

Abstract
Purpose of review
Congenital hyperinsulinism (HI) is the most common cause of persistent hypoglycemia in infants and children. Early and appropriate recognition and treatment of hypoglycemia is vital to minimize neurocognitive impairment.
Recent findings
There are at least 11 known monogenic forms of HI and several associated syndromes. Molecular diagnosis allows for prediction of the effectiveness of diazoxide and the likelihood of focal HI. Inactivating mutations in the genes encoding the ATP-sensitive potassium channel (KATP HI) account for 60% of all identifiable mutations, including 85% of diazoxide-unresponsive cases. Syndromes or disorders associated with HI include Beckwith-Wiedemann syndrome, Kabuki syndrome, Turner syndrome, and congenital disorders of glycosylation. While focal Hi can be cured by resection of the lesion, therapeutic options for non-focal HI remain limited and include diazoxide, octreotide, and long-acting somatostatin analogs, and near-total pancreatectomy. Although sirolimus has been reported to improve glycemic control in infants with diazoxide-unresponsive HI, the extent of improvement has been limited, and significant adverse events have been reported.
Summary
Identification of the etiology of congenital hyperinsulinism helps guide management decisions. Use of therapies with limited benefit and significant potential risks should be used with caution.
Keywords: hypoglycemia, insulin, beta cell, KATP channel, diazoxide, Beckwith-Wiedemann syndrome
Introduction
Congenital hyperinsulinism, the most common cause of persistent hypoglycemia in infants and children, occurs in approximately 1 in 50,000 live births, but the incidence can be as high as 1 in 2,500 in populations with high consanguinity rates. Early recognition and management of severe hypoglycemia is necessary to minimize risk of permanent neurologic damage, which is common among individuals with HI.(1–3) In 2015, the Pediatric Endocrine Society released recommendations for evaluation and management of persistent hypoglycemia (4), and sensitive laboratory-based criteria (Table 1) can be used to confirm the diagnosis.(5) Although persistent hypoglycemia in newborns may also result from other causes, including hypopituitarism, our review focuses on monogenic and syndromic forms of HI and their management.
Table 1.
Sensitivity of measures obtained at the time of hypoglycemia to diagnose congenital hyperinsulinism
| Definition | Sensitivity, % | Specificity, % | |
|---|---|---|---|
| Detectable insulin | any detectable value | 82.2% | 100% |
| Elevated C-peptide | ≥0.5 ng/mL | 88.5% | 100% |
| Suppressed IGFBP1 | ≤ 110 ng/mL | 85% | 96.5% |
| Suppressed β-hydroxybutyrate | <1.8 mM | 100% | 100% |
| Suppressed free fatty acids | <1.7 mM | 86.9% | 100% |
| Positive glycemic response to 1 mg glucagon injection | Increased in plasma glucose ≥30 mg/dL | 88.9% | 100% |
Reproduced with permission from (5).
Molecular Genetics
There are currently at least 11 known monogenic causes of HI.(6) However, a genetic etiology remains unknown in approximately 40–50% of affected children even after extensive genetic evaluation.(7, 8) More recently, it has become apparent that in some children, hyperinsulinism is only one feature of more complex syndromes, such as Beckwith-Wiedemann syndrome. The most common monogenic form of hyperinsulinism is due to inactivating mutations in the genes encoding the ATP-sensitive potassium channel (KATP HI), representing approximately 60% of all identifiable mutations, including 85% of diazoxide-unresponsive cases and 17% of diazoxide-responsive cases.(7) We discuss aspects of the most common monogenic (Figure 1) and syndromic forms of HI below.
Figure 1. Beta-cell depicting the impact of known monogenic causes of HI on insulin secretion.
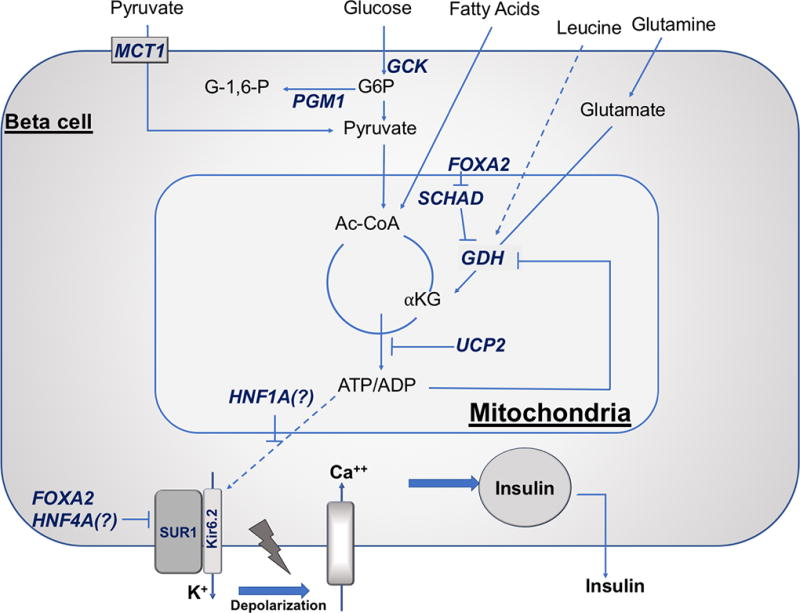
Glucose-stimulated insulin secretion is triggered by an increased in ATP/ADP ratio resulting from glucose metabolism. Glucokinase (GCK) is the β-cell glucose sensor setting the threshold for insulin secretion. The increased in ATP/ADP ratio results in closing of the ATP-sensitive KATP channels (SUR1/Kir6.2), with subsequent plasma membrane depolarization, activation of voltage-gated calcium channels, cytosolic calcium increase, and insulin release from stored intracellular granules. Eleven known β-cell genes are responsible for monogenic HI: ABCC8 (encoding sulfonylurea receptor 1, SUR1), KCNJ11 (encoding inwardly rectifying potassium channel 6.2, Kir6.2), GCK (encoding glucokinase), SLC16A1 (encoding monocarboxylate transporter 1, MCT1), FOXA2 (forkhead box A2), HADH (encoding short-chain 3-hydroxyacyl-CoA dehydrogenase, SCHAD), GLUD1 (encoding glutamate dehydrogenase, GDH), PGM1 (encoding phosphoglucomutase 1), HNF1A (hepatocyte nuclear factor 1A), HNF4A (hepatocyte nuclear factor 4A), and UCP2 (uncoupling protein 2). Other abbreviations: αKG, α-ketoglutarate; Ac-CoA, acetyl-CoA; G6P, glucose 6-phosphate; G-1,6-P, glucose 1,6-bisphosphate; INS, insulin.
KATP Hyperinsulinism
The most common and most severe form of HI is due to inactivating mutations in ABCC8 and KCNJ11, which encode the two subunits of the beta-cell ATP-sensitive potassium channel (SUR-1 and Kir6.2, respectively).(9, 10) KATP HI is usually unresponsive to diazoxide due to the absence or deficiency of functional KATP channels. KATP HI presents in the first few days after birth, and affected infants are born large for gestational age. Recessive KATP mutations, including missense, nonsense, or splicing defects, lead to diazoxide-unresponsiveness through complete absence of KATP channels. In contrast, dominant KATP mutations (missense) lead to mutant KATP subunits that impair channel activity when assembled into the KATP complex, and the degree of diazoxide-responsiveness is determined by the relative impairment in channel activity.(11, 12) Hypoglycemia due to KATP HI occurs in both the fasting state and after protein load, likely due to glutamine-stimulated “amplification” of GLP-1 receptor signaling.(13, 14)
There are two distinct histological forms of KATP HI: a diffuse form, in which all pancreatic β-cells are affected, and a focal form, in which only a small area of the pancreas is affected. Diffuse hyperinsulinism results from biallelic recessive mutations in ABCC8 or KCNJ11 but can also result from dominant mutations. Focal hyperinsulinism, which accounts for approximately 50% of cases of severe hyperinsulinism, is the result of a “two hit” mechanism: 1) a paternally-inherited recessive mutation in ABCC8 or KCNJ11, and 2) somatic loss of the maternal inherited 11p15 chromosomal region in which these genes and tumor suppressing maternally imprinted genes are encoded, compensated by paternal uniparental disomy.(15) The focal form of KATP HI can be cured by surgical resection of the lesion; thus, when evaluating children with hyperinsulinism, it is extremely important to identify those that are likely to have focal HI. There are some subtle clinical differences between diffuse and focal cases, but overall it is difficult to predict focal disease from the clinical presentation.(16) However, genetic testing is extremely helpful to predict focal HI: the finding of a single heterozygous recessive mutation in either ABCC8 or KCNJ11 has 94% positive predictive value for focal disease, while two recessive KATP channel mutations predict diffuse disease.(7)
There is marked clinical heterogeneity in the clinical presentation and the clinical course of children with KATP HI (17), and the study of tissue and isolated islets from the pancreas of children who have undergone pancreatectomy is starting to provide some insight into the molecular and cellular mechanisms that may, at least in part, explain this heterogeneity.(18, 19)
GDH Hyperinsulinism
Dominant activating mutations in GLUD1, which encodes for the enzyme glutamate dehydrogenase (GDH), give rise to hyperinsulinism/hyperammonemia (HI/HA) syndrome, the second most common cause of congenital HI. GDH mediates protein-stimulated insulin secretion through allosteric activation by leucine.(20) The functional impact of the most common GDH mutation, GDH-Ser445Leu, was recently characterized by Grimaldi et al (21), who demonstrated glutamine-stimulated mitochondrial activation and ATP rise in a beta cell line expressing mutant enzyme. Based on their findings of high sensitivity to the allosteric activator ADP, mutant GDH-S445L enzyme would be constitutively hyperactive, leading to the hyperinsulinism phenotype.
One recent case report described a functionally homozygous activating mutation of GLUD1 (novel frameshift mutation c.37delC from the asymptomatic mother and a de novo activating mutation p.S445L), which resulted in severe onset hypoglycemia, hyperammonemia and seizures on the first day of life. In lymphoblasts from the described patient, GTP inhibition of GDH activity led to half-maximal inhibitory concentration (IC50) for GTP that was approximately 7 times higher compared to a heterozygote for p.S445L and 200 times wild-type.(22) Notably, the patient’s phenotype was within the range of severity for a heterozygous GLUD1 mutation. This report was the first of a homozygous activating mutation of GLUD1 in a human.
In addition to fasting hypoglycemia due to hyperinsulinism, HI/HA is characterized by hyperammonemia and protein-induced hypoglycemia, and the clinical phenotype correlates with the degree of impaired responsiveness to GTP inhibition.(23) Hyperammonemia occurs due increased renal ammoniagenesis.(24) Individuals with HI/HA syndrome are diazoxide-responsive, have protein-sensitive hypoglycemia, normal birth weight, and later-onset hypoglycemia (median 4 months).(20) Hyperammonemia appears asymptomatic and unnecessary to treat.(20) Individuals with HI/HA have increased risk of seizures, in particular absence seizures, unrelated to hypoglycemia, as well as higher rates of developmental delay that appear to be independent of hypoglycemic brain damage.(25)
A similar phenotype of fasting and protein-induced hyperinsulinism, but without hyperammonemia, is due to inactivating mutations in HADH, which encodes the mitochondrial enzyme short-chain 3-hydroxyacyl-CoA dehydrogenase, known as SCHADHI.(26) In addition to its primary role in the catalysis of fatty acid oxidation of medium and short chain 3-hydroxy fatty acyl-CoAs, SCHAD is an allosteric inhibitor of GDH.(25) With SCHAD deficiency, loss of GDH inhibition leads to protein-sensitive hyperinsulinism.(25, 27) Camtosun et al recently reported the long-term clinical course of a patient with a deep intronic HADH splicing mutation (c.636+471G>T). The patient was macrosomic at birth and was diagnosed with HI at 30 days of life and treated with diazoxide. By the age of 20 years, she continued to require a low dose of diazoxide (2–3 mg/kg/day) to maintain euglycemia.(28) The case highlights the persistence of abnormal glucose and insulin regulation into adulthood.
HNF4A and HNF1A Hyperinsulinism
Heterozygous mutations in genes encoding transcription factors hepatocyte nuclear factors 4α or 1 α (HNF4A, HNF1A) lead to both congenital hyperinsulinism and subsequent monogenic diabetes.(29–32) Together, they account for approximately 6% of all diazoxide-responsive cases.(33) Although the precise mechanism by which mutations in HNF4A or HNF1A lead to the phenotypes of both HI and diabetes is unknown, HNF4α binds to the promoters of 11% of islet genes, and its deficiency likely impacts one or more of these downstream targets.(32)
HI due to HNF4A or HNF1A mutations tends to be diazoxide-responsive (31, 34), and HNF4A HI represented the third most common cause of HI in a cohort of 220 diazoxide-responsive HI patients studied by Flanagan et al.(34) Of note, HNF1A mutations were not evaluated in this cohort. Limited case reports and case series of HNF4A and HNF1A HI have demonstrated the variable duration of hypoglycemia phenotype and subsequent development of diabetes as early as the first decade of life.(35, 36)
GCK Hyperinsulinism
Dominant activating mutations in glucokinase (encoded by GCK), a hexokinase expressed in β-cells that sets the threshold for glucose-stimulated insulin secretion, is a less common cause of HI. The severity of GCK HI is highly variable: while some children can be managed medically, others may require pancreatectomy.(37)
UCP2 Hyperinsulinism
Diazoxide-responsive HI due to dominant inactivating mutations in UCP2 (uncoupling protein 2) were first described in 2008 (38), and a total of 9 patients have been described to date.(7, 38, 39) Functional data suggests that the phenotype results from excessive glucose-stimulated insulin secretion through enhanced glucose oxidation.(39) Of the cases reported, duration of diazoxide-requirement varied, with apparent resolution occurring between 11 months and 7.5 years.(40)
Since the availability of large-scale population data, doubt has been raised about the role of the reported variants in the HI phenotype described.(41) Using the gnomAD database, Laver et al found that four of the variants reported as pathogenic, accounting for 8/9 reported patients, were present at high frequency in the dataset. One variant reported in three studies (7, 38, 39), p.Ala268Gly, was present at a frequency in gnomAD that would equate to an incidence of 1 in 128, much higher than the known incidence of 1 in 50,000 in outbred populations. Further evaluation of additional cases will be needed to better understand the functional impact of UCP2 mutations.
MCT1 Hyperinsulinism
Mutations in the upstream promoter regions of SLC16A1, which encodes moncarboxylate transporter 1 (MCT1), the pyruvate transporter, have been linked to exercise-induced hypoglycemia.(42) This phenotype results from loss of usual suppression of MCT1 in β-cells, allowing for insulin secretion in response to pyruvate, which rises with anaerobic exercise.(6)
Beckwith-Wiedemann Syndrome (BWS)
The BWS locus on chromosome 11p15.5 includes adjacent imprinted genes that are growth-promoting (IGF2) or growth-inhibitory (non-coding RNA H19 and CDKN1C).(43) Either hypomethylation of the growth-promoting genes or hypermethylation of the growth-inhibitory genes can result in the phenotype. Twenty percent of cases are caused by 11p paternal uniparental isodisomy, resulting in both over-expression of IGF2 and no expression of CDKN1C. Approximately 50% of individuals with BWS have HI, with ~5% that is severe and persistent, which may be related to expanded β-cell mass and abnormal β-cell insulin secretion.(43) As noted by a recent case report of a child with subtle hemihypertrophy and KATP HI, BWS should be considered in the setting of syndromic features and paternally inherited KATP channel mutation.(44)
Kabuki Syndrome (KS)
KS is the second most common syndromic form of HI, with HI occurring in up to 70% of cases.(40) KS results from mutations in one of 2 genes, either autosomal recessive mutations in KMT2D (70–75% of cases) or X-linked mutations in KDM6A (1–9% of cases).(45) Although the mechanism of HI in KS is unknown, it appears to be more common in the X-linked form.(46) Unlike BWS-associated HI, most are diazoxide-responsive.(6)
Congenital disorders of glycosylation and related mutations
Congenital disorders of glycosylation (CDG) have a wide phenotypic spectrum, with three identified to be associated with HI: phosphomannomutase 2 (PMM2) deficiency (CDG1a), mannosephosphate isomerase deficiency (CDG1b), and phosphoglucomutase 1 (PGM1) deficiency (CDG1t). CDG1a can present with significant heterogeneity, from single organ involvement presenting as isolated HI to multivisceral failure.(47) HI associated with CDG1b has been treated with supplemental oral mannose treatment.(48, 49) PGM1 deficiency (CDG1t) also has a wide phenotypic spectrum, which in addition to hyperinsulinemic hypoglycemia includes growth retardation, hepatopathy, dilated cardiomyopathy, hypogonadotropic hypogonadism, myopathy, bifid uvula, and malignant hyperthermia.(50)
Recently, a promoter mutation in PMM2 (c.-167G>T) was identified in 17 children with both HI and congenital polycystic kidney disease from 11 unrelated families.(51) These patients did not exhibit diagnostic features of CDG1a. The authors report that most of the patients were treated with diazoxide and responded, but some did not receive treatment for hypoglycemia. The most common presentation of HI was hypoglycemic seizures, with a median age at HI diagnosis of 10 months of life.
FOXA2 Hyperinsulinism
A novel syndrome of hypopituitarism and hyperinsulinism associated with inactivating mutations in the developmental transcription factor forkhead box A2 (Foxa2) was recently described in two reports.(52, 53) Both mutations were identified through whole exome sequencing. Described characteristics include single median maxillary central incisor, choroidal coloboma, pulmonary stenosis, persistent oxygen requirement, neuro-developmental delay (52), coarse facial features, hypertelorism, thin upper lip, low set ears, and widely spaced nipples.(53) Foxa2 mRNA is expressed in the developing hypothalamus, pituitary, pancreas, lungs, and esophagus of mouse embryos.(52) Transactivation of target genes critical for beta cell function (ABCC8, KCNJ11, HADH) and pituitary development (GLI2, NKX2-2, SHH) is significantly decreased with mutant Foxa2 compared to wild type.(53) These two reports highlight an apparent new etiology of HI that should be considered especially when pituitary deficiencies co-exist.
Turner Syndrome
HI appears to occur at higher rates in infants with TS than expected: 6/678 HI cases had TS, compared to 1:2,500 newborns.(54) As in Kabuki syndrome, KDM6A haploinsufficiency may be implicated, as demonstrated by abnormalities of insulin release that occur in control human islets when treated with KDM6A inhibitor.(54)
Value of a genetic diagnosis
Genetic diagnosis is important for prognostication, genetic counseling, and to anticipate the clinical course and screen for diabetes later in life in individuals with HNFs HI. But perhaps the most critical value of a genetic diagnosis is the ability to predict focal hyperinsulinism. The rapid identification of a KATP mutation has been demonstrated (55) and can have significant clinical impact. In addition to anticipating diazoxide-unresponsiveness, if a KATP mutation is found, whether the mutation is recessive and monoallelic, recessive and biallelic, or dominant can predict whether the disease occurs as a focal area of adenomatosis in the pancreas (focal HI) or as diffuse disease (diffuse HI).(7) The potential to cure patients with focal HI with a limited pancreatic resection highlights the importance of genetic testing to avoid prolonged medical therapy and ongoing risk of hypoglycemia. The most common method of testing for HI mutations remains Sanger or Next Generation sequencing, but as whole exome sequencing becomes lower in cost, it may have a role in HI diagnosis.(56) However, this technique would miss deep intronic mutations, which may also cause HI by pseudoexon activation.(57)
Therapeutic options
One of the major advances in the management of HI has been the development of 18FDOPA PET for localization of focal lesions prior to surgery, which has led to the ability to cure these cases.(58) (N.B. In the United States, 18FDOPA is used under an investigational IND). Therefore, surgical resection of the lesion is the treatment of choice for focal HI. Current therapeutic options (Figure 2) in non-focal HI are limited. In cases unresponsive to all medical management, near-total pancreatectomy may be required. In cases of diazoxide-unresponsiveness, octreotide and longer-acting somatostatin analogs remain first-line therapy and are generally considered to be safe and effective, although they are not approved for this indication in the United States.(59) Unfortunately, a dose-dependent reduction in splanchnic blood flow may increase risk of necrotizing enterocolitis, limiting its use in neonates.(60, 61) The most commonly recognized long-term effects of octreotide include transient transaminitis and asymptomatic gallbladder pathology.(62) Van der Steen et al recently described an international experience with longer-acting somatostatin analogs lanreotide and sandostatin-LAR.(63) They found improved glycemic control in 89% of patients and recommended monitoring liver enzymes every 4–6 weeks and abdominal ultrasound every 3–6 months due to high prevalence of elevated liver enzymes (37%) and asymptomatic cholelithiasis.
Figure 2. Recommended approach to diagnosis and management of congenital hyperinsulinism.
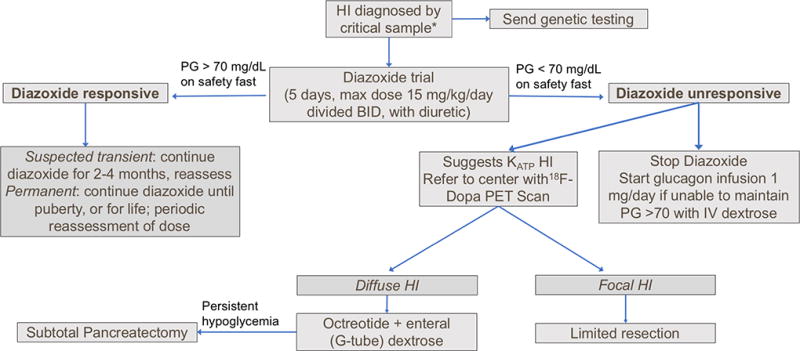
Hypoketotic hypoglycemia, with positive glycemic response to glucagon. Data from (5).
Potential future therapies include GLP-1 receptor antagonists such as Exendin-(9–39) and continuous glucagon.(40) Sirolimus, an mTOR inhibitor, has been investigated for use in diffuse HI that is unresponsive to diazoxide. There have been no controlled trials to date, and experience has been limited to only a handful of infants reported so far.(64, 65) The reported cases have either required no additional medical therapy during sirolimus treatment or ongoing low dose of octreotide. Notably, the largest study of sirolimus for HI therapy showed limited success (euglycemia in only 3/10 patients) and significant adverse events, including elevated triglycerides, anemia, stomatitis, sepsis, varicella zoster, and gut dysmotility in association with exocrine pancreatic insufficiency.(66) The risk for significant adverse events combined with limited apparent benefit have led to a call for extreme caution with sirolimus use as a therapy for HI in infancy.(67)
Conclusion
Management of congenital hyperinsulinism can be highly dependent upon the etiology, including the genetic mutation or associated syndrome. KATP HI is the most common form but is often the most challenging to treat due to diazoxide-unresponsiveness. Potential future therapies are under active investigation, but therapies with limited benefit and potentially significant risks should be used with caution.
Key Points box.
Congenital hyperinsulinism should be suspected in neonates with persistent hypoglycemia beyond 48 hours of life.
Diazoxide is the first-line medication but is often ineffective in HI due to inactivating mutations in the genes encoding the ATP-sensitive potassium channel (KATP HI).
Molecular diagnosis may aid in treatment decisions due to the ability to anticipate the likelihood of diazoxide responsiveness and the possibility of focal lesions.
Therapies for HI remain limited.
Acknowledgments
MEV is a fellow of the Center for Healthcare Improvement and Patient Safety and the Leonard Davis Institute of Health Economics at the University of Pennsylvania.
Financial support and sponsorship
This work was support by National Institutions of Health grants T32 DK07314 (MEV) and DK098517 (DDDL).
Footnotes
Conflicts of interest
The authors have no conflict of interest.
References and Recommended Reading
- 1.Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA, De Leon DD. High Risk of Diabetes and Neurobehavioral Deficits in Individuals With Surgically Treated Hyperinsulinism. J Clin Endocrinol Metab. 2015;100(11):4133–9. doi: 10.1210/jc.2015-2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2**.Ludwig A, Enke S, Heindorf J, Empting S, Meissner T, Mohnike K. Formal Neurocognitive Testing in 60 Patients with Congenital Hyperinsulinism. Horm Res Paediatr. 2018;89(1):1–6. doi: 10.1159/000481774. This prospective study of 60 patients with HI used formal neurocognitive testing to evaluate developmental delay. [DOI] [PubMed] [Google Scholar]
- 3**.Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen AM, et al. Both Low Blood Glucose and Insufficient Treatment Confer Risk of Neurodevelopmental Impairment in Congenital Hyperinsulinism: A Multinational Cohort Study. Front Endocrinol (Lausanne) 2017;8:156. doi: 10.3389/fendo.2017.00156. This report of 75 patients with HI from two European centers describes risk of neurodevelopmental impairment related to both severe hypoglycemia and treatment delay. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thornton PS, Stanley CA, De Leon DD, Harris D, Haymond MW, Hussain K, et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr. 2015;167(2):238–45. doi: 10.1016/j.jpeds.2015.03.057. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara C, Patel P, Becker S, Stanley CA, Kelly A. Biomarkers of Insulin for the Diagnosis of Hyperinsulinemic Hypoglycemia in Infants and Children. J Pediatr. 2016;168:212–9. doi: 10.1016/j.jpeds.2015.09.045. [DOI] [PubMed] [Google Scholar]
- 6.Stanley CA. Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders. J Clin Endocrinol Metab. 2016;101(3):815–26. doi: 10.1210/jc.2015-3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013;98(2):E355–63. doi: 10.1210/jc.2012-2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol. 2013;168(4):557–64. doi: 10.1530/EJE-12-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas P, Ye Y, Lightner E. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet. 1996;5(11):1809–12. doi: 10.1093/hmg/5.11.1809. [DOI] [PubMed] [Google Scholar]
- 10.Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 1995;268(5209):426–9. doi: 10.1126/science.7716548. [DOI] [PubMed] [Google Scholar]
- 11.Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, et al. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the beta-cell sulfonylurea receptor SUR1. Diabetes. 2011;60(6):1797–804. doi: 10.2337/db10-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin GM, Rex EA, Devaraneni P, Denton JS, Boodhansingh KE, DeLeon DD, et al. Pharmacological Correction of Trafficking Defects in ATP-sensitive Potassium Channels Caused by Sulfonylurea Receptor 1 Mutations. J Biol Chem. 2016;291(42):21971–83. doi: 10.1074/jbc.M116.749366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly A, Ng D, Ferry RJ, Jr, Grimberg A, Koo-McCoy S, Thornton PS, et al. Acute insulin responses to leucine in children with the hyperinsulinism/hyperammonemia syndrome. J Clin Endocrinol Metab. 2001;86(8):3724–8. doi: 10.1210/jcem.86.8.7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calabria AC, Li C, Gallagher PR, Stanley CA, De Leon DD. GLP-1 receptor antagonist exendin-(9–39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP-sensitive K+ channel. Diabetes. 2012;61(10):2585–91. doi: 10.2337/db12-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Lonlay P, Fournet JC, Rahier J, Gross-Morand MS, Poggi-Travert F, Foussier V, et al. Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J Clin Invest. 1997;100(4):802–7. doi: 10.1172/JCI119594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lord K, Dzata E, Snider KE, Gallagher PR, De Leon DD. Clinical presentation and management of children with diffuse and focal hyperinsulinism: a review of 223 cases. J Clin Endocrinol Metab. 2013;98(11):E1786–9. doi: 10.1210/jc.2013-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Salomon-Estebanez M, Flanagan SE, Ellard S, Rigby L, Bowden L, Mohamed Z, et al. Conservatively treated Congenital Hyperinsulinism (CHI) due to K-ATP channel gene mutations: reducing severity over time. Orphanet J Rare Dis. 2016;11(1):163. doi: 10.1186/s13023-016-0547-3. This study describes a cohort of children with KATP HI and assesses predictors of improved glycemic control over time. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18*.Han B, Mohamed Z, Estebanez MS, Craigie RJ, Newbould M, Cheesman E, et al. Atypical Forms of Congenital Hyperinsulinism in Infancy Are Associated With Mosaic Patterns of Immature Islet Cells. J Clin Endocrinol Metab. 2017;102(9):3261–7. doi: 10.1210/jc.2017-00158. This study of pancreatic tissue from 3 patients with atypical HI found that atypical HI was associated with an immature delta-cell profile. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19*.Li C, Ackermann AM, Boodhansingh KE, Bhatti TR, Liu C, Schug J, et al. Functional and Metabolomic Consequences of KATP Channel Inactivation in Human Islets. Diabetes. 2017;66(7):1901–13. doi: 10.2337/db17-0029. This study demonstrates the complex pathophysiology of KATP HI by examining cytosolic calcium, insulin secretion, oxygen consumption, and [U-13C]glucose metabolism in islets isolated from the pancreases of children with KATPHI who required pancreatectomy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stanley CA, Fang J, Kutyna K, Hsu BY, Ming JE, Glaser BPM HI/HA Contributing Investigators. Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome: predominance of mutations in exons 11 and 12 of the glutamate dehydrogenase gene. Diabetes. 2000;49:667–73. doi: 10.2337/diabetes.49.4.667. [DOI] [PubMed] [Google Scholar]
- 21*.Grimaldi M, Karaca M, Latini L, Brioudes E, Schalch T, Maechler P. Identification of the molecular dysfunction caused by glutamate dehydrogenase S445L mutation responsible for hyperinsulinism/hyperammonemia. Hum Mol Genet. 2017;26(18):3453–65. doi: 10.1093/hmg/ddx213. This study assesses the molecular mechanism of dysregulated insulin secretion and ammonia production due to the most frequent HI/HA mutation, Ser445Leu. [DOI] [PubMed] [Google Scholar]
- 22*.Barrosse-Antle M, Su C, Chen P, Boodhansingh KE, Smith TJ, Stanley CA, et al. A severe case of hyperinsulinism due to hemizygous activating mutation of glutamate dehydrogenase. Pediatr Diabetes. 2017;18(8):911–6. doi: 10.1111/pedi.12507. This report describes a child with two mutations in GLUD1 as well as the functional impact of the mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glaser BKP, Heyman M, Davis E, Cuesta A, Buchs A, Stanley CA, Thornton PSPM, Matschinsky FM, Herold KC. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338:226–30. doi: 10.1056/NEJM199801223380404. [DOI] [PubMed] [Google Scholar]
- 24.Treberg JR, Clow KA, Greene KA, Brosnan ME, Brosnan JT. Systemic activation of glutamate dehydrogenase increases renal ammoniagenesis: implications for the hyperinsulinism/hyperammonemia syndrome. Am J Physiol Endocrinol Metab. 2010;298(6):E1219–25. doi: 10.1152/ajpendo.00028.2010. [DOI] [PubMed] [Google Scholar]
- 25.Palladino AA, Stanley CA. The hyperinsulinism/hyperammonemia syndrome. Reviews in Endocrine and Metabolic Disorders. 2010;11(3):171–8. doi: 10.1007/s11154-010-9146-0. [DOI] [PubMed] [Google Scholar]
- 26.Clayton PT, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, et al. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of β-oxidation in insulin secretion. Journal of Clinical Investigation. 2001;108(3):457–65. doi: 10.1172/JCI11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heslegrave AJ, Kapoor RR, Eaton S, Chadefaux B, Akcay T, Simsek E, et al. Leucine-sensitive hyperinsulinaemic hypoglycaemia in patients with loss of function mutations in 3-Hydroxyacyl-CoA Dehydrogenase. Orphanet J Rare Dis. 2012;7:25. doi: 10.1186/1750-1172-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camtosun E, Flanagan SE, Ellard S, Siklar Z, Hussain K, Kocaay P, et al. A Deep Intronic HADH Splicing Mutation (c.636+471G>T) in a Congenital Hyperinsulinemic Hypoglycemia Case: Long Term Clinical Course. J Clin Res Pediatr Endocrinol. 2015;7(2):144–7. doi: 10.4274/jcrpe.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pingul MM, Hughes N, Wu A, Stanley CA, Gruppuso PA. Hepatocyte nuclear factor 4alpha gene mutation associated with familial neonatal hyperinsulinism and maturity-onset diabetes of the young. J Pediatr. 2011;158(5):852–4. doi: 10.1016/j.jpeds.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007;4(4):e118. doi: 10.1371/journal.pmed.0040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab. 2012;97(10):E2026–30. doi: 10.1210/jc.2012-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, et al. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes. 2008;57(6):1659–63. doi: 10.2337/db07-1657. [DOI] [PubMed] [Google Scholar]
- 33*.Tung JY-l, Boodhansingh K, Stanley CA, De Leon D. Clinical heterogeneity of hyperinsulinism due to HNF1A and HNF4A mutations. Pediatric Diabetes. 2018 doi: 10.1111/pedi.12655. This study describes the variable phenotype of HNF-HI and highlights the value of recognition, given the impact on management. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol. 2010;162(5):987–92. doi: 10.1530/EJE-09-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arya VB, Rahman S, Senniappan S, Flanagan SE, Ellard S, Hussain K. HNF4A mutation: switch from hyperinsulinaemic hypoglycaemia to maturity-onset diabetes of the young, and incretin response. Diabet Med. 2014;31(3):e11–5. doi: 10.1111/dme.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGlacken-Byrne SM, Hawkes CP, Flanagan SE, Ellard S, McDonnell CM, Murphy NP. The evolving course of HNF4A hyperinsulinaemic hypoglycaemia--a case series. Diabet Med. 2014;31(1):e1–5. doi: 10.1111/dme.12259. [DOI] [PubMed] [Google Scholar]
- 37.Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, et al. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes. 2009;58(6):1419–27. doi: 10.2337/db08-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez-Barroso MM, Giurgea I, Bouillaud F, Anedda A, Bellanne-Chantelot C, Hubert L, et al. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One. 2008;3(12):e3850. doi: 10.1371/journal.pone.0003850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39**.Ferrara CT, Boodhansingh KE, Paradies E, Giuseppe F, Steinkrauss LJ, Topor LS, et al. Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J Clin Endocrinol Metab. 2017;102(3):942–9. doi: 10.1210/jc.2016-3164. This study provides valuable diagnostic testing information for HI, including sensitivity and specificity of multiple critical labs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40*.De Leon DD, Stanley CA. Congenital Hypoglycemia Disorders: New Aspects of Etiology, Diagnosis, Treatment and Outcomes: Highlights of the Proceedings of the Congenital Hypoglycemia Disorders Symposium, Philadelphia April 2016. Pediatr Diabetes. 2017;18(1):3–9. doi: 10.1111/pedi.12453. This summary of the Congenital Hypoglycemia Disorders Symposium (Philadelphia, PA, USA, 2016) provides an overview of mechansisms and management of HI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41*.Laver TW, Weedon MN, Caswell R, Hussain K, Ellard S, Flanagan SE. Analysis of large-scale sequencing cohorts does not support the role of variants in UCP2 as a cause of hyperinsulinaemic hypoglycaemia. Human Mutation. 2017;38(10):1442–4. doi: 10.1002/humu.23289. This Letter to the Editor argues that, based on large-scale sequencing cohort studies, variants in UCP2 are not likely responsible for HI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, et al. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes. 2003;52:199–204. doi: 10.2337/diabetes.52.1.199. [DOI] [PubMed] [Google Scholar]
- 43.Kalish JM, Boodhansingh KE, Bhatti TR, Ganguly A, Conlin LK, Becker SA, et al. Congenital hyperinsulinism in children with paternal 11p uniparental isodisomy and Beckwith-Wiedemann syndrome. J Med Genet. 2016;53(1):53–61. doi: 10.1136/jmedgenet-2015-103394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kocaay P, Siklar Z, Ellard S, Yagmurlu A, Camtosun E, Erden E, et al. Coexistence of Mosaic Uniparental Isodisomy and a KCNJ11 Mutation Presenting as Diffuse Congenital Hyperinsulinism and Hemihypertrophy. Horm Res Paediatr. 2016;85(6):421–5. doi: 10.1159/000446153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bogershausen N, Gatinois V, Riehmer V, Kayserili H, Becker J, Thoenes M, et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum Mutat. 2016;37(9):847–64. doi: 10.1002/humu.23026. [DOI] [PubMed] [Google Scholar]
- 46.Gole H, Chuk R, Coman D. Persistent Hyperinsulinism in Kabuki Syndrome 2: Case Report and Literature Review. Clin Pract. 2016;6(3):848. doi: 10.4081/cp.2016.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shanti B, Silink M, Bhattacharya K, Howard NJ, Carpenter K, Fietz M, et al. Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis. 2009;32(Suppl 1):S241–51. doi: 10.1007/s10545-009-1180-2. [DOI] [PubMed] [Google Scholar]
- 48.De Lonlay P, Cuer M, Vuillaumier-Barrot S, Beaune G, Castelnau P, Kretz M, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: a new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. J Pediatr. 1999;135:379–83. doi: 10.1016/s0022-3476(99)70139-3. [DOI] [PubMed] [Google Scholar]
- 49.Babovic-Vuksanovic D, Patterson M, Schwenk W, O’Brien J, Vockley J, Freeze H, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr. 1999;135(6) doi: 10.1016/s0022-3476(99)70103-4. [DOI] [PubMed] [Google Scholar]
- 50.Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S, et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med. 2014;370(6):533–42. doi: 10.1056/NEJMoa1206605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51*.Cabezas OR, Flanagan SE, Stanescu H, Garcia-Martinez E, Caswell R, Lango-Allen H, et al. Polycystic Kidney Disease with Hyperinsulinemic Hypoglycemia Caused by a Promoter Mutation in Phosphomannomutase 2. J Am Soc Nephrol. 2017;28(8):2529–39. doi: 10.1681/ASN.2016121312. This report describes the co-occurance of HI and polycystic kidney disease in 17 patients from 11 families and identifies a common promoter mutation in PMM2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52*.Giri D, Vignola ML, Gualtieri A, Scagliotti V, McNamara P, Peak M, et al. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum Mol Genet. 2017;26(22):4315–26. doi: 10.1093/hmg/ddx318. This is the first report of a novel mutation in FOXA2 leading to hypopituitarism and hyperinsulinism. [DOI] [PubMed] [Google Scholar]
- 53*.Vajravelu ME, Chai J, Krock B, Baker S, Langdon D, Alter C, et al. Congenital Hyperinsulinism and Hypopituitarism Attributable to a Novel Mutation in FOXA2. J Clin Endocrinol Metab. 2018 doi: 10.1210/jc.2017-02157. This study describes a mutation in FOXA2 that led to both hypopituitarism and hyperinsulinism and the impact of the mutation on transactivation of genes critical for beta cell function and pituitary development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gibson C, Boodhansingh K, Ganguly A, Stanley CA, editors. Congenital Hyperinsulinism in Turner’s syndrome; Poster LBS 024-030 presented at: 97th Annual Meeting of the Endocrine Society; 2015 March 5–8, 2015; San Diego, CA. [Google Scholar]
- 55.Banerjee I, Skae M, Flanagan SE, Rigby L, Patel L, Didi M, et al. The contribution of rapid KATP channel gene mutation analysis to the clinical management of children with congenital hyperinsulinism. Eur J Endocrinol. 2011;164(5):733–40. doi: 10.1530/EJE-10-1136. [DOI] [PubMed] [Google Scholar]
- 56*.Johnson SR, Leo PJ, McInerney-Leo AM, Anderson LK, Marshall M, McGown I, et al. Whole-exome sequencing for mutation detection in pediatric disorders of insulin secretion: Maturity onset diabetes of the young and congenital hyperinsulinism. Pediatr Diabetes. 2018 doi: 10.1111/pedi.12638. This study included 5 subjects with HI with known mutations and conducted whole exom sequencing using two different kits, succsefully identifying all 5 mutations. [DOI] [PubMed] [Google Scholar]
- 57.Flanagan Sarah E, Xie W, Caswell R, Damhuis A, Vianey-Saban C, Akcay T, et al. Next-Generation Sequencing Reveals Deep Intronic Cryptic ABCC8 and HADH Splicing Founder Mutations Causing Hyperinsulinism by Pseudoexon Activation. The American Journal of Human Genetics. 2013;92(1):131–6. doi: 10.1016/j.ajhg.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laje P, States LJ, Zhuang H, Becker SA, Palladino AA, Stanley CA, et al. Accuracy of PET/CT Scan in the diagnosis of the focal form of congenital hyperinsulinism. J Pediatr Surg. 2013;48(2):388–93. doi: 10.1016/j.jpedsurg.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59*.Hosokawa Y, Kawakita R, Yokoya S, Ogata T, Ozono K, Arisaka O, et al. Efficacy and safety of octreotide for the treatment of congenital hyperinsulinism: a prospective, open-label clinical trial and an observational study in Japan using a nationwide registry. Endocr J. 2017;64(9):867–80. doi: 10.1507/endocrj.EJ17-0024. This combined open-label clinical trial (SCORCH study) and observational study (SCORCH registry) evaluated the efficacy and safety of octreotide for diazoxide-unresponsive HI in Japan, concluding it was effective and well tolerated in the majority of patients. [DOI] [PubMed] [Google Scholar]
- 60.Hawkes CP, Adzick NS, Palladino AA, De Leon DD. Late Presentation of Fulminant Necrotizing Enterocolitis in a Child with Hyperinsulinism on Octreotide Therapy. Horm Res Paediatr. 2016;86(2):131–6. doi: 10.1159/000443959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61**.McMahon AW, Wharton GT, Thornton P, De Leon DD. Octreotide use and safety in infants with hyperinsulinism. Pharmacoepidemiol Drug Saf. 2017;26(1):26–31. doi: 10.1002/pds.4144. This study, a collaboration between the FDA and a pediatric hospital, evaluates serious adverse events in 103 children with HI treated with octreotide and highlights the risk of nectrotizing enterocolitis even in full-term infants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Demirbilek H, Shah P, Arya VB, Hinchey L, Flanagan SE, Ellard S, et al. Long-term follow-up of children with congenital hyperinsulinism on octreotide therapy. J Clin Endocrinol Metab. 2014;99(10):3660–7. doi: 10.1210/jc.2014-1866. [DOI] [PubMed] [Google Scholar]
- 63**.van der Steen I, van Albada ME, Mohnike K, Christesen HT, Empting S, Salomon-Estebanez M, et al. A Multicenter Experience with Long-Acting Somatostatin Analogues in Patients with Congenital Hyperinsulinism. Horm Res Paediatr. 2017 doi: 10.1159/000485184. This retrospective cohort study of 27 patients with HI across 6 European centers describes the effectiveness of long-acting somatostatin analogs and highlights the risk for increased liver enzymes. [DOI] [PubMed] [Google Scholar]
- 64.Senniappan S, Brown RE, Hussain K. Sirolimus in severe hyperinsulinemic hypoglycemia. N Engl J Med. 2014;370(25):2448–9. doi: 10.1056/NEJMc1404716. [DOI] [PubMed] [Google Scholar]
- 65*.Unal S, Gonulal D, Ucakturk A, Siyah Bilgin B, Flanagan SE, Gurbuz F, et al. A Novel Homozygous Mutation in the KCNJ11 Gene of a Neonate with Congenital Hyperinsulinism and Successful Management with Sirolimus. J Clin Res Pediatr Endocrinol. 2016;8(4):478–81. doi: 10.4274/jcrpe.2773. This report describes a neonate with a novel KCNJ11 mutation leading to diazoxide-unresponsive HI that was successfully treated with sirolimus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66**.Szymanowski M, Estebanez MS, Padidela R, Han B, Mosinska K, Stevens A, et al. mTOR Inhibitors for the Treatment of Severe Congenital Hyperinsulinism: Perspectives on Limited Therapeutic Success. J Clin Endocrinol Metab. 2016;101(12):4719–29. doi: 10.1210/jc.2016-2711. This observational cohort study of 10 children with severe HI treated with mTOR inhibitors details the limited efficacy and significant adverse effects possible with this therapy. [DOI] [PubMed] [Google Scholar]
- 67*.Banerjee I, De Leon D, Dunne MJ. Extreme caution on the use of sirolimus for the congenital hyperinsulinism in infancy patient. Orphanet J Rare Dis. 2017;12(1):70. doi: 10.1186/s13023-017-0621-5. This Letter to the Editor summarizes concerns related to use of sirolimus for HI in infancy. [DOI] [PMC free article] [PubMed] [Google Scholar]