

Summary
Chimeric antigen receptors (CARs) significantly enhance anti-tumor activity of immune effector cells. While most studies have evaluated CAR-expression in T cells, here we evaluate different CAR constructs that improve natural killer (NK) cell-mediated killing. We identified a CAR containing the transmembrane domain of NKG2D, the 2B4 co-stimulatory domain, and the CD3ζ signaling domain to mediate strong antigen-specific NK cell signaling. NK cells derived from human iPSCs that express this CAR (NK-CAR-iPSC-NK cells) have a typical NK cell phenotype and demonstrate improved anti-tumor activity compared to T-CAR expressing iPSC-derived NK cells (T-CAR-iPSC-NK cells) and non-CAR expressing cells. Using an ovarian cancer xenograft model, NK-CAR-iPSC-NK cells significantly inhibited tumor growth and prolonged survival compared to PB-NK cells, iPSC-NK cells, or T-CAR-iPSC-NK cells. Additionally, NK-CAR-iPSC-NK cells demonstrate similar in vivo activity as T-CAR-expressing T cells, though with less toxicity. These NK-CAR-iPSC-NK cells now provide standardized, targeted “off the shelf” lymphocytes for anti-cancer immunotherapy.
Keywords: natural killer cells, chimeric antigen receptors, iPSCs, immunotherapy, ovarian cancer
eTOC Blurb
Natural killer (NK) cells are a key part of the immune system’s ability to mediate anti-cancer activity. Kaufman and colleagues utilize human iPSCs to produce NK cells with novel chimeric antigen receptors that specifically target cancer cells in an antigen-specific manner to improve survival in an ovarian cancer xenograft model.
Introduction
Immune cell-based therapies have become a promising approach to better treat and potentially cure malignancies that are refractory to other modalities such as chemotherapy, surgery, or radiation therapy. Recent clinical trials include use of T lymphocytes and natural killer (NK) cells for treatment of hematopoietic and solid tumors(Barrett et al., 2014; Handgretinger et al., 2016; Ramos et al., 2016). While use of T cells engineered to express chimeric antigen receptors (CARs) have demonstrated potency against CD19-expressing tumors in several trials(Grupp et al., 2013; Porter et al., 2011; Qasim et al., 2017), the efficacy of CAR-T cells against solid tumors has been more limited(Ahmed et al., 2015; Beatty et al., 2014; Fesnak et al., 2016). Notably, CAR-T cell-based therapies typically require collection and gene modification of patient (autologous) T cells. This process can be time consuming and inefficient. Indeed, not all patients are able to effectively mobilize functional T cells in numbers suitable for this process(Ramos et al., 2016; Themeli et al., 2015).
Natural Killer (NK) cells comprise another important part of the cellular immune system, with potent ability to kill tumor cells and virally-infected cells(Caligiuri, 2008; Guillerey et al., 2016; Morvan and Lanier, 2016). NK cells mediate their activity without requiring HLA-matching (Morvan and Lanier, 2016). Therefore, NK cells function as allogeneic effectors and do not need to be collected from a patient or a specific HLA-matched donor. NK cell-mediated cytotoxicity is regulated by a repertoire of activating and inhibitory receptors(Guillerey et al., 2016; Morvan and Lanier, 2016). Activating receptors includes natural cytotoxic receptors (NCRs), NKG2D, CD16 (FcγRIIIa), FasL, TRAIL and co-stimulatory receptors such as LFA-1, CD244 (2B4), and CD137 (41BB). These activating cell surface receptors have the capacity to trigger cytolytic programs, as well as cytokine and chemokine secretion via intra-cytoplasmic ITAMs such as in 2B4, 41BB and/or via other transmembrane signaling adaptors (Bryceson et al., 2006; Caligiuri, 2008; Guillerey et al., 2016; Lanier, 2008; Long et al., 2013; Morvan and Lanier, 2016; Smyth et al., 2005; Vivier et al., 2004).
Many trials of adoptive NK cell-based immunotherapy have been done over the past decade. These trials have used NK cells isolated from peripheral blood, umbilical cord blood, and the NK cell line, NK92(Bachanova et al., 2014; Dolstra et al., 2017; Handgretinger et al., 2016; Klingemann et al., 2016; Koehl et al., 2016; Liu et al., 2017; Romee et al., 2016; Yang et al., 2016). Trials using adoptive transfer of allogeneic NK cells demonstrate they are safe, with little evidence of toxicities such as graft-versus-host disease (GvHD), cytokine release syndrome, or neurotoxicity(Bachanova and Miller, 2014; Handgretinger et al., 2016; Miller et al., 2005; Morvan and Lanier, 2016; Romee et al., 2016). Typically, these clinical studies use a combination of lympho-depleting chemotherapy and cytokine treatment to enable in vivo survival and expansion of the allogeneic NK cells for 1–3 weeks post treatment. While NK cells isolated from peripheral blood (PB-NK cells) or cord blood (UCB-NK cells) demonstrate efficacy against acute myelogenous leukemia (AML), there has been less activity against solid tumors(Geller et al., 2011; Geller and Miller, 2011; Sakamoto et al., 2015; Yang et al., 2016). While the NK92 cell line has also been used in clinical trials, these cells are aneuploid and must be irradiated before being administered to patients (Klingemann et al., 2016). This limits the survival and proliferation of the NK cells- two key criteria that are known to correlate improved efficacy of NK cell-based immunotherapy(Bachanova et al., 2014; Miller et al., 2005; Romee et al., 2016). In contrast, iPSC-NK cells can be produced from a standardized cell population to provide a homogeneous NK cell population that can be grown to clinical scale (Hermanson et al., 2016; Knorr et al., 2013b).
Engineering NK cells with CARs to improve killing of solid tumors has been previously proposed(Cheng et al., 2013; Figueroa et al., 2015; Hermanson and Kaufman, 2015; Zhang et al., 2017), and clinical trials utilizing CAR-expressing NK cells for the treatment of both hematological malignancies and refractory solid tumors have been initiated (Bollino and Webb, 2017) [ClinicalTrials.gov: NCT00995137, NCT01974479, NCT02839954, NCT02892695, NCT02742727, and NCT02944162]. However, these trials all utilize either PB-NK cells or NK92 cells, as well as CARs designed for T cells and not optimized for NK cell signaling.
Here, we hypothesized that engineering NK cell activation domains(Altvater et al., 2009; Chang et al., 2013; Song et al., 2011; Topfer et al., 2015; Zhang and Sentman, 2011) into CARs would improve the anti-tumor efficacy of NK cells, especially against solid tumors that are more resistant to NK cell-mediated killing(Bollino and Webb, 2017; Geller et al., 2011; Geller and Miller, 2011; Hermanson and Kaufman, 2015; Sakamoto et al., 2015; Yang et al., 2016). Primary NK cells are relatively less efficient to genetically modify compared to T cells(Carlsten and Childs, 2015). However, human induced pluripotent stem cells (iPSCs) can efficiently produce NK cells(Hermanson et al., 2016; Knorr et al., 2013b; Ni et al., 2014) and be genetically-modified using both viral and non-viral methods(Giudice and Trounson, 2008; Wilber et al., 2007; Xie et al., 2014). Here, we demonstrate that iPSCs engineered to express CAR constructs specifically designed to enhance NK cell activity can derive CAR-expressing antigen-specific NK cells with potent ability to kill mesothelin-expressing tumors both in vitro and in vivo. Therefore, NK-CAR-expressing iPSC-derived NK cells provide a potential strategy to produce “off-the-shelf”, targeted allogeneic lymphocytes suitable to treat otherwise refractory malignancies.
Results
Screening CAR constructs with enhanced NK cell activity
Our initial studies screened 9 different CAR constructs optimized for activity in NK cells. All CARs were targeted to the tumor-associated antigen mesothelin (meso) (Hassan and Ho, 2008) via the anti-meso scFv SS1(Chowdhury et al., 1998; Hassan and Ho, 2008). We designed NK cell-specific CARs that consisted of a transmembrane domain and 1 or 2 co-stimulatory domains (CD/s) typically expressed in NK cells, as well as the ζ stimulatory domain that is common to both the CD3 complex in T cells(Love and Hayes, 2010) and CD16 in NK cells(Lanier et al., 1989) (Table 1). Initial studies expressed these constructs in NK92 cells to efficiently test the ability of these CARs to mediate increased killing of meso+ cells.
Table 1.
Construct of Chimeric Antigen Receptors (CARs)
| CARs | Target | Construct (-TM-CD/s-SD) |
|---|---|---|
| T-CAR | hMesothelin | scFv-CD28-CD28-CD137-CD3ζ |
| CAR1 | hMesothelin | scFv-CD16-2B4-CD3ζ |
| CAR2 | hMesothelin | scFv-NKp44-DAP10-CD3ζ |
| CAR3 | hMesothelin | scFv-NKp46-2B4-CD3ζ |
| CAR4 | hMesothelin | scFv-NKG2D-2B4-CD3ζ |
| CAR5 | hMesothelin | scFv-NKG2D-CD137-CD3ζ |
| CAR6 | hMesothelin | scFv-NKG2D-2B4-DAP12-CD3ζ |
| CAR7 | hMesothelin | scFv-NKG2D-2B4-DAP10-CD3ζ |
| CAR9 | hMesothelin | scFv-NKG2D-CD137-2B4-CD3ζ |
| CAR10 | hMesothelin | scFv-NKG2D-CD3ζ |
CAR-expressing-NK92 cells (Supplemental Figures S1A and S1B) were tested for their ability to kill K562 cells (mesoneg), MA148 ovarian cancer cells (mesolo), meso-expressing K562 cells (mesohigh) and A1847 ovarian cancer cells (mesohigh) (Figure 1A). NK92 cells expressing NK cell-specific CAR4, CAR7, and CAR9 had the greatest increase in anti-tumor activity when stimulated by mesohigh targets (Figure 1B–1E). These CARs also specifically enhanced CD107a expression (indicative of cytotoxic granule release) after meso+ target stimulation (Figure 1F and 1G). Interestingly, these NK-CARs mediated killing of mesohigh tumors more than NK92 cells expressing the 3rd generation T cell CAR (T-CAR) expressed in NK92 cells. Importantly, this was not dependent on the expression level of T-CAR vs NK-CAR, as each vector was expressed at similar levels across all of the lines (Supplemental Figures S1A and S1B).
Figure 1. CAR mediated anti-tumor activity in NK92 cells.

(A) Surface expression of mesothelin (meso) in leukemia cell lines K562 and K562meso, ovarian cancer cell lines MA148 and A1847. (B–E) NK92 cells that express the indicated CARs were co-cultured with (B) mesoneg target cells of K562, (C) mesohigh target cells of K562meso, (D) mesolow target cells of MA148 cells, and (E) mesohigh target cells of A1847 at indicated effector to target ratios. The mean of % specific tumor cell lysis ± S.D are shown. (F and G) CD107a expression was determined in anti-CD56 labeled effector cells after stimulation by (F) K562 and K562meso cells, and (G) MA148 and A1847. Data were normalized to the percentage of media-treated effector cells, shown as mean ± S.D.
Statistical analysis by two-tailed Student t-test, * P<0.05, ** P<0.001.
See also Figure S1.
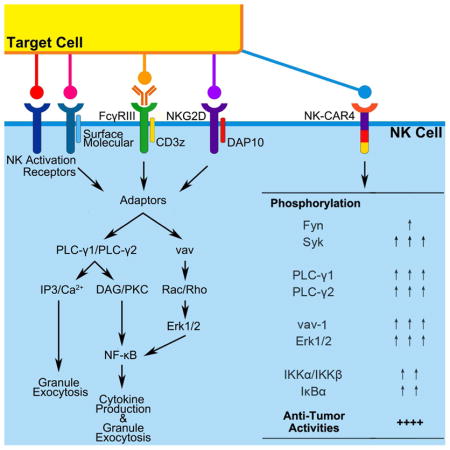
We next did a more detailed comparison of NK92 cells expressing anti-meso CAR4 (SS1-NKG2D-2B4ζ) and the anti-meso T-CAR (SS1-CD28-41BBζ) (Supplemental Figures S1C and S1D). Again, CAR4-expressing cells demonstrated greater cytotoxicity against meso+ targets (Supplementary Figure S1E and S1F), and also a corresponding increase in expression of CD107a and IFN-γ (Supplemental Figures S1G and S1H). To better define cell-signaling pathways that mediate CAR-induced cellular activation, we evaluated phosphorylation of key NK cell signaling mediators, including Syk, Erk1, and Erk2. For these studies, we compared phosphorylation of these proteins in NK92 cells expressing CAR4 and T-CAR (Supplementary Figure S2). CAR4-NK92 cells stimulated by A1847 (mesohigh) demonstrated an early increase of Syk phosphorylation, and later in p-Erk1 and p-Erk2 (Supplemental Figure S2A). Notably, CAR4-NK92 cells demonstrated significantly increased ratio of phosphorylation in both Syk and Erk1/2 compared to T-CAR-NK92 cells (Supplemental Figures S2B and S2C).
We next tested the role of each domain in the NK-CAR4 construct to mediate improved NK cell activity by introducing site-specific mutations known to decrease function of each domain of CAR4: NKG2D (CAR4-NKG2DΔ consists of a Arg to Ala point mutation in transmembrane section(Garrity et al., 2005)), 2B4 (CAR4-2B4Δ consists of Tyr to Phe point mutations in all 3 ITAMs and 1 ITSM of cytoplasmic domains(Lanier, 2008)), and CD3ζ (CAR4-CD3zΔ consists of Tyr to Phe point mutations in all 3 ITAMs of cytoplasmic domains(Love and Hayes, 2010)). An additional construct has all three domains mutated [CAR4(meso) ALLΔ] (Figure 2A and 2B). We examined NK cell-mediated cytotoxicity by the CAR4 domain-specific mutant constructs in CAR-expressing NK92 cells. Initial studies demonstrated that each vector was expressed at similar levels in all CAR-expressing NK92 cell populations tested (Figure 2A). As expected, NK-CAR4 consistently mediated improved killing of mesohigh targets (Figure 2C–2F). In contrast, under the same conditions, NK-CAR4-NKG2DΔ and NK-CAR4-2B4Δ resulted in decreased cytolytic ability, though NK-CAR4-CD3zΔ had relatively little effect. Both cytotoxic granule release (CD107a expression, Figure 2G) and IFN-γ production (Figure 2H) in NK-CAR4 was weakened by NKG2DΔ and 2B4Δ, and abolished in ALLΔ. Additionally, we investigated signaling pathway of domain mutated CAR4 in response to target stimulation (Figure 2I). When stimulated by mesohigh targets, both NKG2DΔ, 2B4Δ had less phosphorylation of Syk (Figure 2I), and less phosphorylation ratio in PLC-γ2, and Erk1/2 compared to the wild-type CAR4 expressing cells (Figure 2J and 2K). Interestingly, CD3zΔ lead to a decrease in Syk and PLC-γ2 phosphorylation, but not Erk1/2 phosphorylation. ALLΔ also eliminated these CAR4-NK signaling events. These studies demonstrate that each NK-specific domain of NKG2D-2B4ζ facilitated CAR activity to promote antigen-induced NK cell-mediated cytotoxicity.
Figure 2. Domain-deficient CAR4-mediated NK cell activation response and cytolysis in NK92 cells.

(A) Expression of GFP and surface expression of CAR4 in NK92 cells assessed by flow cytometry. (B) Schematic representation of the transposon vector encoding the domain-deficient CAR4 mutation TM: NKG2D (NKG2DΔ), CD: 2B4 (2B4Δ), SD: CD3ζ (CD3zΔ), and all domain-deficient mutated (ALLΔ). (C–F) NK92 cells were co-cultured with europium-loaded target cells of (C) mesoneg K562, (D) mesohigh K562meso, (E) mesolow MA148, and (F) mesohigh A1847 at indicated effector to target ratios. The mean of % specific tumor cell lysis ± S.D are shown. (G and H) CD107a expression, and IFN-γ production was analyzed in anti-CD56 labeled effector cells after co-culture with target cells. (I) Protein analysis of total protein and phospho-protein molecular of Syk, PLC-γ2 and Erk1/2 in cell lysate of NK92 cells by immunoblots in different conditions. Left: unstimulated; middle: 1-hour mesolow MA148 cell stimulation; right: 1-hour mesohigh A1847 cell stimulation. Anti-CD3ζ antibody was used to determine CAR expression with estimated molecular size. GAPDH and P-CK were used as loading control. (J and K) Quantification of phosphorylated level over total protein level in NK effector cells co-cultured with mesohigh A1847 target cells were performed in protein lysates stained for of (J) PLC-γ2, (K) Erk1/2. Data were shown as mean ± S.D.
Statistics by two-tailed Students t-test: NS: not significant. * P<0.05, ** P<0.01,
Expression and function of NK-CARs in iPSC-derived NK cells
To provide a source of CAR-expressing NK cells similar to peripheral blood NK cells, we stably expressed T-CAR(meso), “empty” (no scFv) version of NK-CAR(-)s and anti-meso NK-CAR(meso)s in human iPSCs. Previous studies demonstrate human iPSCs (and hESCs) can be differentiated into NK cells with phenotypic and functional similarities to NK cells isolated from peripheral blood PB-NK cells(Hermanson et al., 2016; Knorr et al., 2013b; Ni et al., 2011). For these studies, CARs were subcloned into a PiggyBac transposon vector with cHS4 insulator element(Burgess-Beusse et al., 2002; Moriarity et al., 2013; Yahata et al., 2007). These CAR-expressing iPSCs demonstrated similar efficiency in NK cell production as non-CAR-expressing iPSCs (Supplemental Figure S3) (Knorr et al., 2013b; Ng et al., 2008). Moreover, the iPSC-NK cell phenotype with or without CAR-expression was similar to PB-NK cells (Figure 3A). Notably, NKG2D expression was not compromised in NK-CAR expressing iPSC-NK cells.
Figure 3. Phenotype and anti-tumor activities of CAR-expressing iPSC derived NK cells.

(A) Flow cytometric analysis of GFP, and NK cell surface receptors in the gate of CD56+ NK cell populations. (B) iPSC-NK cells derived from pooled or clonal CAR4(meso)-iPSC (#1 and #4), CAR4(-)-iPSC (#2 and #3) were co-cultured with europium-loaded K562meso cells (left), or A1847 cells (right) as different effector to target ratios. The mean of % specific tumor cell lysis ± S.D are shown. (C and D) CD107a expression (C), and IFN-γ production (D) was accessed by flow cytometery in anti-CD56 labeled iPSC-NK populations after the stimulation of K562meso or A1847. Data were plotted and shown as mean ± S.D. (E) Membrane protein analysis in cell lysate of iPSC-NK populations by immunoblots. NCAM and GAPDH were used as loading controls. (F) Co-IP was performed by using an anti-DAP-10 antibody in cell lysate of A1847 cell-stimulated iPSC-NK populations. Protein was subjected to the analysis of DAP-10, NKG2D, and CD3z by immunoblots. (G) Total and phospho-protein analysis of Fyn-PLC pathway (15 min A1847 stimulation); Syk-Vav1-Erk pathway and NF-κB (IKKα/β and IκBα) pathway (30 min A1847 stimulation) in cell lysate of iPSC-NK populations by immunoblots. Lane 1–4: unstimulated iPSC-NK cell populations as in (E); Lane 5–8: A1847 stimulated iPSC-NK cell populations as in (F). (H and I) Cytolysis ability of iPSC-NK populations, or T cell populations against (H) K562meso or (I) A1847 cells were quantified using the IncuCyte real-time imaging system over a 24 hour time-course. Percentage of caspase 3/7 event stained cells over the total pre-labeled cells were measured.
Statistics by two-tailed Student t-test, * P<0.05, ** P<0.01.
See also Figure S4 and S6.
We next investigated the function of CAR-expressing iPSC-derived NK cells. Here, we again tested our lead NK-CAR constructs (CAR4, CAR7, CAR9) now expressed in iPSC-NK cells. To confirm that CAR4 activity was antigen-specific and not due to non-specific NK cell activation via overexpression of the signaling domains, we also tested “empty” CAR [CAR(-)] constructs that contains the same CD8hinge-NKG2D-2B4ζ domains without the anti-meso-scFv. CAR expression was initially evaluated by immunoblots and flow cytometry (Supplemental Figures S4A and S4B). In iPSC-NK cells, NK-CAR4(meso), NK-CAR7(meso), and NK-CAR9(meso) had similar expression level as well as the expression of the empty (no scFv) version of these CARs: NK-CAR4(-), NK-CAR7(-), and NK-CAR9(-) based on GFP expression and detection of CAR-expressed zeta in the membrane fraction of cell lysates. We again tested function using cytotoxicity and CD107a (granule release) assays via stimulation by K562 and K562meso cells (Supplemental Figure S4C–S4E). As expected, all three NK-CARs demonstrated antigen-specific increase in cytotoxicity and CD107a expression. In contrast, the “empty” NK-CAR(-) expressing iPSC-NK cells had limited cytotoxicity and CD107a expression, similar to iPSC-NK cell with no CAR expression.
We next compared the function of NK-CAR4 with the T-CAR expressed in iPSC-NK cells. Here, we extended our studies to evaluate iPSC-NK cells derived from a clonal population of CAR4-iPSCs. Clones of CAR4(meso) and CAR4(-) iPSCs were validated by vector copy number and similar CAR expression in flow cytometry (GFP) and immunoblots (Supplemental Figures S5A–S5C). Clonally-derived CAR4(meso)-iPSC-NK cells recapitulate the strong cytolytic activity as pooled (non-clonal) CAR4(meso)-iPSC-NK cells against mesohigh targets (Figure 3B). Consistent with our findings in NK92 cells, T-CAR(meso)-iPSC-NK cells were less able to kill mesohigh targets compared to NK-CAR4(meso). Again, the A1847 cells (mesohigh) stimulated CD107a (granule release) and IFN-γ expression in both the CAR4-iPSC-NK cells and the T-CAR-iPSC NK cells, though the responses were greater in the CAR4-iPSC-NK cells (Figure 3C and 3D). Furthermore, we investigated the NK cell activation signaling pathway mediated by CARs in iPSC-NK cells in response to mesohigh A1847 stimulation (Figure 3E–3G). First we verified the similar surface expression of CARs by membrane protein immunoblot (Figure 3E). The ligation of NK receptor NKG2D leads to the activating signaling transduction by the recruitment of endogenous DAP10(Eagle and Trowsdale, 2007; Garrity et al.; Rosen et al.). Similarly, under the stimulation of A1847 cells, which express NKG2D ligands such as ULBP(Song et al.), the endogenous DAP10 was recruited with endogenous NKG2D molecular in all iPSC-NK population (Figure 3F). Notably, NK-CAR4(meso) (NKG2D-2B4ζ), but not other CARs, was involved in the recruitment of endogenous DAP10 in the condition of mesohigh A1847 stimulation (Figure 3F). We next investigated the down-stream activation signaling mediated by CAR expression in iPSC-NK cells (Figure 3G). With meso stimulation, CAR(meso) mediated an early phosphorylation increase of PLC-γ1 and PLC-γ2, though without the activation of Fyn (Supplemental Figures S5D and S5E). Moreover, CAR4(meso)-iPSC-NK cells demonstrated phosphorylated activation in the Syk-vav1-Erk pathway (Figure 3G and Supplemental Figures S5F). There is also activation of the NF-κB pathway (Figure 3G) demonstrated by phosphorylation increase of IκBα (Supplemental Figure S5G) compared to T-CAR(meso)-iPSC-NK cells. In contrast, the CAR4(-)-iPSC-NK cells and non-CAR-expressing-iPSC-NK cells did not activate these intracellular signaling events when stimulated by meso-expressing tumor cells.
To further evaluate the anti-tumor activity of NK-CAR4 and T-CAR, we compared these CAR-expressing iPSC-NK cells and CAR-expressing primary T cells. Here, human primary CD3+ T cells were isolated from 3 different donors and engineered to express the 3rd generation anti-meso T-CAR(meso)(SS1-CD28-41BBζ). Stable vector expression was accessed by GFP expression (Supplemental Figure S5H). And again, CAR4(meso)-expressing iPSC-NK cells demonstrated superior anti-tumor activity against either meso-expressing K562 or A1847 cells in compared to T-CAR(meso)-expressing iPSC-NK cells. Notably, T-CAR(meso)-expressing T cells exhibited better anti-tumor activities than NK-CAR4(meso)-expressing T cells, especially with enhanced the cytolytic activities against K562(meso) cells (Figures 3H and 3I).
NK cell-specific CAR4-iPSC-NK cells have improved activity against ovarian cancer in vivo
To evaluate activity of the CAR4-iPSC-NK cells in vivo, we tested killing of the A1847 ovarian cancer cells in a mouse xenograft model, as previously utilized (Hermanson et al., 2016; Knorr et al., 2013a). Here, we initially compared PB-NK cells, iPSC-NK cells, CAR4(meso)-iPSC-NK cells, and T CAR(meso)-iPSC-NK cells. Mice were inoculated intraperitoneally (ip) with luciferase-expressing A1847 cells and 4 days later received a single injection of 1.5 × 107 NK cells (Figure 4A). As in previous studies of NK cell-mediated anti-tumor activity, IL-2 and IL-15 were administrated for 21 days to promote in vivo NK survival and expansion (Hermanson et al., 2016; Knorr et al., 2013a; Woll et al., 2009). Mice were monitored by bioluminescent imaging (BLI) (Figure 4B). Compared to untreated tumor-bearing mice, treatment with NK cells produced a significant reduction in tumor burden after 7 days (P<0.0001, Figure 4C), with maximal anti-tumor activity seen with the CAR4-iPSC-NK cells after 28-day (P=0.0044 compared to iPSC-NK cells, Figure 4D). This improved anti-tumor activity of the CAR4-iPSC-NK cells compared to PB-NK cells, iPSC-NK cells, and T-CAR-iPSC-NK cells (Figure 4E, with analysis of individual mice in Supplemental Figure S6A) and lead to markedly improved survival compared to iPSC-NK cells (P=0.0017, HR=0.2236, 95%CI: 0.009016–0.2229) and T-CAR-iPSC-NK cells (P=0.0018, HR=0.2153, 95%CI: 0.009771–0.2511), as well as compared to PB-NK cells (P=0.0018, Figure 4F).
Figure 4. CAR4-expressing iPSC derived NK cells display superior anti-ovarian cancer activity in vivo.

(A) Schematic of in vivo studies using luciferase (luc)-expressing mesohigh A1847 cells in a mouse xenograft model treated with PB-NK cells and iPSC-NK populations and cytokine administration. (B) Tumor burden was determined by weekly bioluminescent imaging (BLI). (C and D) Images of representative time points are shown. Quantification of tumor burden on (C) day 7, and (D) day 28 was plotted based on BLI total flux (photons/sec). (E) Tumor burden of each group was monitored for 49 days post NK population infusion. The BLI data is plotted, mean ± S.D are shown. Statistic: two-tailed Student t-test, T-CAR(meso)-iPSC-NK vs. CAR4(meso)-iPSC-NK. (F) Kaplan-Meier curve representing the percent survival of the experimental groups: Tumor only, or treated with PB-NK cells, iPSC-NK cells, T-CAR(meso)-iPSC-NK cells, or CAR4(meso)-iPSC-NK cells. Statistic: two-tailed Log-rank test. (G–I) Flow cytometric quantification of CD45+CD56+CD3- NK cell population from (G) peripheral blood, (H) spleen, and (I) peritoneal fluid. Each dot represents one recipient mouse. Median ± S.D. is shown.
Statistical analysis by two-tailed one-way ANOVA. *P<0.05, ** P<0.01, *** P<0.001.
See also Figure S7A.
We next investigated the in vivo persistence of NK cells in a separate group of tumor-bearing mice post-injection by examining blood, spleen, and peritoneal fluid for the presence of NK cells. At day 10, CAR4-iPSC-NK cells were significantly increased in the circulation (Figure 4G, mean 4.28%, P=0.0143), spleen (Figure 4H, mean 7.22%, P=0.0066), and peritoneal fluid (Figure 4I, mean 9.80%, P=0.0410) compared to PB-NK cells and iPSC-NK cells. By 21 days when exogenous cytokine administration ended, the CAR4-iPSC-NK cell levels returned to the same as PB-NK cells and iPSC-NK cells. By day 28, few NK cells are seen in these tissues, as previously demonstrated(Hermanson et al., 2016; Knorr et al., 2013a). Together, these studies demonstrate that NK-CAR-expressing iPSC-derived NK cells mediate improved anti-tumor activity compared to non-CAR expressing PB-NK cells or iPSC-NK cells, as well as improved activity compared to T-CAR-expressing iPSC-NK cells.
An additional comparison of the anti-tumor activity was done to directly compare NK-CARs expressed in iPSC-NK cells to T-CARs expressed in primary T cells. The same A1847 xenograft model was established and tested with one dose of 1.0 × 107 NK or T cell population (Figure 5A). Both unmodified iPSC-NK cells and T cells show persistence early after infusion and reduced after day 10, though both unmodified iPSC-NK cells and CAR4-iPSC-NK cells demonstrated persistence at day 21(Figure 5B and 5C). Both T-CAR-expressing T cell and NK-CAR4 expressing iPSC-NK cells demonstrated in significant in vivo persistence post-injection (Figure 5B and 5C). Compared to unmodified T cells, T-CAR-expressing T cells were significantly increased in circulation at day 10 (Figure 5B, mean 2.394%, P=0.0163), and continued to proliferate at day 21 (Figure 5B, mean 25.28%, P=0.0009). Similarly, NK-CAR4-iPSC-NK cells exhibited increase in circulation at day 10 (Figure 5C, mean 1.81%, P=0.0049), and returned to basal levels (similar to non-CAR-expressing iPSC-NK cells) at day 21. The in vivo expansion and persistence corresponded to the anti-tumor activity of the NK-CAR4-iPSC-NK cells and T-CAR-expressing T cells (Figure 5A and D). Both T-CAR expressing T cells and NK-CAR4-expressing iPSC-NK cells demonstrated potent ability to reduce tumor burden at day 21. Detail of anti-tumor activity in individual mice is shown in Supplemental Figure S6B.
Figure 5. Comparison of CAR-expressing T cells to CAR-expressing NK cells.

(A) NSG mice were inoculated intraperitoneally with 2 × 105 luc+ A1847 cells. 3 days later, mice received 225cGy radiation following with 1.0 × 107 of iPSC-NK or T cell populations infusion intraperitoneally. Cytokines were administrated as indicated in previous for the following 21 days in mice that received iPSC-NK population treatment only. Tumor burden was determined by weekly BLI imaging until day 21. Images of representative time points are shown. (B and C) Flow cytometric quantification of (C) CD45+CD3+ T cell populations and (B) CD45+CD56+CD3- iPSC-NK populations from peripheral blood. Each dot represents one recipient mouse. (D) BLI data of tumor burden of each group monitored for 21 days post immune effector cell infusion (dpi). (E) Body weight of each group was monitored and plotted weekly in the time-course for 21 days post immune effector cell infusion. Mean ± S.D. is shown. Statistical analysis by two-tailed one-way ANOVA, T cells vs. iPSC-NK cells, T CAR(meso)-T cells vs. CAR4(meso)-iPSC-NK. (F–H) Levels of (F) hIFN-γ, (G) hTNF-α, and (H) hIL-6 were measured on plasma collected from peripheral blood in different days post immune effector cells infusion by ELISA assay. Median ± S.D. is shown. Statistic: two-tailed one-way ANOVA. (I) Kaplan-Meier curve represents the percent survival of the experimental groups.
Statistical analysis by two-tailed Log-rank test.
n.d.: not detected, NS: not significant, *P<0.05, ** P<0.01, *** P<0.001.
See also Figure S7B–S7D.
Notably, mice in both groups that received primary T cells demonstrated loss of body weight, more so for the T-CAR expressing T cells treated group (Figure 5E). T-CAR expressing T cells treated mice demonstrated significant body weight loss (> 50%, n=2 at days 23 and 39) and/or severe visceral hemorrhage and ischemia (n=3 at days 35, 39 and 52), leading to deaths between days 23–52. Examination of these mice demonstrated T-CAR-expressing T cells lead to an enlarged spleen and infiltrated organs with pathogenic damage, such as liver, lung, kidney, gut, but not in cardiac muscle (Supplemental Figure S6C). Notably, the NK-CAR4-iPSC-NK cells treated mice did not demonstrate weight loss (Figure 5E) or early non-tumor mediated deaths (Supplemental Figure S6D). In the course of immune effector cell treatment, we also evaluated the cytokine production release in plasma such as hIFN-γ (Figure 5F), hTNF-α (Figure 5G), and hIL-6 (Figure 5H). Both CAR expressing iPSC-NK cells and T cells produced IFN-γ detected in plasma (Figure 5F), but T-CAR-T cell treatment results to the increase of IFN-γ at 10 days post cell infusion and remained elevated until day 20 (Figure 5F). Similarly, NK-CAR4-iPSC-NK cells had increases of hTNF-α at 2 days post cell infusion, but this level returned to baseline by day 10. However, T-CAR-T cells lead to a more persistent increase in both hTNF-α and IL-6 levels from Day 2 to Day 20 that is tightly associated with cytokine release syndrome (CRS) in clinic practice(Maude et al.; Teachey et al.) (Figure 5G and 5H).
Direct comparison of survival of mice treated with NK-CAR4-iPSC-NK cells and 3rd generation T-CAR-expressing T cells demonstrated markedly improved survival of the NK-CAR-expressing NK cell treated mice (P=0.0316, HR=0.1372, 95%CI: 0.02011–0.7854) (Figure 5I), with 4 of 5 mice that received NK-CAR4-iPSC-NK cells still alive 70 days post-treatment, whereas all the T-CAR-expressing T cell-treated mice had a median survival of 39 days, 4 of 5 mice dead by Day 52, and one mouse still alive at 70 days post-treatment. Again, the non-CAR-expressing iPSC-derived NK cells mediated improved anti-tumor activity, as in previous studies(Hermanson et al., 2016) (Figure 4), with the anti-meso CAR4 further improving survival in this model (P=0.0064, HR=0.0979, 95%CI: 0.01566–0.4724).
Discussion
Here, we were able to design and test CAR constructs with NK cell-specific activating domains to promote improved anti-tumor activity. We combined the potency of CAR-mediated tumor cell targeting with the ability to efficiently derive large numbers of engineered NK cells from human iPSCs to generate NK-CAR expressing, iPSC-derived human NK cells. NK-CAR-iPSC-NK cells display a similar phenotype to PB-NK cells and unmodified iPSC-NK cells. Expression of an NK-CAR construct consisting of NKG2D-2B4ζ in iPSC-NK cells markedly improved NK-cell mediated cytotoxicity against antigen-expressing leukemia targets and ovarian cancer targets. Moreover, NKG2D-2B4ζ-iPSC-NK cells were able to effectively inhibit growth and prolong survival in an ovarian cancer xenograft model. Indeed, this anti-tumor activity was greater than T-CAR(CD28-41BBζ)-expressing iPSC-derived NK cells or non-targeted NK cells isolated from PB or derived from iPSCs. Direct comparison with T-CAR expressing T cells demonstrated similar anti-tumor activity at Day 21 of treatment, though markedly improved survival of the NK-CAR-iPSC-NK cells due to less toxicity of the NK cell-based treatment (less cytokine release and lymphocyte-related pathological damage). Mechanistic studies demonstrate the NKG2D-2B4ζ domains stimulate activation of known NK cell signaling pathways that mediate key NK cell effector activity(Bryceson et al., 2006; Kwon et al.; Lanier, 2008; Long et al., 2013). This NK-CAR-mediated signaling activity promotes increased in vivo NK cell expansion and survival that likely leads to improved anti-tumor activity of compared the PB-NK cells and iPSC-NK cells (Figure 4G–4I). Indeed, several clinical studies demonstrate that the expansion and persistence of NK population tightly corresponds to the outcome of adoptive NK cell therapy (Bachanova and Miller, 2014; Childs and Carlsten, 2015; Miller et al., 2005; Romee et al., 2016; Sakamoto et al., 2015). Therefore, this strategy now provides an enticing approach to create lymphocytes from a standardized cell source for a targeted “off-the-shelf” anti-cancer immunotherapy.
Many pre-clinical and clinical studies demonstrate the utility of adoptive NK cell-based immunotherapy to treat cancers with minimal toxicities such as GvHD or cytokine-release syndrome (Bachanova and Miller, 2014; Handgretinger et al., 2016; Miller et al., 2005; Romee et al., 2016). To date, PB-NK cells have been most effective in treatment of AML, with relatively little activity against solid tumors. Most recent trials have used PB-NK cells derived from allogeneic donors, resulting in cell products that are heterogeneous, differ between donors, and are limited to one patient per donor (Cheng et al., 2013; Geller et al., 2011; Handgretinger et al., 2016; Koehl et al., 2016; Miller et al., 2005; Romee et al., 2016). Here, we utilized an efficient and well-defined system to produce homogeneous and well-characterized human NK cells from iPSCs that demonstrate similar anti-tumor activity to PB-NK cells, even without CAR-expression. Notably, iPSC-NK cell production can be done at a clinical scale(Knorr et al., 2013b; Ni et al., 2013). Additionally, iPSC-NK cells engineered to express an optimized CAR maintain a typical NK cell phenotype and NK cell-mediated cytolytic machinery (Figure 3).
NK cell-mediated degranulation and transcription of cytokine and chemokine genes are initiated by intracellular signaling event delivered by activating and co-activating receptors(Guillerey et al., 2016; Long et al., 2013; Morvan and Lanier, 2016). Specifically, phosphorylation of activating pathways such as Syk-PLC-γ-DAG/IP3 and Rac1-PAK1-MEK-ERK result in actin polymerization and granule polarization (Lanier, 2008; Vivier et al., 2004). Phosphorylation of NF-κB pathway leads to activate synthesis of inflammatory cytokine genes (Kwon et al.; Lanier, 2008). We tested multiple NK-CAR constructs to identify an optimal NKG2D-2B4ζ combination. NKG2D is a type II transmembrane-anchored C-type lectin-like protein that is a key activation receptor of NK cells. Ligation of NKG2D associated with DNAX-activating protein 10 (DAP10), NKG2D signal results to promote the cytotoxic ability of NK cells(Dhar and Wu, 2018; Ho et al., 2002). Engagement of endogenous 2B4 by its ligand CD48 triggers phosphorylation of ITAMs in the cytoplasmic tail of 2B4. This signaling is required for optimal NK cell function(Nakajima et al., 1999) and potently enhances activation induced by other NK cell receptors(Sivori et al., 2000). In NK cells, CD3ζ also transmits intracellular signaling via cytoplasmic ITAMs after binding of the Fc receptor (CD16) to IgG-opsonized targets, thereby inducing antibody-dependent cellular cytotoxicity (ADCC)(Lanier et al., 1989). Interestingly, mutation of ITAMs in cytoplasmic CD3ζ domain of NKG2D-2B4ζ did not significantly weaken the cytotoxicity performing in CAR-expressing NK92 cell. Incorporation of additional co-stimulatory domains such as DAP10, DAP12, or CD137 in the NKG2D-2B4ζ CAR construct did not improve CAR-mediated killing in this system, at least as assessed by in vitro studies (Supplemental Figure S4). Together, these studies demonstrate the combination of NKG2D-2B4ζ in a CAR confers potent up-regulation in phosphorylated activation of PLC-γ, Syk-vav1-Erk pathway and also NF-κB pathway, that is sufficient to promote improved NK cell-mediated granulation, cytokine production, and cytotoxicity of antigen-expressing tumor cells. This activation also leads to increased expansion and survival of the NK-CAR-iPSC-NK cells enabling improved anti-tumor activity.
CAR-expressing NK cells are being developed for clinical trials, though most of these studies use CARs designed for T cells that are expressed in NK cells(Bollino and Webb, 2017; Hermanson and Kaufman, 2015). Indeed, clinical trials with haploidentical PB-NK cells that express an anti-CD19-41BBζ construct are underway for treatment of relapsed or refractory B-lineage acute lymphoblastic leukemia [ClinicalTrials.gov: NCT01974479 and NCT00995137]. Of course, these engineered PB-NK cell products must be manufactured on a donor/patient-specific basis. Umbilical cord blood-NK cells expressing anti-CD19 CARs are also being developed (Liu et al.). NK92 cells that express 41BB-CD28-ζ intracellular domains are also now in clinical trials for both hematologic malignancies and solid tumors [ClinicalTrials.gov: NCT02742727, NCT02944162, NCT02839954]. In contrast, iPSC-derived NK cells have normal karyotype and while these cells can persist in vivo post-infusion(Knorr et al., 2013a), our pre-clinical in vivo studies do not demonstrate evidence of malignant NK cell transformation when hESC or iPSC-derived NK cells are injected in immunodeficient mice.
Use of iPSCs (or hESCs) for NK cell production provides a more efficient means for gene modification compared to primary NK cells isolated from peripheral blood(Childs and Carlsten; Hermanson and Kaufman, 2015). In addition to CAR expression, other modifications such as deletion of inhibitory receptors or expression of cytokines can potentially be engineered into these cells to further enhance anti-tumor activity(Imai et al., 2005; Koneru et al., 2015; Long, 2008). Additionally, this can be done as a one-time genetic modification event, rather than requiring patient-specific gene modification, as done with current CAR-T cell studies. iPSC-derived NK cells can also be produced in large scale as a standardized cell product (Knorr et al., 2013b). This will allow for multiple doses to be administered, rather than the single cell dosing done with current CAR-T cell-based therapies. This repeat dosing, possibly combined with cycles of chemotherapy, may mediate more effective therapy against refractory solid tumors. Of course, CARs have been designed to target many tumor antigens with many ongoing clinical trials(Ahmed et al., 2015; Beatty et al., 2014; Bollino and Webb, 2017; Fesnak et al., 2016; Grupp et al., 2013; Qasim et al., 2017). Additionally, hESC and iPSC-derived NK cells can be engineered to express chimeric receptors to target chronic infectious diseases such as HIV(Ni et al., 2014; Ni et al., 2011). Therefore, NK cell-specific CAR constructs may also be valuable for treatment of chronic infectious diseases.
Together, these studies demonstrate the ability to use human pluripotent stem cells as a platform to create a homogeneous population of CAR-targeted NK cells to improve in vivo anti-tumor activity. Use of NK cell-specific signaling domains mediates antigen-specific activation of specific signaling pathways to improve function of these cells. This approach combined with clinical scale production of iPSC-derived NK cells now enables clinical trials of CAR-targeted NK cells for treatment of refractory malignancies.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC-CD34 | BD Biosciences | Cat#345804 RRID: AB_2686894 |
| PE-CD45 | BD Biosciences | Cat#555483 RRID: AB_395875 |
| PE-CD43 | BD Biosciences | Cat#560199 RRID:AB_1645655 |
| APC-CD56 | BD Biosciences | Cat#555518 RRID: AB_398601 |
| PE-CD117 | BD Biosciences | Cat#340529 RRID: AB_400044 |
| PE-CD3 | BD Biosciences | Cat#552127 RRID: AB_394342 |
| PE-CD94 | BD Biosciences | Cat#555889 RRID: AB_396201 |
| PE-NKG2D | BD Biosciences | Cat#561815 RRID: AB_10896282 |
| PE-NKp46 | BD Biosciences | Cat#557991 RRID: AB_396974 |
| PE-NKp44 | BD Biosciences | Cat#558563 RRID: AB_647239 |
| PE-TRAIL | BD Biosciences | Cat#565499 RRID: N/A |
| PE-FAS ligand | BD Biosciences | Cat#564261 RRID:N/A |
| PE-CD16 | BD Biosciences | Cat#555407 RRID: AB_395807 |
| APC-anti-human MSLN | R&D Systems | Cat#FAB32652A RRID: AB_2298058 |
| PE-NKG2A | R&D Systems | Cat#FAB1059P RRID: AB_2132978 |
| PE-goat-anti-mouse IgG F(ab)2- | Affymetrix eBioscience Inc. | Cat#15208759 RRID:N/A |
| APC-CD107a | Affymetrix eBioscience Inc. | Cat#14-1079-80 RRID: AB_467426 |
| PacBlue-IFN-γ | Biolegend | Cat#502522 RRID: AB_893525 |
| PacBlue-CD45 | Biolegend | Cat#103126 RRID: AB_493535 |
| Rabbit monoclonal anti-Fyn [EPR5500] | abcam | Cat#ab125016 RRID: AB_10972030 |
| Rabbit monoclonal anti-Fyn (phospho Y530) + Yes (phospho Y537) | abcam | Cat# b188319 RRID:N/A |
| Rabbit polyclonal anti-PLCγ2 | cell signaling technology | Cat#3872 RRID: AB_2299586 |
| Rabbit polyclonal anti-phospho-PLCγ2 (Tyr1217) | cell signaling technology | Cat#3871 RRID: AB_2299548 |
| Mouse monoclonal anti-PLCγ1 [M156] | abcam | Cat#ab41433 RRID: AB_777246 |
| Rabbit monoclonal anti-phospho-PLCγ1 (Ser1248) | cell signaling technology | Cat#8713 RRID: AB_10890863 |
| Rabbit monoclonal anti-Syk | cell signaling technology | Cat#2712 RRID: AB_10828107 |
| Rabbit monoclonal anti-phospho-Syk (Tyr525/526) | cell signaling technology | Cat#2710 RRID: AB_2197222 |
| Rabbit polyclonal anti- VAV1 | abcam | Cat#ab97574 RRID: AB_10680940 |
| Rabbit monoclonal anti-phospho-VAV1 (Y174) | abcam | Cat#ab76225 RRID: AB_1524546 |
| Mouse monoclonal anti-p44/42 MAPK (Erk1/2) (3A7) | cell signaling technology | Cat#9107 RRID: AB_10695739 |
| Rabbit polyclonal anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | cell signaling technology | Cat#9101 RRID: AB_331646 |
| Rabbit polyclonal-anti-IKKβ | cell signaling technology | Cat#2684 RRID: AB_10699025 |
| Rabbit monoclonal anti-phospho-IKKα/β (Ser176/180) | cell signaling technology | Cat#2697 RRID: AB_2079382 |
| Rabbit monoclonal anti-IκBα | cell signaling technology | Cat#9242 RRID: AB_823540 |
| Mouse monoclonal anti-phospho-IκBα (Ser32/36) (5A5) | cell signaling technology | Cat#9246 RRID: AB_2267145 |
| Goat polyclonal-anti-NKG2D, | abcam | Cat#ab36136 RRID: AB_776794 |
| Rabbit monoclonal anti-CD3zeta | abcam | Cat#ab40804 RRID: AB_726343 |
| Mouse monoclonal anti-DAP10 (H-3) | santa cruz biotechnology | Cat#sc-374196 RRID: AB_10987875 |
| Mouse monoclonal anti-GAPDH | abcam | Cat#ab8245 RRID: AB_2107448 |
| Rabbit monoclonal anti-NCAM | abcam | Cat#ab215981 RRID:N/A |
| Mouse monoclonal anti-pan Cytokeratin | abcam | Cat#ab6401 RRID: AB_305450 |
| Rabbit polyclonal anti-CD45 | abcam | Cat#ab10559 RRID: AB_442811 |
| Bacterial and Virus Strains Cat# | ||
| pKT2-mCAG-IRES-GFPZEO | Branden Moriarity lab | N/A |
| PiggyBac-mCAG-IRES-GFPZEO | Branden Moriarity lab | N/A |
| pCMV(CAT)T7-SB100 | Addgene | Cat#34879 |
| Super piggyBac Transposase expression vector | System Biosciences Inc. | Cat#PB210PA-1 |
| pCDH-CMV-MCS-EF1α-GreenPuro | System biosciences Inc. | Cat#CD513B-1 |
| Biological Samples | ||
| Human Serum AB | Sigma-Aldrich | Cat#H4522 |
| Peripheral blood buffy coat | San Diego Blood Bank (https://www.sandiegobloodbank.org/) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| α-MEM culture medium | Fisher Scientific | Cat#12634 |
| Horse serum | Fisher Scientific | Cat#16050130 |
| AIM V medium | Fisher Scientific | Cat# 31035025 |
| Fetal bovine serum | Fisher Scientific | Cat# 10437010 |
| TransDux MAX Lentivirus Transduction Enhancer | System Biosciences Inc. | Cat#LV860A-1 |
| Chromium-51 | PerkinElmer | Cat#NEZ030S001MC |
| Europium | PerkinElmer | Cat# AD0024 |
| CellEvent™ Caspase-3/7 Green Detection Reagent | Thermo fisher | Cat#C10423 |
| IncuCyte® Caspase-3/7 Green Apoptosis Assay Reagent | Essenbioscience | Cat#4440 |
| zeomycin | Thermo Fisher | Cat#r25001 |
| GolgiStop | BD Biosciences | Cat#554724 |
| GolgiPlug | BD Biosciences | Cat#555029 |
| NgoMIV | New England Biotechnology | Cat#R0564 |
| SbfI | New England Biotechnology | Cat#R0642 |
| Gateway LR Clonase II Enzyme mix | Thermo Fisher | Cat# 11791100 |
| Gateway BP Clonase II Enzyme mix | Thermo Fisher | Cat#11789020 |
| Recombinant human IL-2 | Proleukin | Cat#NDC-66483-116- 07 |
| Recombinant human IL-3 | Pepro Technology Inc. | Cat#200-03 |
| Recombinant human IL-7 | R&D Systems | Cat#207-IL |
| Recombinant human IL-15 | R&D Systems | Cat#247-IL |
| Recombinant human FLT-3 Ligand | Pepro Technology Inc. | Cat#300-19 |
| Recombinant human SCF | R&D Systems | Cat#255-SC/CF |
| Recombinant human VEGF | R&D Systems | Cat#293-VE |
| Recombinant human BMP-4 | R&D Systems | Cat#314-BP |
| Recombinant human bFGF basic | R&D Systems | Cat#4114-TC |
| Critical Commercial Assays | ||
| Pierce™ Classic Magnetic IP/Co-IP Kit | Thermo Fisher | Cat#88804 |
| Dynabeads Human T-Activator CD3/CD28 | Thermo Fisher | Cat#11132D |
| EasySep™ Human T Cell Isolation Kit | Stemcell technologies | Cat#17951 |
| Mem-PER™ Plus Membrane Protein Extraction Kit | Thermo Fisher | Cat#89842 |
| Human IFN-gamma Quantikine ELISA Kit | R&D Systems | Cat#SIF50 |
| Human TNF-alpha Quantikine ELISA Kit | R&D Systems | Cat#STA00C |
| IL-6 Human ELISA Kit | Thermo Fisher | Cat#EH2IL6 |
| Cell Line Optimization 4D-Nucleofector® X Kit | Lonza | Cat#V4XC-9064 |
| P3 Primary Cell 4D-Nucleofector® X Kit L | Lonza | Cat#V4XP-3012 |
| Experimental Models: Cell Lines | ||
| Human: iPS cells | Dan Kaufman lab | N/A |
| Human: NK 92 cells | ATCC | Cat#CRL-2407 |
| Human: K-562 cells | ATCC | Cat#CCL-243 |
| Human: MA148 cells | Sundaram Ramakrishnan lab | N/A |
| Human: A1847 cells | Reuben Harris lab | N/A |
| Human: artificial Antigen Presenting cells | Dean A. Lee lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson lab | Cat#005557 |
| Oligonucleotides | ||
| Primer for human RNase gene (Forward :5′-GCG GAG GGA AGC TCA TCA G-3′; reverse: 5′-CTG GCC CTA GTC TCA GAC CTT-3′) | This paper | N/A |
| Primer for GFP:zeo (Forward :5′-ATT CTG GTT GAG CTG GAT GG-3′; reverse: 5′-GTC AGG GTG GTC ACC AGA GT-3′: | This paper | N/A |
| Recombinant DNA | ||
| Sequence (g-block): anti-mesothelin scFv | Chowdhury et al., 1998 | N/A |
| gBlocks gene fragment: human CD8a extraocular domain | NCBI Reference Sequence: NM_001768 | N/A |
| gBlocks gene fragment: human CD28 transmembrane domain | NCBI Reference Sequence: NM_006139.3 | N/A |
| gBlocks gene fragment: human CD28 intraocular domain | NCBI Reference Sequence: NM_006139.3 | N/A |
| gBlocks gene fragment: human CD137 intraocular domain | NCBI Reference Sequence: NM_001561.5 | N/A |
| gBlocks gene fragment: human CD3zata intraocular domain | NCBI Reference Sequence: NM_198053.2 | N/A |
| gBlocks gene fragment: human CD16 transmembrane domain | NCBI Reference Sequence: NM_001127593.1 | N/A |
| gBlocks gene fragment: human NKp44 transmembrane domain | NCBI Reference Sequence: NM_001199509.1 | N/A |
| gBlocks gene fragment: human NKp46 transmembrane domain | NCBI Reference Sequence: NM_001145457.2 | N/A |
| gBlocks gene fragment: human NKG2D transmembrane domain | NCBI Reference Sequence: NM_007360.3 | N/A |
| gBlocks gene fragment: human 2B4 intraocular domain | NCBI Reference Sequence: NM_001166663.1 | N/A |
| gBlocks gene fragment: human DAP10 intraocular domain | NCBI Reference Sequence: NM_001007469.1 | N/A |
| gBlocks gene fragment: human DAP12 intraocular domain | NCBI Reference Sequence: NM_198125.2 | N/A |
| Software and Algorithms | ||
| Prism version7 | Prism Graphpad | N/A |
| FlowJo | https://www.flowjo.com/ | N/A |
| Odyssey imaging systems | IL-DOR | N/A |
| IncuCyte real-time image system | Essenbioscience | N/A |
| Xenogen IVIS imaging system | Caliper Life Science | N/A |
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Dan Kaufman (dskaufman@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
NOD/SCID/g (NSG) mice were purchased from Jackson Laboratories and used for ovarian cancer xenografts with NK cell treatment studies. Mice were housed in individually ventilated cages (IVC) under controlled climate and enrichment environmental conditions (bubble material) with unlimited access to sterile food and water. 5 female mouse (8–12 week-old) mice were used per group for ovarian cancer xenografts experiments. with immune cell treatment. Mice were randomized to each experimental group. Animal health was monitored weekly then more frequently as tumors progressed until being sacrificed before the disease was evident. All animal use protocols were pre-approved by the UCSD Institutional Animal Care and Use Committee
Cell Culture
Induced pluripotent stem cells (iPSCs, UCBiPS7), derived from umbilical cord blood CD34+ cells were maintained on mouse embryonic fibroblasts (MEF) as described previously(Ni et al., 2013). K562 cell line (ATCC, Manassas, VA), leukemia cell line established from a patient with chronic myelogenous leukemia. NK92 cell line, leukemia cell line established from a patient with non-Hodgkin’s lymphoma, was cultured in complete α-MEM culture medium containing 200u/ml hIL-2, 10% horse serum, 10% fetal bovine serum. CARs, cloned into the transposon plasmids, were co-nucleofected with transposase such as SB100X or super piggyback transposes at transposon vs. transposes ratio of 3:1 into iPSCs or NK92 cells by using a Lonza 4D-nucleofector device as previous described(Wilber et al., 2007). Primary T cell is isolated from PBMC purchased from San Diego Blood Bank by using EasySep™ Human T Cell Isolation Kit. Isolated T cell was culture in AIM V medium with 10% Human Serum AB. CARs were cloned into Lentivirus vector, then, virus packaging was performed in System Biosciences. T cell was transduced in ROI of 8 by using TransDux MAX ™. Transfected cells were under drug selection of 10ug/ml zeomycin and/or, GFP+ detection/sorting accessed by flow cytometry. Cells were incubated in a dark humidity 37°C incubator with 5% CO2.
METHOD DETAILS
NK cell derivation from CAR-expressing iPSCs
The derivation of NK cells from iPSCs and CAR transfected iPSCs have been previously described(Knorr et al., 2013b; Ng et al., 2008). Briefly, 3,000 TrypLE-adapted iPSCs were seeded in 96-well round-bottom plates with APEL culture(Ng et al., 2008) containing 40 ng/ml human Stem Cell Factor (SCF), 20 ng/ml human Vascular Endothelial Growth Factor (VEGF), and 20 ng/ml recombinant human Bone Morphogenetic Protein 4 (BMP-4). After day 11 of hematopoietic differentiation, spin embryoid bodies (EBs) were then directly transferred into each well of uncoated 24-well plates under a condition of NK cell culture. Cells were then further differentiated into NK cells as previously reported(Bachanova et al., 2014; Ni et al., 2013) using 5 ng/mL IL-3 (first week only), 10 ng/mL IL-15, 20 ng/mL IL-7, 20 ng/mL SCF, and 10 ng/mL flt3 ligand for 28–32 days. Half-media changes were performed weekly. NK cells were harvested for irradiated mbIL-21 expressing artificial antigen presenting cells (aAPCs) expansion(Denman et al., 2012) with 50 units/mL of hIL-2. aAPCs were kindly provided by Dr. Dean A. Lee(Denman et al., 2012; Knorr et al., 2013b) (Nationwide Children’s Hospital.)
Molecular Constructs
Transposon vectors of pKT2-mCAG-IRES-GFP:zeo and PiggyBac mCAG-IRES-GFP:zeo were designed and reconstructed as previously describe(Moriarity et al., 2013). In which, transgene expression is driven by the mCAG promoter. Anti-mesothelin antibody scFv (SS1)-PE38(Chowdhury et al., 1998), extracellular domain of CD8a extracellular domain, transmembrane domain of CD28, CD16, NKp44, NKp46, NKG2D; cytoplasmic signaling domain of CD28, CD137, 2B4, DAP10, DAP12, and CD3ζ were used to construct NK-specific CARs. CARs containing/or not anti-mesothelin scFv, and various combinations of wild type and mutant domains of CD8 hinge-TM-CDs-SD (CD3ζ) were synthesized as gBlocks gene fragment by Integrated DNA Technologies, Inc. and cloned into the NgoMIV and SbfI site of pKT2/mCAG-antiMESOCAR-IRES-GFP:Zeo using restriction enzyme cloning and ligation. Correct CARs’ sequences were confirmed by restriction enzyme digest and sequencing analyses. Insulated PiggyBac vectors were generated by PCR of CAR expression cassettes from SB transposon vectors (i.e. mCAG-antiMESOCAR-IRES-GFP:Zeo) and subsequent BP Clonase reaction into pDONR221 to generate pENTR221-CAR cassette plasmids. pENTR221-CAR cassette plasmids were subsequently used for LR Clonase reaction into PB-I-DEST-I to generate final PiggyBac expression vectors. The PB-I-DEST-I vector contains a 2.4kb cHS4 insulator (I) flanking the Gateway destination cassette (DEST) used for LR clonase cloning. Generation of stable clone of CAR transfected NK cells was performed as described above. To determine the copy numbers of integrated vector, we isolated genomic DNA from the iPSCs and NK92 cells and performed quantitative PCR using sets of primers specific for GFP:zeo region of vector and for the human RNase gene. To determine absolute value, we generated a standard curve using serial dilutions of a plasmid containing GFP:zeo region. Reactions were carried out in triplicate in CFX384 Touch™ Real-Time PCR Detection System.
Cell lysis assay
Human erythroleukemia targets (K562, K562meso) and human ovarian cancer targets (MA148 cells or A1847 cells) were incubated with 51chromium (51Cr) or europium for 1 hour at 37°C, washed three times, and cocultured with NK cells at the indicated effector to target (E:T) ratios. Total lysis (test release) was achieved with the use of 5% Triton-X 100. After a period of incubation, cells were harvested and analyzed. Specific 51Cr or europium/ lysis was determined following the equation: Percentage of specific lysis = 100 × (Test release − Spontaneous release)/(Maximal release − Spontaneous release).
CD107a expression and IFN-γ staining
NK cells were incubated with or without cancer targets (K562 cells, K562meso cells, MA148, cells, or A1847 cells) at 1:2 effector to target ratios. CD107a-APC antibody was added to each well and allowed to incubate for 1 hour, following by adding GolgiStop and GolgiPlug for additional 2 hours incubation. At the completion of incubation, cells were washed with FACS buffer, were stained with CD56-PE and live/dead Aqua staining. Cells were then fixed with fixation buffer for 10 minutes on ice, following by permeabilization with perm/wash buffer for 10 minutes at 4°C. Cells were washed and stained with interferon-γ (IFN-γ)-Pacific Blue for 30min at 4°C, then final washed for analysis. CD107a expression and intracellular IFN-γ production were evaluated by normalization data of NK cell without target cell co-culture.
Flow Cytometry
The antibodies were listed in KEY RESOURCES TABLE. Flow cytometry was done on a BD FACSCalibur or LSRII, and data were analyzed using FlowJo.
Immunoblot
Suspension cells were lysed in RIPA lysis buffer with fresh protease inhibitor cocktail (Roche) on ice for 20 min and sonicated for 2 seconds on ice. Membrane protein was extracted using Mem-PER™ Plus Membrane Protein Extraction Kit. Sample protein was measured by a standard bicinchoninic acid assay, size fractioned by polyacrylamide gel electrophoresis (PAGE), and transferred to nitrocellulose membrane. Nonspecific binding was blocked by incubating in TBST 5% BSA plus 1% Triton X-100 solution for 1 hours, followed by incubation with primary antibodies listed in key resource table, overnight at 4°C. Species specific IRDye–conjugated secondary antibodies (1:10, 000,) were applied to membranes for 1 hour at room temperature. Immunoreactive products were visualized in Odyssey Imaging Systems. All loading samples were normalized by staining of GAPDH.
Ovarian Cancer Xenografts with NK cell treatment
Mesohigh A1847 ovarian cancer cells were incorporated into a previously described NK cell xenogeneic mouse model system(Hermanson et al., 2016). NOD-scid IL2rγ null (NSG, n=5/group) mice were obtained from Jackson Laboratories for all in vivo experiments. Mice were given 2 × 105 of luciferase expressing ovarian cancer cell in the route of I.P. 4 days prior to NK cell infusion (Day −4). On Day −1 mice were conditioned with 225 cGy, and bioluminescent imaging (BLI) was used to normalize tumor engraftment burden in each group. 1.5 × 107 or 1.0 × 107 cells per mouse NK cells or T cells were then given intraperitoneally on Day 0. Cytokine administration of hIL-2 (10,000 unit/mouse, every 2–3 day for 21 days) and hIL-15 (10ng/mouse for 7 days) was initiated on mice under NK cell therapy after day 1. Tumor aggressiveness was determined by BLI weekly using the Xenogen IVIS Imaging system.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantifications and statistics
The student T test was used to test for the significance. Mean values ± S.D. are shown. Differences between more than 2 groups were tested by one-way ANOVA analysis. P-value for pairwise comparisons is conservatively adjusted for multiple comparisons using a Bonferroni correction. All data was presented of 3 independent experiments. Overall survival (OS) analysis was calculated using Kaplan-Meier methods and compared to treatment group using log rank tests; median survival and 95% confidence intervals (CI) are presented. P-values <0.05, two-side, were considered statistically significant. Statistical analysis is performed in the environment of GraphPad Prism Statistical.
Supplementary Material
Highlights.
Human iPSC-derived NK cells provide an “off the shelf” resource for cancer therapy
New CAR constructs optimize targeted anti-tumor activity of iPSC-derived NK cells
Optimized NK-CAR expressing iPSC- NK cells activate specific intracellular signals
NK-CAR iPSC-NK cells demonstrate increased in vivo expansion and improved activity
Acknowledgments
We thank Jacqueline K Le and Natalie K. Wolf for technical assistance. The serous epithelial ovarian tumor cell lines MA-148 and A1847 were kindly provided by Sundaram Ramakrishnan (University of Minnesota) and Reuben Harris (University of Minnesota), respectively. We appreciate helpful discussions with Dr. Bob Valamehr and colleagues at Fate Therapeutics. We gratefully acknowledge support and assistance from the Sanford Consortium for Regenerative Medicine Flow Cytometry, Histology cores and pathologist Dr. Kent Osborn. These studies were supported by funding from the NIH (R01CA203348 and U01 CA217885), CIRM (DISC2-09615), and Fate Therapeutics to DSK.
Footnotes
Author Contributions: Conceptualization: YL, DLH, and DSK. Methodology: YL, DLH, BSM, and DSK. Investigation: YL and DLH. Resources: BSM and DSK. Writing-original draft: YL. Writing- review and editing: DSK. Supervision and funding acquisition: DSK.
Competing Financial Interests
DSK has research funding and serves as a consultant for Fate Therapeutics. DSK, DLH, and BSM have filed a patent related to these studies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, Campana D, Juergens H, Pule M, Rossig C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. 2009;15:4857–4866. doi: 10.1158/1078-0432.CCR-08-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lewis D, Hippen K, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123:3855–3863. doi: 10.1182/blood-2013-10-532531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachanova V, Miller JS. NK cells in therapy of cancer. Crit Rev Oncog. 2014;19:133–141. doi: 10.1615/critrevoncog.2014011091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–347. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollino D, Webb TJ. Chimeric antigen receptor-engineered natural killer and natural killer T cells for cancer immunotherapy. Transl Res. 2017;187:32–43. doi: 10.1016/j.trsl.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214:73–91. doi: 10.1111/j.1600-065X.2006.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess-Beusse B, Farrell C, Gaszner M, Litt M, Mutskov V, Recillas-Targa F, Simpson M, West A, Felsenfeld G. The insulation of genes from external enhancers and silencing chromatin. Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16433–16437. doi: 10.1073/pnas.162342499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–469. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsten M, Childs RW. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front Immunol. 2015;6:266. doi: 10.3389/fimmu.2015.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777–1786. doi: 10.1158/0008-5472.CAN-12-3558. [DOI] [PubMed] [Google Scholar]
- Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10:230–252. doi: 10.1038/cmi.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs RW, Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov. 2015;14:487–498. doi: 10.1038/nrd4506. [DOI] [PubMed] [Google Scholar]
- Chowdhury PS, Viner JL, Beers R, Pastan I. Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc Natl Acad Sci U S A. 1998;95:669–674. doi: 10.1073/pnas.95.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, Singh H, Hurton L, Maiti SN, Huls MH, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One. 2012;7:e30264. doi: 10.1371/journal.pone.0030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. 2018;51:55–61. doi: 10.1016/j.coi.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, Leenders M, Bohme F, Kok N, Trilsbeek C, et al. Successful Transfer of Umbilical Cord Blood CD34+ Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin Cancer Res. 2017;23:4107–4118. doi: 10.1158/1078-0432.CCR-16-2981. [DOI] [PubMed] [Google Scholar]
- Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nat Rev Immunol. 2007;7:737–744. doi: 10.1038/nri2144. [DOI] [PubMed] [Google Scholar]
- Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa JA, Reidy A, Mirandola L, Trotter K, Suvorava N, Figueroa A, Konala V, Aulakh A, Littlefield L, Grizzi F, et al. Chimeric antigen receptor engineering: a right step in the evolution of adoptive cellular immunotherapy. Int Rev Immunol. 2015;34:154–187. doi: 10.3109/08830185.2015.1018419. [DOI] [PubMed] [Google Scholar]
- Garrity D, Call ME, Feng J, Wucherpfennig KW. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci U S A. 2005;102:7641–7646. doi: 10.1073/pnas.0502439102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, Jonson AL, Panoskaltsis-Mortari A, Curtsinger J, McKenna D, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. 2011;13:98–107. doi: 10.3109/14653249.2010.515582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller MA, Miller JS. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy. 2011;3:1445–1459. doi: 10.2217/imt.11.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice A, Trounson A. Genetic modification of human embryonic stem cells for derivation of target cells. Cell Stem Cell. 2008;2:422–433. doi: 10.1016/j.stem.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025–1036. doi: 10.1038/ni.3518. [DOI] [PubMed] [Google Scholar]
- Handgretinger R, Lang P, Andre MC. Exploitation of natural killer cells for the treatment of acute leukemia. Blood. 2016;127:3341–3349. doi: 10.1182/blood-2015-12-629055. [DOI] [PubMed] [Google Scholar]
- Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008;44:46–53. doi: 10.1016/j.ejca.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson DL, Bendzick L, Pribyl L, McCullar V, Vogel RI, Miller JS, Geller MA, Kaufman DS. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells. 2016;34:93–101. doi: 10.1002/stem.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol. 2015;6:195. doi: 10.3389/fimmu.2015.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho EL, Carayannopoulos LN, Poursine-Laurent J, Kinder J, Plougastel B, Smith HR, Yokoyama WM. Costimulation of multiple NK cell activation receptors by NKG2D. J Immunol. 2002;169:3667–3675. doi: 10.4049/jimmunol.169.7.3667. [DOI] [PubMed] [Google Scholar]
- Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–383. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingemann H, Boissel L, Toneguzzo F. Natural Killer Cells for Immunotherapy - Advantages of the NK-92 Cell Line over Blood NK Cells. Front Immunol. 2016;7:91. doi: 10.3389/fimmu.2016.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr DA, Bock A, Brentjens RJ, Kaufman DS. Engineered human embryonic stem cell-derived lymphocytes to study in vivo trafficking and immunotherapy. Stem Cells Dev. 2013a;22:1861–1869. doi: 10.1089/scd.2012.0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, Lee DA, Kaufman DS. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013b;2:274–283. doi: 10.5966/sctm.2012-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl U, Kalberer C, Spanholtz J, Lee DA, Miller JS, Cooley S, Lowdell M, Uharek L, Klingemann H, Curti A, et al. Advances in clinical NK cell studies: Donor selection, manufacturing and quality control. Oncoimmunology. 2016;5:e1115178. doi: 10.1080/2162402X.2015.1115178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4:e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HJ, Choi GE, Ryu S, Kwon SJ, Kim SC, Booth C, Nichols KE, Kim HS. Stepwise phosphorylation of p65 promotes NF-kappaB activation and NK cell responses during target cell recognition. Nat Commun. 2016;7:11686. doi: 10.1038/ncomms11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL, Yu G, Phillips JH. Co-association of CD3 zeta with a receptor (CD16) for IgG Fc on human natural killer cells. Nature. 1989;342:803–805. doi: 10.1038/342803a0. [DOI] [PubMed] [Google Scholar]
- Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2017 doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. 2008;224:70–84. doi: 10.1111/j.1600-065X.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love PE, Hayes SM. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect Biol. 2010;2:a002485. doi: 10.1101/cshperspect.a002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017–4023. doi: 10.1182/blood-2014-12-580068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- Moriarity BS, Rahrmann EP, Keng VW, Manlove LS, Beckmann DA, Wolf NK, Khurshid T, Bell JB, Largaespada DA. Modular assembly of transposon integratable multigene vectors using RecWay assembly. Nucleic Acids Res. 2013;41:e92. doi: 10.1093/nar/gkt115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. doi: 10.1038/nrc.2015.5. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Cella M, Langen H, Friedlein A, Colonna M. Activating interactions in human NK cell recognition: the role of 2B4-CD48. Eur J Immunol. 1999;29:1676–1683. doi: 10.1002/(SICI)1521-4141(199905)29:05<1676::AID-IMMU1676>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Ng ES, Davis R, Stanley EG, Elefanty AG. A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nat Protoc. 2008;3:768–776. doi: 10.1038/nprot.2008.42. [DOI] [PubMed] [Google Scholar]
- Ni Z, Knorr DA, Bendzick L, Allred J, Kaufman DS. Expression of chimeric receptor CD4zeta by natural killer cells derived from human pluripotent stem cells improves in vitro activity but does not enhance suppression of HIV infection in vivo. Stem Cells. 2014;32:1021–1031. doi: 10.1002/stem.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z, Knorr DA, Clouser CL, Hexum MK, Southern P, Mansky LM, Park IH, Kaufman DS. Human pluripotent stem cells produce natural killer cells that mediate anti-HIV-1 activity by utilizing diverse cellular mechanisms. J Virol. 2011;85:43–50. doi: 10.1128/JVI.01774-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z, Knorr DA, Kaufman DS. Hematopoietic and nature killer cell development from human pluripotent stem cells. Methods Mol Biol. 2013;1029:33–41. doi: 10.1007/978-1-62703-478-4_3. [DOI] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- Ramos CA, Heslop HE, Brenner MK. CAR-T Cell Therapy for Lymphoma. Annu Rev Med. 2016;67:165–183. doi: 10.1146/annurev-med-051914-021702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8:357ra123. doi: 10.1126/scitranslmed.aaf2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DB, Araki M, Hamerman JA, Chen T, Yamamura T, Lanier LL. A Structural basis for the association of DAP12 with mouse, but not human, NKG2D. J Immunol. 2004;173:2470–2478. doi: 10.4049/jimmunol.173.4.2470. [DOI] [PubMed] [Google Scholar]
- Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, Sakai F, Kato A, Tanabe M, Enoki T, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med. 2015;13:277. doi: 10.1186/s12967-015-0632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivori S, Parolini S, Falco M, Marcenaro E, Biassoni R, Bottino C, Moretta L, Moretta A. 2B4 functions as a co-receptor in human NK cell activation. Eur J Immunol. 2000;30:787–793. doi: 10.1002/1521-4141(200003)30:3<787::AID-IMMU787>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H, Takeda K, van Dommelen SL, Degli-Esposti MA, Hayakawa Y. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42:501–510. doi: 10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, Figini M, June CH, Coukos G, Powell DJ., Jr In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB) Cancer Res. 2011;71:4617–4627. doi: 10.1158/0008-5472.CAN-11-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song DG, Ye Q, Santoro S, Fang C, Best A, Powell DJ., Jr Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum Gene Ther. 2013;24:295–305. doi: 10.1089/hum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themeli M, Riviere I, Sadelain M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 2015;16:357–366. doi: 10.1016/j.stem.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, Bachmann M, Fussel M, Schackert G, Temme A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194:3201–3212. doi: 10.4049/jimmunol.1400330. [DOI] [PubMed] [Google Scholar]
- Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science. 2004;306:1517–1519. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- Wilber A, Linehan JL, Tian X, Woll PS, Morris JK, Belur LR, McIvor RS, Kaufman DS. Efficient and stable transgene expression in human embryonic stem cells using transposon-mediated gene transfer. Stem Cells. 2007;25:2919–2927. doi: 10.1634/stemcells.2007-0026. [DOI] [PubMed] [Google Scholar]
- Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, Kaufman DS. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009;113:6094–6101. doi: 10.1182/blood-2008-06-165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Ye L, Chang JC, Beyer AI, Wang J, Muench MO, Kan YW. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24:1526–1533. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahata K, Maeshima K, Sone T, Ando T, Okabe M, Imamoto N, Imamoto F. cHS4 insulator-mediated alleviation of promoter interference during cell-based expression of tandemly associated transgenes. J Mol Biol. 2007;374:580–590. doi: 10.1016/j.jmb.2007.09.054. [DOI] [PubMed] [Google Scholar]
- Yang Y, Lim O, Kim TM, Ahn YO, Choi H, Chung H, Min B, Her JH, Cho SY, Keam B, et al. Phase I Study of Random Healthy Donor-Derived Allogeneic Natural Killer Cell Therapy in Patients with Malignant Lymphoma or Advanced Solid Tumors. Cancer Immunol Res. 2016;4:215–224. doi: 10.1158/2326-6066.CIR-15-0118. [DOI] [PubMed] [Google Scholar]
- Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, Wels WS. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front Immunol. 2017;8:533. doi: 10.3389/fimmu.2017.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Sentman CL. Cancer immunotherapy using a bispecific NK receptor fusion protein that engages both T cells and tumor cells. Cancer Res. 2011;71:2066–2076. doi: 10.1158/0008-5472.CAN-10-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.