

Abstract
Quinoline compounds have been extensively explored as anti-malaria and anti-cancer agents for decades and show profound functional bioactivities, however, the studies of these compounds in other medicinal fields have lagged dramatically. In this study, we report the development of a series of facilely accessible quinoline derivatives that display potent antibacterial activity against a panel of multidrug-resistant Gram-positive bacterial strains, especially C. difficile. We also demonstrated that these molecules are effective in vivo against C. difficile. These results revealed that these types of quinoline compounds could serve as prototypes for the development of an appealing class of antibiotic agents used to combat Gram-positive drug-resistant bacterial strains, including C. difficile.
Keywords: quinoline, antibacterial, Gram-positive, Clostridium difficile, in vivo
TOC image
1. Introductions
Methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant Staphylococcus epidermidis (MRSE), Vancomycin-Resistant Enterococci faecalis (VRE), and Clostridium difficile (C. difficile) are Gram-positive bacteria that cause severe concerns to public health. The superbug MRSA is among the most common of several difficult-to-treat infections,1 leading to high morbidity and mortality in the United States.2, 3 MRSE is also known as an opportunistic pathogen of humans and causes various diseases that could also be life-threatening.4 As one of the predominant enterococcal species, VRE has been recognized as a common cause of endocarditis as well as the second most common cause of wound and nosocomial urinary tract infections in the United States, mainly due to the ability to acquire resistance to the majority of currently available antibiotics.5 More recently, studies showed that C. difficile is the most common cause of hospital-associated diarrhea and could induce other related complications,6–8 which lead to 29,000 deaths in the United States in 2011.7 C. difficile has been implicated as the leading cause of gastroenteritis-associated death and is emerging as a major enteric pathogen worldwide.9–11 These Gram-positive bacterial infections have led to incredible expenditures and mortality in the United States and other countries.12 To tackle infections from these antibiotic-resistant Gram-positive pathogens, persistent efforts have been made among the scientific community,13–18 however, novel antibiotics that inhibit highly lethal Gram-positive bacterial infections are still urgently needed.
The quinolones, (Figure 1a) such as ciprofloxacin, levofloxacin, lomefloxacin etc. have been one of the largest families of synthetic antibiotic drugs clinically,19, 20 however, extensive use has resulted in the development of resistance to these antibiotics.21 With similar core structures, quinazolines (Figure 1b) have also shown potential as an antibacterial with anti-MRSA activity, as demonstrated by several research groups.22–24 In contrast, the quinolines, with slight difference in the core scaffold compared with quinolones and quinazolines, have been mainly reported to have antimalarial activity (e.g., chloroquine, mefloquine, primaquine, amodiaquine, etc.), as well as analgesic, anti-inflammatory, and antineoplastic activities.25 The antibacterial study of quinoline compounds have significantly lagged behind quinolones and quinazolines in terms of antibacterial studies. There is limited literature that documents quinoline activity against Mycobacterium tuberculosis,26–29 whose cell wall has characteristics of both Gram-positive and Gram-negative bacteria.30 These compounds potentially target the proton pump of adenosine triphosphate (ATP) synthase.26 To the best of our knowledge, quinoline derivatives have been rarely reported to be active against Gram-positive MRSA,31 MRSE, VRE, and C. difficile. We envisioned that quinoline compounds might be active against other bacterial strains, although the exact antibacterial target remains unclear. Herein, we synthesized a focused library of quinoline compounds, and explored their activity against a panel of Gram-positive bacteria.
Figure 1.
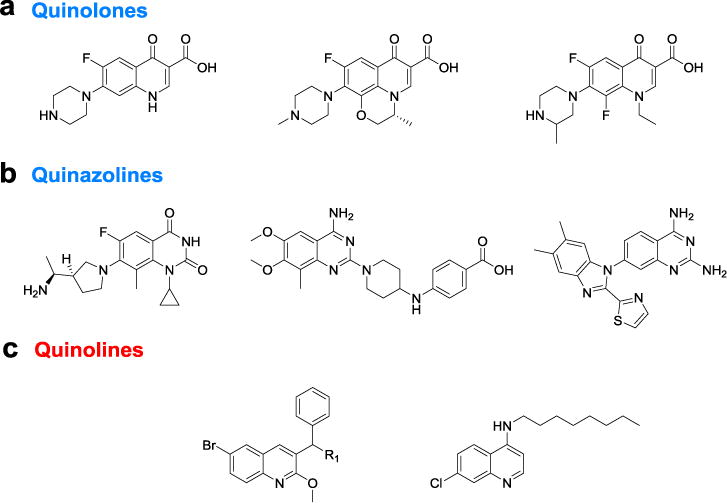
Examples of quinolones, quinazolines, and quinolines with reported antibacterial activity.
2. Results and discussion
2.1. Synthesis of quinoline compounds
The quinoline library was synthesized through a three-step reaction by using commercially available starting materials. As briefly shown in Scheme 1, commercially available 4-chloro 6,7-dimethoxyquinoline (A) was treated with methanesulfonic acid in the presence of L-methionine to selectively remove 6-methoxy group.32 The afforded mono-hydroxy intermediate B was reacted with benzyl bromide to give the ether C, which could be used without any further purification. Amine substitution occurred at position-4 in the presence of catalytic concentrated HCl to give the 4-amino-6-ether-substitued quinoline. Recrystallization from ethyl acetate/dichloromethane gave the final compounds without further column purification. Throughout this synthetic sequence, only B needed to be purified by column chromatography, hence large scale of molecules could be easily obtained for further investigations in the future.
Scheme 1. Synthesis of quinoline derivatives a.
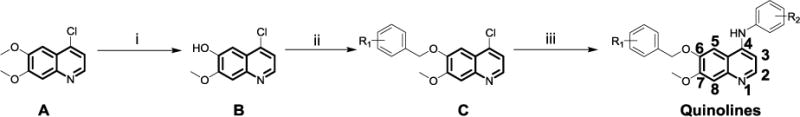
aReagents and conditions: (i) L-methionine, methanesulfonic acid, 120 °C, yield 57.4%; (ii) benzyl bromide, DMF, K2CO3, 60 °C, yield 84.1‒91.6%; (iii) substituted aniline, HCl (cat.), n-butanol, 120 °C, yield 71.6‒93.2%.
2.2. Potency of compounds against MRSA, MRSE, and VRE
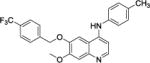
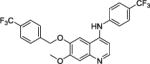
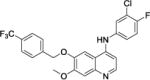
Two areas of substitution were assessed, including N4-substitution and O6-substitution (O7-substitution was not preferred here because the intermediate B is much easier accessed). Eight compounds were synthesized and tested against three clinically relevant, multidrug resistant Gram-positive bacterial pathogens, namely MRSA, MRSE, and VRE. The last-resort antibiotic, daptomycin,33 was employed as a control. As shown in Table 1, firstly, when R1 and R2 were both kept as para-methyl group, only weak activities were detected for compound 1, with minimum inhibitory concentration (MIC) of 12 μg/mL against MRSA. Replacement of R1 by a trifluoromethyl group afforded compound 2 with improved activity, exhibiting a MIC of 3.0 μg/mL for all three bacterial strains. We then kept R1 as CF3 group and modified R2 as m-trifluoromethyl, p-trifluoromethyl, and 3-chloro-4-fluoro to furnish compounds 3, 4, and 5, respectively. Compared with 2, the activity of 3 against MRSA and VRE did not change, whereas that against MRSE decreased one-fold. Compound 4 had enhanced activity, with MICs of 0.75 μg/mL for both MRSA and VRE, showing that p-CF3 derivation leads to a more active compound than p-CH3 derivation. The substitution of 3-chloro-4-fluoro group on compound 5 also had better activity against MRSA and VRE, compared to compound 2.
Table 1.
The structure of quinoline compounds 1–5 and their antibacterial activity against MRSA, MRSE, and VRE.
| Cpd | Structure | MIC (μg/mL) | VRE | |
|---|---|---|---|---|
| MRSA | MRSE | |||
| 1 |
|
12 | NDa | ND |
| 2 |
|
3.0 | 3.0 | 3.0 |
| 3 |

|
3.0 | 6.0 | 3.0 |
| 4 |
|
0.75 | 3.0 | 0.75 |
| 5 |
|
1.5 | 3.0 | 1.5 |
| Daptomycin | 0.5 | 0.5 | 1.0 | |
ND, not determined because compounds are not active.
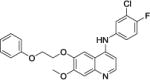
Following this data, three compounds with R2 modified as 3-chloro-4-fluoro group but with various of R1 groups, were evaluated. As shown in Table 2, compound 6, with a p-isopropyl phenyl ring substituted quinoline, also demonstrated potent activity against MRSA with MIC of 1.5 μg/mL, although the potency against MRSE and VRE moderately dropped at 6.0 and 3.0 μg/mL, respectively. In the case of compound 7, we chose to substitute the phenyl ring in ortho position. 7 also displayed potent activity against the three bacteria, with MICs of 1.5, 3.0, and 1.5 μg/mL, respectively. The introduction of an ethoxy spacer between the benzene ring and oxygen at 4-position resulted in compound 8, which did not show compromised activity, and also resulted in potent antibacterial activity of 3.0 μg/mL. Based on the preliminary data, it is still early to draw conclusion on the relationship of chemical substitutions and the antibacterial activity, more systematic works need to be done to clearly discuss the structure-activity relationship in the future.
Table 2.
The structure of quinoline compounds 6–8 and their antibacterial activity against MRSA, MRSE, and VRE.
| Cpd | Structure | MIC (μg/mL) | VRE | |
|---|---|---|---|---|
| MRSA | MRSE | |||
| 6 |

|
1.5 | 6.0 | 3.0 |
| 7 |

|
1.5 | 3.0 | 1.5 |
| 8 |
|
3.0 | 3.0 | 3.0 |
| Daptomycin | 0.5 | 0.5 | 1.0 | |
2.3. Potency of compounds against C. difficile in vitro and in vivo
C. difficile infection (CDI) has emerged as the most common care-associated infection and the most frequent hospital-acquired intestinal infection worldwide,34 leading to heavy economic burden to the society.35 Currently, standard therapy depends on treatment with vancomycin, metronidazole or fidaxomicin. None of these is fully effective.36, 37 Moreover, an estimated 15‒35% of those infected with C. difficile relapse following treatment.38, 39 New, effective antibiotics against C. difficile are urgently needed.
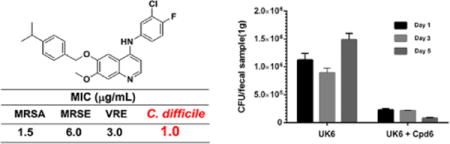
We therefore evaluated the antibacterial potency of our quinoline compounds on the hypervirulent C. difficile UK6. As shown in Table 3, the majority of these quinoline compounds displayed potent in vitro activity, with MICs ≤ 4.0 μg/mL, except for compound 1 which did not show any activity under the assay conditions (MIC > 16 μg/mL). Compound 7 could eradicate bacteria at the minimum concentration of 8.0 μg/mL. More intriguingly, compound 6 had the best bactericidal activity against C. difficile, with MIC as low as 1.0 μg/mL, which is comparable to that of the positive control, Vancomycin, which has been routinely used in the treatment of CDI patients.
Table 3.
The antibacterial activity of compounds 1–8 against C. difficile.
| C. difficile |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Vancomycin | |
| MIC (μg/mL) | >16 | 4.0 | 4.0 | 2.0 | 4.0 | 1.0 | 8.0 | 4.0 | 0.5 |
To determine the cytotoxicity of frontrunner compounds 4 and 6, the human liver cancer cell HepG2 and the human embryonic kidney cell HEK-293 were employed, using the MTT method. As shown in Figure 2, neither of the compounds showed cytotoxicity at the concentration of 20 μg/mL. Compound 4 had 15-fold selectivity over the MIC of C. difficile, with half maximal inhibitory concentration (IC50) values of 34.8 and 28.6 μg/mL (70.7 and 58.1 μM) against HepG2 and HEK-293 cells, respectively. More encouragingly, compound 6 was much less cytotoxic, with IC50 of 66.7 and 65.5 μg/mL (148.2 and 145.5 μM) against HepG2 and HEK-293 cells, respectively, which indicated more than 60-fold selectivity over the MIC value against C. difficile.
Figure 2.
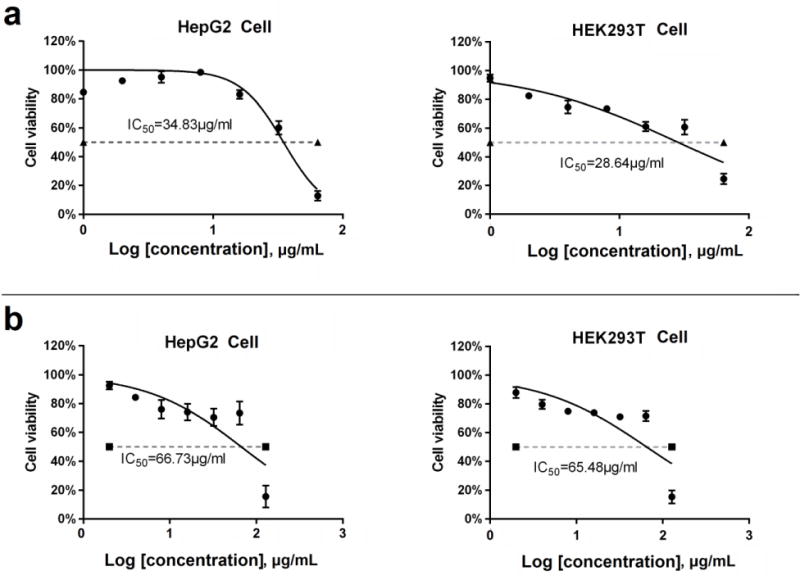
The cytotoxicity of compounds 4 (a) and 6 (b) on human liver cancer cell HepG2 and human embryonic kidney cell HEK-293. The cell viability was determined by MTT assay (three replicates), and recorded as a percentage of compound-treated cells compared to untreated cells (DMSO). Error bars are shown as ± SEM.
Given the good selectivity index of compound 6, we conducted an in vivo assay to evaluate the treatment efficacy of compound 6 in a mouse model of CDI. To this end, twenty-five mice were divided into three groups. Group UK6 (n=10) were challenged with spores of C. difficile UK6. Group UK6+Cpd6 (n=10) were challenged with spores of C. difficile UK6, and treated with compound 6. Group Cpd6 (n=5) were only treated with compound 6 without C. difficile infection. At 4 hours post challenge, the mice in UK6+Cpd6 were given one dose of compound 6 (50 mg/kg) via gavage. From the first day post challenge (day 1), mice in group UK6+Cpd6 received one dose of compound 6 twice a day (50 mg/kg/day) for five days. Meanwhile, the mice in group Cpd6 were also given compound 6 at the same time with the same dose to determine the toxicity of the compound to the mice. After C. difficile challenge /or compound treatment, mice were monitored daily for five days after challenge with C. difficile spores for weight changes, survival, diarrhea, and other symptoms of the disease. As shown in Figure 3a, 20% increase of survival was observed with the administration of compound 6 (group UK6+Cpd6) from day 3 to day 5, compared with control (C. difficile UK6 only) group. Over the entire experimental period (five days), a significant decrease of diarrhea was observed in the mouse treated with compound 6 (group UK6+Cpd6) (Figure 3b). After 2 days, the administration of 6 lead to 32% decrease of diarrhea compared to the control (group UK6). The data indicated that compound 6 could improve the diarrhea and survival of mice after CDI. No mice developed diarrhea and CDI symptoms in the Cpd6-only group without C. difficile challenge, however one mouse among 5 died, indicating potential toxicity of Cpd6, but we did not look into the cause of death.
Figure 3.

In vivo efficacy of the compound 6 (Cpd 6) in the mouse model of C. difficile-infection. Treatment and infection of mice were described in detail in Supporting Information. Mice were divided into three groups. Group 1 (UK6, n = 10) and group 2 (UK6 + Cpd 6, n = 10) were challenged with C. difficile spores; Group 3 (Cpd 6, n = 5) was treated with Cpd 6 only, without C. difficile challenge. Survival rate (a) and percent of diarrhea (b) were shown. Results were analyzed by the two-way ANOVA and Pearson Chi-Square.
The amount of C. difficile in fecal samples after treatment were also determined. As shown in Figure 4, the C. difficile in feces from mice treated with compound 6 (group UK6+Cpd6) showed more than 80% decrease compared with that of the UK6 UK6-only group. After 5 days, the amount of C. difficile in feces of the control group (group UK6) continued to increase, while mice treated with compound 6 (group UK6+Cpd6) were observed with a 14-fold decrease of C. difficile compared with control (group UK6). This result demonstrated that compound 6 was effective on inhibition of C. difficile in mice.
Figure 4.
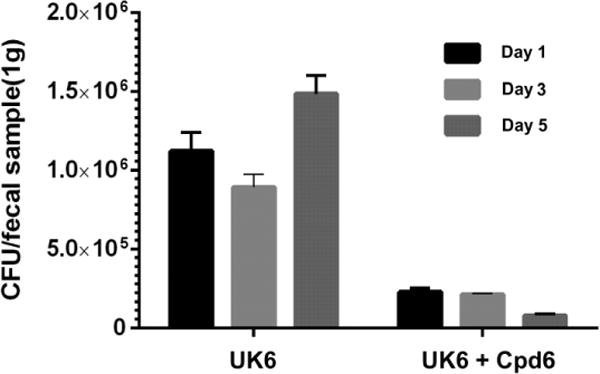
C. difficile accounts in fecal samples from treated and control mice challenged with 106 colony-forming units (CFU) of C. difficile UK6 spores. C. difficile accounts were reported as CFU per gram of fecal samples. Feces were collected on days 1, 3, and 5 post-challenge from every mouse in each cohort containing 10 mice. C. difficile accounts in feces from mice treated with compound 6 showed significantly decrease in FCU compared with that of the UK6 challenged group (P<0.001). Error bars are shown as ± SEM.
3. Conclusions
To summarize, we have reported a series of quinoline compounds which can be accessed in a straightforward manner, and display good potency against multidrug-resistant Gram-positive bacteria including MRSA, MRSE, and VRE. Furthermore, the majority of the quinolines displayed very potent activity against C. difficile, an emerging hypervirulent bacterium. One of the quinoline compounds showed MIC of 1.0 μg/mL against C. difficile, very close to that of Vancomycin (MIC = 0.5 μg/mL). Moreover, the quinoline also revealed good efficacy in an in vivo mouse model of CDI. Our results provide an alternative way to combat gram-positive resistant bacterial pathogens, especially C. difficile, and shed light on the development of more potent compounds. Compared to vancomycin and fidaxomicin, the facile synthesis and high cost-effectiveness of these compounds make them an appealing class of antibiotic agents combating drug-resistant bacterial strains. Further studies on the optimization of activity and selectivity, as well as the mechanism of actions are underway in our lab.
4. Experimental section
4.1. General Information
The starting material 4-chloro-6,7-dimethoxyquinoline (A) was purchased from Matrix Scientific. Solvents and other reagents were purchased from either Sigma-Aldrich or Fisher Scientific and were used without further purification. The purity of the compounds was determined to be >95% by analytical HPLC (1 mL/min flow, 5% to 100% linear gradient of solvent B (0.1% TFA in acetonitrile) in A (0.1% TFA in water) over 50 min was used). The NMR spectra were obtained on a Varian Inova 500 instrument.
4.2. Synthesis of Compounds
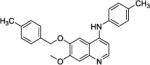
Typical synthesis procedures of compound 1: To a solution of compound A (2.23 g, 10 mmol) in 10 mL of methanesulfonic acid was added L-Methionine (3.0 g, 20 mmol) to form a faint brown clear solution, which was heated up at reflux for 12 h until starting material was consumed completely. After cooled down to room temperature, the reaction mixture was carefully poured into 100 g ice, the pH of the mixture was adjusted to 8 with addition of NH4OH solution. The crude was obtained after extraction of the base mixture with EtOAc, drying over anhydrous Na2SO4, filtration and concentration. After silica gel column chromatography, 1.2 g of pure intermediate B was furnished as pale yellow solid (57.4%). To a solution of B (50 mg, 0.239 mmol) in DMF (2 mL) was added K2CO3 (69 mg, 0.5 mmol) at room temperature, after stirring for 10 min, 4-methylbenzyl bromide (42.3 mg, 0.25 mmol) was added into the mixture. The suspension was heated at 50 °C for 6 h, then poured in water, and extracted with EtOAc (10 mL ×3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced vacuum to give faint yellow crude C-1 (67 mg, yield 89.8%), which was pure enough for the next reaction. Compound C-1 (65 mg, 0.21 mmol) and p-toluidine (25 mg, 0.23 mmol) were dissolved in 1-butanol (2 mL) with 2 drops of concentrated HCl, the afforded solution was refluxed overnight to give a gray suspension, LCMS indicated that the reaction was done. The volatiles were removed under reduced vacuum to give faint gray crude, which was recrystallized from EtOAc to give 73 mg of compound 1 (>96% purity, faint gray solid, yield 91.2%). 7-methoxy-6-((4-methylbenzyl)oxy)-N-(p-tolyl)quinolin-4-amine, compound 1, 1H NMR (600 MHz, CD3OD) δ 8.14 (d, J = 7.2 Hz, 1H), 8.00 (s, 1H), 7.36‒7.38 (m, 4H), 7.30‒7.32 (m, 2H), 7.27 (s, 1H), 7.21 (d, J = 7.8 Hz, 2H), 6.74 (d, J = 7.2 Hz, 1H), 5.22 (s, 2H), 4.04 (s, 3H), 2.42 (s, 3H), 2.35 (s, 3H). 13C NMR (150 MHz, CD3OD) δ 156.1, 154.0, 149.4, 139.0, 137.9, 137.6, 135.6, 134.7, 132.9, 130.3 (2C), 128.8 (2C), 127.7 (2C), 125.2 (2C), 110.6, 102.7, 99.3, 99.0, 70.9, 53.4, 19.8, 19.7. HRMS (ESI) C25H25N2O2 [M+H]+ calcd = 385.1911; found = 385.1919.
Compounds 2‒8 were synthesized according to the same procedures of that of compound 1 except for using different benzyl bromide or aniline reagents accordingly.
7-methoxy-N-(p-tolyl)-6-((4-(trifluoromethyl)benzyl)oxy)quinolin-4-amine
7-methoxy-N-(p-tolyl)-6-((4-(trifluoromethyl)benzyl)oxy)quinolin-4-amine, compound 2, faint gray solid, overall yield 83.7% from compound B. 1H NMR (600 MHz, CD3OD) δ 8.17 (d, J = 7.2 Hz, 1H), 8.10 (s, 1H), 7.71 (s, 4H), 7.36 (d, J = 6.6 Hz, 2H), 7.30‒7.32 (m, 3H), 6.75 (d, J = 7.2 Hz, 1H), 5.37 (s, 2H), 4.07 (s, 3H), 2.42 (s, 3H). 13C NMR (150 MHz, CD3OD) δ 156.0, 154.5, 149.1, 140.6, 139.2, 137.7, 135.8, 134.6, 130.2 (2C), 129.8 (JC-C-F = 31.5 Hz), 127.6 (2C), 125.2, 125.1 (JC-C-F = 12.0 Hz), 125.1, 125.0, 123.8 (JC-F = 114.0 Hz), 111.5, 103.0, 99.5, 99.0, 69.9, 55.7, 19.7. HRMS (ESI) C25H22F3N2O2 [M+H]+ calcd = 439.1628; found = 439.1626.
7-methoxy-6-((4-(trifluoromethyl)benzyl)oxy)-N-(3-(trifluoromethyl)phenyl)quinolin-4-amine
7-methoxy-6-((4-(trifluoromethyl)benzyl)oxy)-N-(3-(trifluoromethyl)phenyl)quinolin-4-amine, compound 3, faint yellow solid, overall yield 78.9% from compound B. 1H NMR (600 MHz, CD3OD) δ 8.28 (d, J = 6.6 Hz, 1H), 8.04 (s, 1H), 7.76‒7.78 (m, 4H), 7.72 (s, 4H), 7.35 (s, 1H), 6.90 (d, J = 6.6 Hz, 1H), 5.40 (s, 2H), 4.09 (s, 3H). 13C NMR (150 MHz, CD3OD) δ 156.3, 153.8, 149.4, 140.5, 139.7, 138.4, 136.1, 132.1 (JC-C-F = 33.0 Hz), 130.8 (2C), 130.0 (JC-C-F = 37.5 Hz), 128.5, 127.6 (3C), 125.1, 125.0, 123.6, 121.7, 112.1, 102.9, 99.8, 99.5, 69.9, 56.7. HRMS (ESI) C25H19F6N2O2 [M+H]+ calcd = 493.1345; found = 493.1358.
7-methoxy-6-((4-(trifluoromethyl)benzyl)oxy)-N-(4-(trifluoromethyl)phenyl)quinolin-4-amine
7-methoxy-6-((4-(trifluoromethyl)benzyl)oxy)-N-(4-(trifluoromethyl)phenyl)quinolin-4-amine, compound 4, faint gray solid, overall yield 82.5% from compound B. 1H NMR (600 MHz, CD3OD) δ 8.29 (d, J = 6.6 Hz, 1H), 8.05 (s, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.72 (s, 4H), 7.67 (d, J = 8.8 Hz, 2H), 7.35 (s, 1H), 7.01 (d, J = 7.2 Hz, 1H), 5.40 (s, 2H), 4.09 (s, 3H). 13C NMR (150 MHz, CD3OD) δ 156.3, 153.5, 149.4, 141.2, 140.5, 139.7, 136.1, 130.0 (JC-C-F = 33.0 Hz), 128.4 (JC-C-F = 31.5 Hz), 127.6 (3C), 126.8, 126.7, 125.1, 125.0 (2C), 124.8 (2C), 112.3, 102.9, 99.8, 99.5, 69.9, 56.8. HRMS (ESI) C25H19F6N2O2 [M+H]+ calcd = 493.1345; found = 493.1346.
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-((4-(trifluoromethyl)benzyl)oxy)quinolin-4-amine
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-((4-(trifluoromethyl)benzyl)oxy)quinolin-4-amine, compound 5, faint gray solid, overall yield 81.1% from compound B. 1H NMR (600 MHz, CD3OD) δ 8.25 (d, J = 7.2 Hz, 1H), 7.99 (s, 1H), 7.72 (s, 4H), 7.63 (dt, J = 6.6, 1.2 Hz, 1H), 7.44 (dd, J = 6.6, 1.2 Hz, 1H), 7.33 (s, 1H), 6.81 (d, J = 6.6 Hz, 1H), 5.38 (s, 2H), 4.08 (s, 3H). 13C NMR (150 MHz, CD3OD) δ 156.9 (JC-F = 247.5 Hz), 156.2, 154.2, 149.3, 140.5, 139.6, 135.9, 134.4, 130.0 (JC-C-F = 28.5 Hz), 127.8, 127.6 (2C), 126.0 (JC-C-C-F = 7.5 Hz), 125.1, 125.0, 121.6 (JC-C-F = 22.5 Hz), 117.7, 117.5, 111.8, 102.9, 99.5, 99.3, 69.9, 55.7. HRMS (ESI) C24H18ClF4N2O2 [M+H]+ calcd = 477.0987; found = 477.0997.
N-(3-chloro-4-fluorophenyl)-6-((4-isopropylbenzyl)oxy)-7-methoxyquinolin-4-amine
N-(3-chloro-4-fluorophenyl)-6-((4-isopropylbenzyl)oxy)-7-methoxyquinolin-4-amine, compound 6, white solid, overall yield 62.7% from compound B. 1H NMR (600 MHz, CD3OD) δ 8.23 (d, J = 7.2 Hz, 1H), 7.96 (s, 1H), 7.63 (d, J = 6.6 Hz, 1H), 7.43 (dd, J = 6.6, 1.2 Hz, 2H), 7.41 (d, J = 7.8 Hz, 2H), 7.30 (s, 1H), 7.27 (d, J = 8.4 Hz, 2H), 6.80 (d, J = 7.2 Hz, 1H), 5.23 (s, 2H), 4.06 (s, 3H), 2.92 (p, J = 7.2 Hz, 1H), 1.25 (d, J = 7.2 Hz, 6H). 13C NMR (150 MHz, CD3OD) δ 157.3 (JC-F = 255 Hz), 156.3, 156.1, 154.1, 149.7, 149.1, 139.4, 135.7, 134.4, 133.2, 127.8 (3C), 126.2 (2C), 125.9, 121.6 (JC-C-F = 19.5 Hz), 117.6 (JC-C-F = 22.5 Hz), 111.9, 102.5, 99.3, 99.2, 70.9, 55.6, 33.8, 23.0 (2C). HRMS (ESI) C26H25ClFN2O2 [M+H]+ calcd = 451.1583; found = 451.1582.
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-((2-methylbenzyl)oxy)quinolin-4-amine
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-((2-methylbenzyl)oxy)quinolin-4-amine, compound 7, white solid, overall yield 61.5% from compound B. 1H NMR (600 MHz, CD3OD) δ 8.23 (d, J = 7.2 Hz, 1H), 7.96 (s, 1H), 7.63 (dt, J = 7.8, 1.2 Hz, 1H), 7.44 (dd, J = 7.2, 1.2 Hz, 1H), 7.31 (d, J = 3.6 Hz, 2H), 7.28 (dd, J = 7.2, 2.4 Hz, 2H), 7.17 (t, J = 3.6 Hz, 1H), 6.80 (d, J = 7.2 Hz, 1H), 5.24 (s, 2H), 4.06 (s, 3H), 2.36 (s, 3H). 13C NMR (150 MHz, CD3OD) δ 157.3 (JC-F = 247.5 Hz), 156.3, 154.1, 149.7, 139.4, 138.0, 135.8, 134.4, 130.0, 128.6, 128.1 (2C), 128.0, 127.8, 125.9, 124.6, 121.6 (JC-C-F = 19.5 Hz), 117.6 (JC-C-F = 21.0 Hz), 111.9, 102.5, 99.3, 99.2, 70.0, 55.7, 20.0. HRMS (ESI) C24H21ClFN2O2 [M+H]+ calcd = 423.1270; found = 423.1272.
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(2-phenoxyethoxy)quinolin-4-amine
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(2-phenoxyethoxy)quinolin-4-amine, compound 8, white solid, overall yield 67.8% from compound B. 1H NMR (600 MHz, CD3OD) δ 8.24 (d, J = 7.2 Hz, 1H), 7.91 (s, 1H), 7.62 (dt, J = 6.0, 1.2 Hz, 1H), 7.44 (dd, J = 6.0, 1.2 Hz, 2H), 7.29 (s, 1H), 7.26‒7.29 (m, 2H), 6.96‒6.98 (m, 2H), 6.94 (tt, J = 7.8, 1.2 Hz, 1H), 6.80 (d, J = 6.6 Hz, 1H), 4.54‒4.57 (m, 2H), 4.43‒4.45 (m, 2H), 4.05 (s, 3H). 13C NMR (150 MHz, CD3OD) δ 158.6, 157.3 (JC-F = 243 Hz), 156.1, 154.1, 149.7, 139.5, 135.8, 134.4, 129.1 (2C), 127.8, 125.9, 121.1 (JC-C-F = 133.5 Hz), 117.7, 117.5, 114.2 (2C), 111.8, 102.3, 99.4, 99.2, 68.1, 66.0, 55.6. HRMS (ESI) C24H21ClFN2O3 [M+H]+ calcd = 439.1219; found = 439.1221.
4.3. Minimum Inhibitory Concentrations (MICs) Against Bacteria
The antimicrobial activity of the quinoline compounds was tested according to the standard protocol as described previously.14 Four bacteria strains: MRSA (ATCC 33591), MRSE (RP62A), and VRE (ATCC 700802) were employed. The antimicrobial activities of the cyclic guanidine dimers against C. difficile UK6 were tested using media and methods recommended by the Clinical and Laboratory Standards Institute for susceptibility testing of anaerobes.40 The MICs were determined as the lowest concentration that completely inhibits the bacteria growth. Compounds were dissolved in DMSO to make stock drug solution, and the final concentration of DMSO in drug-treated group was less than 0.5%. The experiments were repeated at least three times with duplicates each time.
4.4. MTT assay
MTT (3-(4,5-dimethylthiazol-2yl)-2,5-dipheynyltetrazolium bromide; Sigma–Aldrich, St. Louis, MO) cell viability assay was performed to evaluate the cytotoxicity of the compounds on human HepG2 and HEK293T cell lines. HepG2 is an immortalized cell line consisting of human liver carcinoma cells. HEK293T is a specific cell line originally derived from human embryonic kidney cells grown in tissue culture. Both cells were maintained in a Dulbecco’s Modified Eagle Medium (DMEM with, 4.5g/L Glucose, L-Glutamine and Sodium Pyruvate, Corning; Corning, Manassas, VA) containing 10 % FBS (Thermo Scientific) and 1 % penicillin/streptomycin at 37°C in 5% CO2. Cells (104 cells/well) were plated in 96-well plates. After incubation overnight, cells were treated with the compounds at concentrations from 128 μg/ml to 0.125 μg/ml or 1% DMSO (as a control reagent) for 24 h at 37°C. Then 10μl of MTT stock solution (5 mg/ml) were added to cells in each well, and further incubated for 4 h at 37°C. After careful removal of media from each well without disturbing cells, 100μl of DMSO was added to each well, and incubated for 15 min at 37°C. Absorbance at 540 nm was read in a Synergy HTX multi-mode reader (Bio Tek Instruments, Inc. Winooski VT). Data were analyzed using Graphpad PRISM 6 (GraphPad Software, Inc., La Jolla, CA), and the half maximal inhibitory concentration (IC50) was reported as a concentration of compound that is required for 50% inhibition in vitro against cells treated with a control reagent.
4.5. In vivo Evaluation of C. difficile UK6 Induced Infection
C57BL/6 female mice (6-week-old) were purchased from Charles River Laboratories, MA. During the experiment, the mice were housed in groups of 5 animals per cage under the same conditions. All animal experiments were approved by the institutional committee for animal care and use at the University of South Florida. The experimental design is illustrated in Figure S1. Twenty-five mice were divided into three groups (group 1‒3). Group 1 (UK6, n = 10) were challenged with spores of C. difficile UK6. Groups 2 (UK6+Cpd6, n = 10) were challenged with spores of C. difficile UK6, and treated by compound 6. Group 3 (Cpd6, n = 5) were only treated with compound 6 without C. difficile infection. The mice were given drinking water containing a mixture of six antibiotics including ampicillin (200 mg/kg), kanamycin (40 mg/kg), gentamycin (3.5 mg/kg), colistin (4.2 mg/kg), metronidazole (21.5 mg/kg) and vancomycin (4.5 mg/kg) for 5 days, and then received autoclaved water for 2 days, followed by a single dose of clindamycin (10 mg/kg) intraperitoneally 1 day before (day 1) challenge day. In the challenge day (day 0), mice in groups 1‒2 were challenged with C. difficile UK6 spores at 106 CFU by gavage. At 4 hours post challenge, the mice in groups 2 were given one dose of compound 6 (50 mg/kg) via gavage. From the first day post challenge (day 1), mice in group 2 received one dose of compound 6 twice a day (50 mg/kg/day) for five days. Meanwhile, the mice in group 3 were also given compound 6 at the same time with the same dose to determine the toxicity of the compound to the mice. After C. difficile challenge /or compound treatment, mice were monitored daily for five days after challenge with C. difficile spores for weight changes, survival, diarrhea, and other symptoms of the disease. Diarrhea was defined as soft or watery feces. Results were analyzed by the two-way ANOVA and Pearson Chi-Square. The differences between the UK6 group and the treatment group (UK6 + Cpd 6) are statistically significant, p < 0.05 (Daily survival rate showed the Row Factor P = 0.0389, the Column Factor P = 0.0493 and Percent of diarrhea showed the Row Factor P = 0.028, the Column Factor P = 0.0519. At time point of day 2 post challenge the diarrhea rate was significant different (P = 0.006, Pearson Chi-Square)).
Fecal samples were collected at the 1st, 3rd, and 5th day post challenge for C. difficile spore enumeration. The percent of diarrhea was defined number of mice that developed diarrhea divided by total number of the mice in a given group. Fecal samples were collected at the 1st, 3rd, and 5th day post challenge for numeration of C. difficile spores.
Fecal samples were weight and shocked in 95% ethanol (0.1g/ml) for 1 hour followed by serial dilution in PBS, spreading on BHI plates supplemented with 10% taurocholic acid, and incubation in an anaerobic chamber. After incubation for 48 hours, the colonies on plates in three duplicates for the selected dilutions were counted.
Supplementary Material
Acknowledgments
This work was supported by NSF CAREER 1351265, NIH 1R01GM112652-01A1, NIH K01-DK092352, NIH R21-AI113470, NIH R03-DK112004, and NIH R01-AI132711.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
A. Supplementary Information
Supplementary Information associated with this article can be found in the online version, at http://dx.doi.org/.
References
- 1.Watkins RR, David MZ, Salata RA. Current concepts on the virulence mechanisms of meticillin-resistant Staphylococcus aureus. J Med Microbiol. 2012;61:1179–1193. doi: 10.1099/jmm.0.043513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MRSA may kill more US citizens than HIV. BMJ (Br Med J) 2007;335:850–850. [Google Scholar]
- 3.Klein E, Smith DL, Laxminarayan R. Hospitalizations and Deaths Caused by Methicillin-Resistant Staphylococcus aureus, United States, 1999–2005. Emerg Infect Dis. 2007;13:1840–1846. doi: 10.3201/eid1312.070629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gill SR, Fouts DE, Archer GL, Mongodin EF, DeBoy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, Dodson RJ, Daugherty SC, Madupu R, Angiuoli SV, Durkin AS, Haft DH, Vamathevan J, Khouri H, Utterback T, Lee C, Dimitrov G, Jiang L, Qin H, Weidman J, Tran K, Kang K, Hance IR, Nelson KE, Fraser CM. Insights on Evolution of Virulence and Resistance from the Complete Genome Analysis of an Early Methicillin-Resistant Staphylococcus aureus Strain and a Biofilm-Producing Methicillin-Resistant Staphylococcus epidermidis Strain. J Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cetinkaya Y, Falk P, Mayhall CG. Vancomycin-Resistant Enterococci. Clin Microbiol Rev. 2000;13:686–707. doi: 10.1128/cmr.13.4.686-707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault A-M, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, René P, Monczak Y, Dascal A. A Predominantly Clonal Multi-Institutional Outbreak of Clostridium difficile–Associated Diarrhea with High Morbidity and Mortality. N Engl J Med. 2005;353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 7.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartlett JG. Antibiotic-Associated Diarrhea. N Engl J Med. 2002;346:334–339. doi: 10.1056/NEJMcp011603. [DOI] [PubMed] [Google Scholar]
- 9.Kwon JH, Olsen MA, Dubberke ER. The Morbidity, Mortality, and Costs Associated with Clostridium difficile Infection. Infect Dis Clin North Am. 2015;29:123–134. doi: 10.1016/j.idc.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Leffler DA, Lamont JT. Clostridium difficile infection. N Engl J Med. 2015;372:1539–1548. doi: 10.1056/NEJMra1403772. [DOI] [PubMed] [Google Scholar]
- 11.Peng Z, Jin D, Kim HB, Stratton CW, Wu B, Tang YW, Sun X. Update on antimicrobial resistance in Clostridium difficile: resistance mechanisms and antimicrobial susceptibility testing. J Clin Microbiol. 2017;55:1998–2008. doi: 10.1128/JCM.02250-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimlichman E, Henderson D, Tamir O, et al. Health care–associated infections: A meta-analysis of costs and financial impact on the us health care system. JAMA Internal Medicine. 2013;173:2039–2046. doi: 10.1001/jamainternmed.2013.9763. [DOI] [PubMed] [Google Scholar]
- 13.O’Connell KM, Hodgkinson JT, Sore HF, Welch M, Salmond GP, Spring DR. Combating multidrug-resistant bacteria: current strategies for the discovery of novel antibacterials. Angew Chem Int Ed. 2013;52:10706–10733. doi: 10.1002/anie.201209979. [DOI] [PubMed] [Google Scholar]
- 14.Teng P, Huo D, Nimmagadda A, Wu J, She F, Su M, Lin X, Yan J, Cao A, Xi C, Hu Y, Cai J. Small Antimicrobial Agents Based on Acylated Reduced Amide Scaffold. J Med Chem. 2016;59:7877–7887. doi: 10.1021/acs.jmedchem.6b00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stokes JM, MacNair CR, Ilyas B, French S, Cote JP, Bouwman C, Farha MA, Sieron AO, Whitfield C, Coombes BK, Brown ED. Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat Microbiol. 2017;2:17028. doi: 10.1038/nmicrobiol.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin S, Koh JJ, Aung TT, Lim F, Li J, Zou H, Wang L, Lakshminarayanan R, Verma C, Wang Y, Tan DT, Cao D, Beuerman RW, Ren L, Liu S. Symmetrically Substituted Xanthone Amphiphiles Combat Gram-Positive Bacterial Resistance with Enhanced Membrane Selectivity. J Med Chem. 2017;60:1362–1378. doi: 10.1021/acs.jmedchem.6b01403. [DOI] [PubMed] [Google Scholar]
- 17.LaMarche MJ, Leeds JA, Brewer J, Dean K, Ding J, Dzink-Fox J, Gamber G, Jain A, Kerrigan R, Krastel P, Lee K, Lombardo F, McKenney D, Neckermann G, Osborne C, Palestrant D, Patane MA, Rann EM, Robinson Z, Schmitt E, Stams T, Tiamfook S, Yu D, Whitehead L. Antibacterial and Solubility Optimization of Thiomuracin A. J Med Chem. 2016;59:6920–6928. doi: 10.1021/acs.jmedchem.6b00726. [DOI] [PubMed] [Google Scholar]
- 18.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Camara M. Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev. 2011;35:247–274. doi: 10.1111/j.1574-6976.2010.00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Bambeke F, Michot JM, Van Eldere J, Tulkens PM. Quinolones in 2005: an update. Clin Microbiol Infect. 2005;11:256–280. doi: 10.1111/j.1469-0691.2005.01131.x. [DOI] [PubMed] [Google Scholar]
- 21.Ruiz J. Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. J Antimicrob Chemother. 2003;51:1109–1117. doi: 10.1093/jac/dkg222. [DOI] [PubMed] [Google Scholar]
- 22.Lam T, Hilgers MT, Cunningham ML, Kwan BP, Nelson KJ, Brown-Driver V, Ong V, Trzoss M, Hough G, Shaw KJ, Finn J. Structure-Based Design of New Dihydrofolate Reductase Antibacterial Agents: 7-(Benzimidazol-1-yl)-2,4-diaminoquinazolines. J Med Chem. 2014;57:651–668. doi: 10.1021/jm401204g. [DOI] [PubMed] [Google Scholar]
- 23.Huband MD, Cohen MA, Zurack M, Hanna DL, Skerlos LA, Sulavik MC, Gibson GW, Gage JW, Ellsworth E, Stier MA, Gracheck SJ. In vitro and in vivo activities of PD 0305970 and PD 0326448, new bacterial gyrase/topoisomerase inhibitors with potent antibacterial activities versus multidrug-resistant gram-positive and fastidious organism groups. Antimicrob Agents Chemother. 2007;51:1191–1201. doi: 10.1128/AAC.01321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kung PP, Casper MD, Cook KL, Wilson-Lingardo L, Risen LM, Vickers TA, Ranken R, Blyn LB, Wyatt JR, Cook PD, Ecker DJ. Structure−Activity Relationships of Novel 2-Substituted Quinazoline Antibacterial Agents. J Med Chem. 1999;42:4705–4713. doi: 10.1021/jm9903500. [DOI] [PubMed] [Google Scholar]
- 25.Marella A, Tanwar OP, Saha R, Ali MR, Srivastava S, Akhter M, Shaquiquzzaman M, Alam MM. Quinoline: A versatile heterocyclic. Saudi Pharm J. 2013;21:1–12. doi: 10.1016/j.jsps.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 27.Diacon AH, Pym A, Grobusch M, Patientia R, Rustomjee R, Page-Shipp L, Pistorius C, Krause R, Bogoshi M, Churchyard G, Venter A, Allen J, Palomino JC, De Marez T, van Heeswijk RPG, Lounis N, Meyvisch P, Verbeeck J, Parys W, de Beule K, Andries K, Neeley DFM. The Diarylquinoline TMC207 for Multidrug-Resistant Tuberculosis. N Engl J Med. 2009;360:2397–2405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 28.Upadhayaya RS, Vandavasi JK, Vasireddy NR, Sharma V, Dixit SS, Chattopadhyaya J. Design, synthesis, biological evaluation and molecular modelling studies of novel quinoline derivatives against Mycobacterium tuberculosis. Bioorg Med Chem. 2009;17:2830–2841. doi: 10.1016/j.bmc.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 29.de Souza MVN, Pais KC, Kaiser CR, Peralta MA, de L, Ferreira M, Lourenço MCS. Synthesis and in vitro antitubercular activity of a series of quinoline derivatives. Bioorg Med Chem. 2009;17:1474–1480. doi: 10.1016/j.bmc.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 30.Fu LM, Fu-Liu CS. Is Mycobacterium tuberculosis a closer relative to Gram-positive or Gram–negative bacterial pathogens? Tuberculosis. 2002;82:85–90. doi: 10.1054/tube.2002.0328. [DOI] [PubMed] [Google Scholar]
- 31.Hoemann MZ, Kumaravel G, Xie RL, Rossi RF, Meyer S, Sidhu A, Cuny GD, Hauske JR. Potent In vitro methicillin-resistant Staphylococcus aureus activity of 2-(1H-indol-3-yl)quinoline derivatives. Bioorg Med Chem Lett. 2000;10:2675–2678. doi: 10.1016/s0960-894x(00)00542-4. [DOI] [PubMed] [Google Scholar]
- 32.He LCH, Zhou Q, Pan X. Preparation of quinoline derivatives as antitumor agents. 101362719A. C.N. Patent. 2009 Feb 11;
- 33.Weis F, Beiras-Fernandez A, Schelling G. Daptomycin, a lipopeptide antibiotic in clinical practice. Curr Opin Investig Drugs. 2008;9:879–884. [PubMed] [Google Scholar]
- 34.O’Donoghue C, Kyne L. Update on Clostridium difficile infection. Curr Opin Gastroenterol. 2011;27:38–47. doi: 10.1097/MOG.0b013e3283411634. [DOI] [PubMed] [Google Scholar]
- 35.McGlone SM, Bailey RR, Zimmer SM, Popovich MJ, Tian Y, Ufberg P, Muder RR, Lee BY. The economic burden of Clostridium difficile. Clin Microbiol Infect. 2012;18:282–289. doi: 10.1111/j.1469-0691.2011.03571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zar FA, Bakkanagari SR, Moorthi KM, Davis M. A Comparison of Vancomycin and Metronidazole for the Treatment of Clostridium difficile–Associated Diarrhea, Stratified by Disease Severity. Clin Infect Dis. 2007;45:302–307. doi: 10.1086/519265. [DOI] [PubMed] [Google Scholar]
- 37.Koo HL, Garey KW, Dupont HL. Future novel therapeutic agents for Clostridium difficile infection. Expert Opin Investig Drugs. 2010;19:825–836. doi: 10.1517/13543784.2010.495386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbut F, Richard A, Hamadi K, Chomette V, Burghoffer B, Petit JC. Epidemiology of recurrences or reinfections of Clostridium difficile-associated diarrhea. J Clin Microbiol. 2000;38:2386–2388. doi: 10.1093/gao/9781884446054.article.t031141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tonna I, Welsby PD. Pathogenesis and treatment of Clostridium difficile infection. Postgrad Med J. 2005;81:367–369. doi: 10.1136/pgmj.2004.028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hecht DH, Onderdonk OA, Citron DM, Roe-Carpenter D, Cox M, Rosenblatt JE, Jacobus N, Wexler HM, Jenkins SG. Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria—. Seventh. Wayne, PA, USA: 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.