas G., Fernandez‐Puente P., Gil C., Haynes P. A., Hernáez M. L., Kim J. Y., Kopylov A., Marcilla M., McKay M. J., Mirzaei M., Molloy M. P., Ohlund L. B., Paik Y.‐K., Paradela A., Raftery M., Sabidó E., Sleno L., Wilffert D., Wolters J. C., Yoo J. S., Zgoda V., Parker C. E., Borchers C. H., EuPA Open Proteomics 2015, 8, 6. [Google Scholar]
- 49. Mohammed Y., Domański D., Jackson A. M., Smith D. S., Deelder A. M., Palmblad M., Borchers C. H., J. Proteomics 2014, 106, 151. [DOI] [PubMed] [Google Scholar]
- 50. Kuzyk M. A., Parker C. E., Domanski D., Borchers C. H., Methods Mol. Biol. 2013, 1023, 53. [DOI] [PubMed] [Google Scholar]
- 51.U.S. Food and_Drug_Administration, US Department of Health and Human Services, Food and Drug Administration. Guidance for Industry Bioanalytical Method Validation. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070107.pdf 2001.
- 52.U.S. Food and Drug Administration, Guidance for Industry Bioanalytical Method Validation ‐ September 2013 Biopharmaceuticals http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf 2013, July 26, 2015.
- 53. Mani D. R., Abbatiello S. E., Carr S. A., BMC Bioinfo. 2012, 13, S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li W., Cohen L. H., Anal. Chem. 2003, 75, 5854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information