

Abstract
Summary
We present POPdemog, an R package which converts coalescent simulation program input parameters into a visual representation of the demographic model. This package is useful for preparing figures, for checking that demographic simulation parameters have been correctly specified, and for understanding demographic models that other researchers have used to simulate genetic data. The POPdemog package supports the ms, msa, msHot, MaCS, msprime, scrm and Cosi2 programs, and includes options for customizing the output figures.
Availability and implementation
The POPdemog package and its tutorial can be freely downloaded from https://github.com/YingZhou001/POPdemog.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Many simulation tools have been developed to support population and evolutionary genetic studies, and many of these programs are able to generate genetic data under complex demographic models (Hoban et al., 2012). Often the demographic models include several populations with time-varying population sizes and multiple migration events. In this case, the large number of demographic parameters makes it difficult to understand the demographic model directly from the simulation program command line (see Box 1).
Box 1. MaCS command (Vernot et al., 2016) for modified Tennessen model (Tennessen et al., 2012):
macs 2025 15000000 -i 10 -r 3.0e-04 -t 0.00069 -T -I 4 10 1006 1008 1 0 -n 4 0.205 -n 1 58.00274 -n 2 70.041 -n 3 187.55 -eg 0.9e-10 1 482.46 -eg 1.0e-10 2 570.18 -eg 1.1e-10 3 720.23 -em 1.2e-10 1 2 0.731 -em 1.3e-10 2 1 0.731 -em 1.4e-10 3 1 0.2281 -em 1.5e-10 1 3 0.2281 -em 1.6e-10 2 3 0.9094 -em 1.7e-10 3 2 0.9094 -eg 0.007 1 0 -en 0.007001 1 1.98 -eg 0.007002 2 89.7668 -eg 0.007003 3 113.3896 -eG 0.031456 0 -en 0.031457 2 0.1412 -en 0.031458 3 0.07579 -eM 0.031459 0 -ej 0.03146 3 2 -en 0.0314601 2 0.2546 -em 0.0314602 2 1 4.386 -em 0.0314603 1 2 4.386 -eM 0.0697669 0 -ej 0.069767 2 1 -en 0.0697671 1 1.98 -en 0.2025 1 1 -ej 0.9575923 4 1 -em 0.06765 2 4 32 -em 0.06840 2 4 0
We have developed the POPdemog R package to capture the complex demographic information from simulation scripts or parameter files, and output the demographic model as a figure (Fig. 1). The main output figure shows the changes in population sizes and migrations events. When multiple migrations happen at a particular time, additional figures can be generated to give fine-scale representation of the overlapping migration events. Currently, POPdemog supports the input to the ms and msa (Hudson, 2002), msHot (Hellenthal and Stephens, 2007), MaCS (Chen et al., 2009), scrm (Staab et al., 2015), msprime (Kelleher et al., 2016) and Cosi2 (Shlyakhter et al., 2014) programs.
Fig. 1.
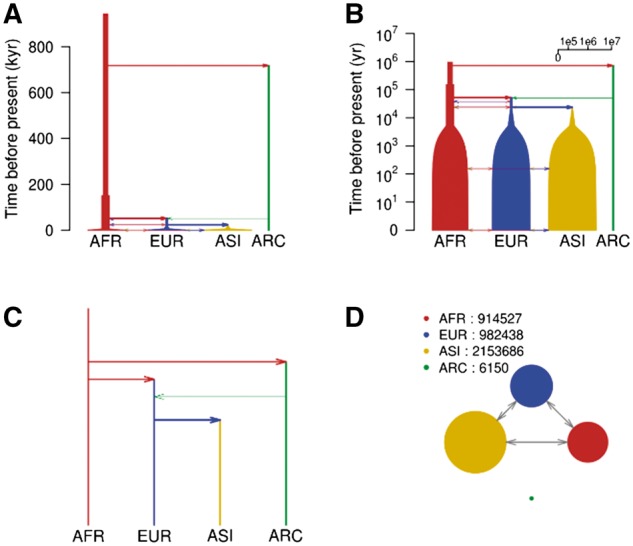
Visual representations of demography in MaCS simulation script for a modified Tennessen model (Box 1) (Vernot et al., 2016). AFR: Africans; EUR: Europeans; ASI: Asians; ARC: Archaic humans. (A) Use a time unit of 1000 years and scale the population size logarithmically. (B) Zoom in on the recent events and scale the population size logarithmically. (C) ‘topology’ of migrations with rates ≥ 0.001 per generation. (D) Migrations and population sizes 1000 years ago, with population sizes scaled linearly. A–C are generated by the PlotMS() function, D is generated by the PlotMig() function
2 Input and output
The input to our program is the command line or parameter file for invoking the simulation program, the simulation program name, and the baseline effective population size. Many coalescent-based simulation programs allow for arbitrary scaling of simulation parameters relative to an unspecified baseline effective population size; thus the effective population size is required by our program in order to adjust the scale. We also provide options to customize the appearance of the output figure such as the time scale, branch positions, branch colors and branch widths for each population. Options for controlling scaling of time and population size are described in Table 1. For example, setting size.scale = ‘log’ shows population sizes on a log scale, which is helpful when population size has undergone exponential growth (Fig. 1A), and setting time.scale = ‘log10year’ allows more detailed visualization of recent events (Fig. 1B). Setting size.scale = ‘topology’ gives an overview of the relationships among the simulated populations ignoring event times and population sizes (Fig. 1C), which permits demographic events to be displayed in time order even when the time between events is extremely small. There are also options for adjustment of branch positions which can minimize the number of crossings of migration arrows and population branches. One can also plot the population sizes and migrations at a particular point in time (Fig. 1D). The scripts used to run the POPdemog program and generate Figure 1 are provided in the Supplementary Information. The POPdemog program includes an online tutorial which contains examples for different simulators and demographic models, as well as an example showing how migration plots can be overlaid onto a world map.
Table 1.
Options for the parameters controlling scaling of population size and event time
| size.scale | ‘log’ | Scale the population size logarithmically, with the base parameter log.base [default] |
| ‘linear’ | Scale population size linearly, with scale parameter linear.scale | |
| ‘topology’ | Ignore population size and event times, output only the topological relationship | |
| time.scale | ‘4Ne’ | Set time unit to 4Ne [default] |
| ‘kyear’ | Set time unit to 1000 years | |
| ‘generation’ | Set time unit to generations | |
| ‘years’ | Set time unit to years | |
| ‘log10year’ | Scale time with function ‘’ |
Note: Further details can be found in the program documentation and the tutorial file.
Funding
Research reported in this publication was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Numbers R01HG005701 and R01HG008359. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest: none declared.
Supplementary Material
References
- Chen G.K. et al. (2009) Fast and flexible simulation of DNA sequence data. Genome Res., 19, 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellenthal G., Stephens M. (2007) msHOT: modifying Hudson’s ms simulator to incorporate crossover and gene conversion hotspots. Bioinformatics, 23, 520–521. [DOI] [PubMed] [Google Scholar]
- Hoban S. et al. (2012) Computer simulations: tools for population and evolutionary genetics. Nat. Rev. Genet., 13, 110–122. [DOI] [PubMed] [Google Scholar]
- Hudson R.R. (2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics, 18, 337–338. [DOI] [PubMed] [Google Scholar]
- Kelleher J. et al. (2016) Efficient Coalescent Simulation and Genealogical Analysis for Large Sample Sizes. PLoS Comput. Biol., 12, e1004842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyakhter I. et al. (2014) Cosi2: an efficient simulator of exact and approximate coalescent with selection. Bioinformatics, 30, 3427–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staab P.R. et al. (2015) Scrm: efficiently simulating long sequences using the approximated coalescent with recombination. Bioinformatics, 31, 1680–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen J.A. et al. (2012) Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science, 337, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernot B. et al. (2016) Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science, 352, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.