

Abstract
Motivation
Mass spectrometry (MS) based quantification of proteins/peptides has become a powerful tool in biological research with high sensitivity and throughput. The accuracy of quantification, however, has been problematic as not all peptides are suitable for quantification. Several methods and tools have been developed to identify peptides that response well in mass spectrometry and they are mainly based on predictive models, and rarely consider the linearity of the response curve, limiting the accuracy and applicability of the methods. An alternative solution is to select empirically superior peptides that offer satisfactory MS response intensity and linearity in a wide dynamic range of peptide concentration.
Results
We constructed a reference database for proteome quantification based on experimental mass spectrum response curves. The intensity and dynamic range of over 2 647 773 transitions from 121 318 peptides were obtained from a set of dilution experiments, covering 11 040 gene products. These transitions and peptides were evaluated and presented in a database named SCRIPT-MAP. We showed that the best-responder (BR) peptide approach for quantification based on SCRIPT-MAP database is robust, repeatable and accurate in proteome-scale protein quantification. This study provides a reference database as well as a peptides/transitions selection method for quantitative proteomics.
Availability and implementation
SCRIPT-MAP database is available at http://www.firmiana.org/responders/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Mass spectrometry-based protein quantification has become an increasingly popular method in biological research, providing high throughput and sensitivity (Bensimon et al., 2012; Picotti and Aebersold, 2012). MS1-based peptide extracted ion chromatograms (XIC) and MS2-based multiple reaction monitoring (MRM) are two approaches employed in quantitative proteomics (Addona et al., 2009; Mueller et al., 2008). The MS1-based quantification relies on accurate mass and peptide elution time, in which all peptides are detected and then aligned across several LC-MS/MS experiments to use all features of the peptides (Cox and Mann, 2008; Tsou et al., 2010). In the MS2-based quantification, in the case for MRM, the first quadrupole is used as a mass filter to selectively inject the target peptide ion, and one or several fragment ions generated by collision dissociation are sequentially observed by the detector over time, resulting in a chromatogram-like trace with retention time and signal intensity as coordinates (Anderson and Hunter, 2006; Picotti et al., 2009).
It is generally accepted that the kernel for accurate MS1 and MS2 quantification is the selection of peptides and transitions, which provide high signal intensity and low level of interfering signals. We previously reported that a successful MS1 absolute quantification largely depends on the characteristics of the tryptic peptides that are influenced by the extent of trypsin digestion of the protein, MS signal intensity, and the response linearity when the concentration of the peptide is varied (Ding et al., 2011). In the MS2-based quantification, fragment ions are used, the intensities of fragments derived from one precursor ion vary substantially (Lange et al., 2008). Several tools and databases were developed to facilitate target selection, particularly for MS2-based approaches (Deutsch et al., 2008; Martin et al., 2008; Mead et al., 2009; Sherwood et al., 2009). The SRMAtlas project led by Ruedi Aebersold’s team (Deutsch et al., 2008) is based on high-quality experimental proteome identification and is the most influential resource for selected/multiple reaction monitoring (SRM/MRM)-based proteomic workflow. The standard deviation of AUC (area under the curve) for marker ions can be obtained within the range of ∼10 to 30% when the selection rules are followed (Xie et al., 2011). Nevertheless, current proteomics quantification as a whole (Lange et al., 2008; Mann, 2006; Ong et al., 2002) is based on the premise that selected targets should display a linear response within the experimental range (Whiteaker et al., 2010) to ensure the accuracy of direct comparison of peptide/transition signals. This is not verified and the available quantification resources lack well-defined response curves and relative signal indices for peptide/transition sets on the proteome scale.
In this study, we constructed an experimental mass spectrum response curve database for both data dependent acquisition (DDA) and data independent acquisition in the form of SWATH, which includes absolute MS signal response curves for all measurable peptides and their fragment ion transitions from the tryptic digests of the HeLa cell line. MS measurements were carried out using up to 2.5 orders of magnitude of serial concentration dilution to cover a reasonable dynamic range. We extracted XIC data for MS1 quantification and utilized SWATH to acquire transition XIC data for MS2 quantification. The abundances and response curves of over 2 647 773 transitions from 121 318 peptides were calculated and stored in a database named SCRIPT-MAP (http://www.firmiana.org/responders), covering 11 040 gene products. A scoring algorithm incorporating MS intensity, correlation coefficient (R2), linear range, slope, and the detection limit in the low abundance range was developed to determine a quantitative index () for all identified peptides and transitions. Best responder peptides were chosen according to the indices and could be used for robust MS-based proteome quantification. The SCRIPT-MAP may advance the accuracy of quantitative proteomics in biological research.
2 Materials and methods
2.1 Sample preparation
For protein profiling, proteins from HeLa cells were extracted using 8 M urea and reduced using dithiothreitol for 4 h at 37°C followed by alkylation using iodoacetamide for 60 min at room temperature in the dark. Samples were digested using trypsin at a mass ratio of 1:50 enzyme/protein overnight at 37°C.
For better quantification of transcriptional factors (TFs), nuclear extracts were first enriched by catTFRE, a DNA affinity reagent that can effectively enrich endogenous transcription factors and co-regulators (Ding et al., 2013a). Briefly, biotinylated catTFRE DNA was immobilized on Dynabeads and then mixed with nuclear extracts (NE). The DNA-NE mixture was incubated for 2 h at 4°C; the protein-bound Dynabeads were washed twice with NETN and twice with PBS. Proteins were eluted by SDS-loading buffer and separated on SDS-PAGE. Peptides were prepared by in-gel digestion; the peptide concentration was estimated using Nano-Drop.
For small scale validation, 293T cell were lysed in the lysis buffer (50 mM NH4HCO3, 2% sodium deoxycholate, 25 mM NaCl, pH 8.5) and processed by reductive alkylation and trypsin digestion. A total of 10 routine experiments over a month period, as well as 5 serial dilution experiments (1000, 500, 250, 125 and 62.5 ng of 293T lysate) were analyzed.
Several human tissue samples were measured to demonstrate the utility of this method. Normal heart tissue or non-cancerous tissues adjacent to tumours were obtained from heart transplant operation or gastric, liver, lung cancer surgeries, respectively. This study was approved by the Ethics Committee of Beijing Proteome Research Center and was performed according to the Declaration of Helsinki Principles.
For isotope labelled QconCAT (Beynon et al., 2005) experiments, we chose 32 metabolic enzymes from selected metabolic pathways, including glycolysis, gluconeogenesis, TCA cycle, fatty acid degradation and lipid synthesis. The SCRIPT-MAP database was queried to select BR peptides to assemble a recombinant QconCAT protein. The selected BR peptides were reverse-translated to cDNA sequences, and the assembled cDNA fragment was synthesized and inserted into the pGEX-4T-2 vector for the expression of a GST-fusion protein. The transformed E. coli were cultured overnight in LB media containing ampicillin (100 μg/ml) and then expanded to SILAC D-MEM Flex-medium (Gibco®) containing ampicillin (100 μg/ml) without fetal bovine serum (Ding et al., 2011). Heavy arginine and heavy lysine were added to a final concentration of 100 μg/ml. Fusion proteins were purified by Glutathione Sepharose (Invitrogen) column and eluted with glutathione. The 13C labelling efficiency of the QconCAT was 99.1% for K, and 98.3% for R, respectively. The 13C labelled recombinant QconCAT protein containing BR peptides were spiked into human tissue samples and digested with trypsin together.
A dual-RPLC-MS/MS procedure was used for data acquisition. The first-dimension RP separation was performed on an L-3000 HPLC System (Rigol) employing a Durashell RP column (5 μm, 150 Å, 250 mm × 4.6 mm ID, Agela) with 2% acetonitrile as phase A and 98% acetonitrile as phase B at pH 10. Twenty-four fractions were collected with a gradient from 5 to 35% of phase B at a flow rate of 1 ml/min.
Fractions collected from the first RPLC were dissolved in HPLC loading buffer and were diluted in sequentially by 2-fold, resulting in the amount of 2000, 1000, 500, 250, 125, 62.5, 31.25, 15.63, 7.81, 3.91 ng of proteins/sample, respectively. The second dimension LC system was directly coupled to a hybrid Q-TOF (Triple-TOF 5600, AB SCIEX) MS for either DDA or SWATH data acquisition. The pre-column on AB 5600 was 5 μm, 300 Å, 2 cm × 100 μm ID; the analytical column was 3 μm, 120 Å, 15 cm × 75 μm ID. The mobile phase consisting of 0.1% formic acid in water (phase A) and 0.1% formic acid in acetonitrile (phase B) were run at a flow rate of 350 nl/min in 75 min.
2.2 MS data acquisition
Both data-dependent acquisition (DDA) and independent acquisition (SWATH) mode were applied to compare the different strategies. In the DDA mode, the source was operated at 2.5 kV and a survey MS scan range of m/z 350–1250. The top 50 precursor ions were selected in each MS scan for subsequent MS/MS scans. MS scans were acquired for 0.25 s, and the 50 MS/MS scans were acquired at 0.04 s each. The MS/MS dynamic exclusion was set at 12 s. The CID energy was automatically adjusted using the rolling CID function of Analyst TF 1.5.1. In the SWATH mode, the procedure was carried out as previously described (Gillet et al., 2012). Briefly, the source was operated at 2.5 kV and the MS1 scan range of m/z 350–1250. The MS2 SWATH scans recorded consecutive high-resolution fragment ion spectra of all peptides within a defined 25 Da precursor ion window during the LC separation. The Q1 scan range was set at 400–1000 m/z and the accumulation time of 100 ms for each individual MS2 scan. The total cycle time was 2.65 s (100 ms × 24 SWATH scans + 0.25 s MS1 scan).
In the validation experiments using the 293T lysate, a LTQ Orbitrap Velos mass spectrometer (Thermo Fisher) was used for quantification in the DDA mode. MS1 was scanned by Orbitrap at the resolution of 60 000, AGC target of 1 × 106, maximum ion injection time of 10 ms, in the scan range of 375–1300 m/z. MS2 was scanned by ion trap, with AGC target of 3 × 104, maximum ion injection time is 10 ms, isolation window is 3 m/z. The CID collision energy was 35%, with top 25 ions scanned by MS2 at dynamic exclusion set as 18 s. The XIC of peptides were extracted and calculated manually for abundance determination.
2.3 Data processing
Wiff files from the Triple-TOF 5600 were first searched using ProteinPilot version 4.2 in the Paragon search engine against the human ref-sequence protein database (version 2013.07, 36 229 entries). The false discovery rate (FDR) was set at 1% at the protein level. For peptide XIC extraction, we first transferred the Wiff data to an mzXML-formatted file and then loaded the identification results for MS1 feature references. We extracted XICs of identified peptides by searching against the MS1 results based on the peptide identification information, and estimated the abundance by the area under the extracted XIC curve. By using peptide cross-assignment, we could quantify a peptide identified only once within all LC-MS/MS trials in the dilution set. The PeakView® software with a SWATH plug-in (AB SCIEX) was used for transition XIC extraction. Information obtained in the DDA mode, including RT, m/z and rank patterns of transition intensity of the peptides, were used as a reference map. Transition XICs were extracted accordingly from data-independent scans of SWATH mode results.
The non-redundant peptide list was used to assemble proteins by applying the parsimony principle (Yang et al., 2004). Differentiable, distinct and equivalent proteins with more than one unique peptide were retained. Protein abundances were then estimated using the iBAQ algorithm. MS results from serial dilutions were transferred into SCRIPT-MAP, an in-house MySQL-based relational database.
3 Results
3.1 Data acquisition for best responder database
We previously reported the development of a fast proteome sequencing strategy (Ding et al., 2013b) that enabled us to identify and quantify more than 100 000 non-redundant peptides (representing 8000 gene products) in half a day. This highly efficient approach allows for proteome-wide quantitative serial-dilution screening. We chose HeLa cells as proteome source because they are widely used in research community and could serve as a more representative proteome system. Whole cell extracts were trypsinized and pre-fractionated into 24 fractions using high-pH reverse phase liquid chromatography (RP-LC). Multiple (1:1 to 1:512) serial dilution MS measurements were carried out and analyzed using both DDA and SWATH data acquisition modes (Fig. 1A and B), resulting in 480 MS runs (24 fractions × 10 dilutions × 2 data acquisition modes) and a total measurement time of 240 h. A total of more than 4 800 000 high-resolution fragmentation spectra were submitted to the paragon search engine, resulting in 95 658 non-redundant peptides representing 10 249 gene products. In order to increase the proteome coverage, we also employed the catTFRE (Ding et al., 2013a) approach to enrich low abundance endogenous DNA-binding proteins (transcription factors and transcription co-regulators). MS measurements were carried out on 10 dilutions with 6 fractions following catTFRE pull-down, resulting in a total of 120 MS runs. As a result, 7870 proteins, 41 490 peptides and 264 036 transitions were identified, in which 1442 proteins, 25 660 peptides and 144 864 transitions were exclusively identified by the catTFRE approach (Table 1). All identified peptide ions and fragment ions were analyzed and imported into an in-house database for data integration and processing. A total of 19 815 proteins from 11 040 gene products which covered 322 KEGG pathways were identified and quantified (Fig. 1C).
Fig. 1.
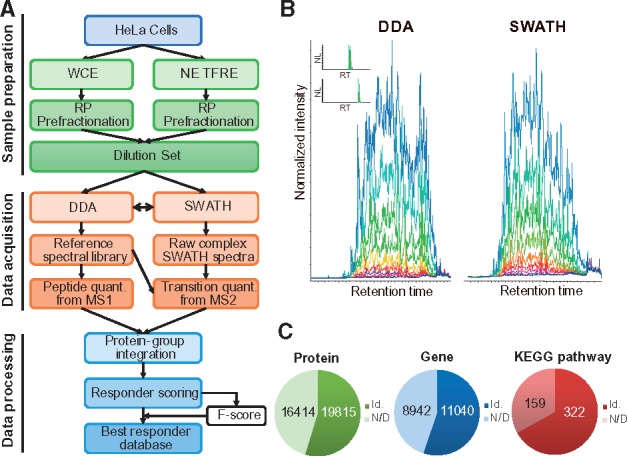
An overview of the experimental workflow. (A) A flowchart of the experimental data acquisition. (B) To increase the accuracy and coverage, both DDA and SWATH data acquisition modes were performed. (C) Number of proteins, genes and KEGG pathways quantified. All identified peptide ions and fragment ions were analyzed and imported into an in-house database for data integration and processing. Id: identified; N/D: not detected
Table 1.
Overview of dataset/database content
| WCE | TFRE | TFRE exclusively | WCE + TFRE | NCBI | KEGG | KEGG coverage | |
|---|---|---|---|---|---|---|---|
| Transition | 2 502 909 | 264 036 | 144 864 | 2 647 773 | |||
| Peptide | 95 658 | 41 490 | 25 660 | 121 318 | |||
| Protein (GI) | 18 373 | 7870 | 1442 | 19 815 | 36 229 | ||
| Gene Symbol | 10 249 | 5049 | 791 | 11 040 | 19 982 | 6816 | 4151/6816 |
| KEGG Pathway | 322 | 301 | 0 | 322 | 481 | 481 | 322/481 |
Note: NCBI GI version: human 2013.07, KEGG version: 2017.12.
3.2 A comprehensive evaluation of MS response curves
We gathered all identified peptides and transitions, and evaluated their MS response curves as x-y graphs of area under the curve (AUC) against loading amount of protein in the set of serial dilution experiments. R2 (correlation between measured AUC against loading amounts), slope and linear range were calculated for each plot to determine the linear performance of the peptides and transitions. Generally, best responders exhibited the best R2 values, widest linear range and lowest MS detection limit. We manually examined the peptide/transition response curves and optimized a formula to match their quantitative features. While slope is an important parameter that measures the response of AUC to the amount variation, using slope alone did not always find the ideal responders (Fig. 2A), R2 and the linear range needed to be considered, too. While combining slope with R2 could be more effective, but simple multiplication did not offer better result than using slope or R2 alone (Fig. 2B). Furthermore, the lower limit of the linear range needed to be considered so that low abundant peptides could be quantified. We operationally defined the linear range as the longest concentration range that yields R2 > 0.9025 (0.95 × 0.95) in the linear regression. We found that superior linear responders could be found based on a Quantification Factors () score (Fig. 2C) using the formula (1):
| (1) |
Fig. 2.
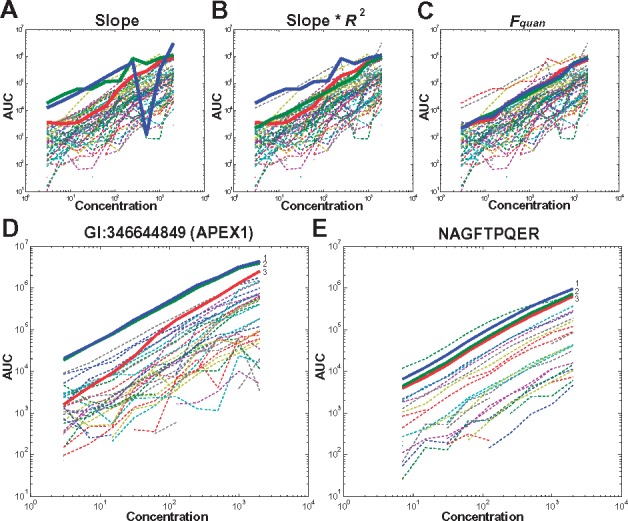
Quantification characteristics of using different parameters. An example from protein GI 51873036 (gene ODGH) when (A) ranked based only on slope. The linearity of the top three peptides with the largest slope (coloured in blue, green and red) is poor. (B) Rank based on slope multiplied by R2. (C) Rank based on , a combination of slope, R2, linear range and the detection limit. (D) An example of best responder peptides of protein GI 346644849 (APEX1) chosen by . (E) An example of best responder transitions from the peptide NAGFTPQER of protein GI 346644849. All of the corresponding peptides/transitions were plotted and the best three linear examples are coloured in blue, green and red, respectively
The quantification factors for peptides and transitions were designated as and , respectively. We then manually evaluated over 1000 peptides and transitions for the effectiveness of the scoring formula. A set of comparison figures were listed in Figure 2D and E, as well as in Supplementary Figure S1.
For a better understanding of how physicochemical properties of the peptides may make them as best responder peptides, we analyzed the distribution of the with respect to parameters such as peptide length, retention time (RT), modification, m/z and charge, and analyzed the distribution of the with respect to parameters such as ion type and amino acid type at the CID fragment site. As shown in Supplementary Figure S2, the features for best responder peptides include: (a) a moderate hydrophobicity (elution at 15–25 min during a 40-min gradient); (b) 8–12 amino acids in length; (c) m/z in the range of 500–750 Da; and (d) doubly charged. For transitions, fragment ions from y4 to y10 made up over 50% of the best-transition responders.
We hosted the data on an in-house MySQL-based relational database SCRIPT-MAP (http://www.firmiana.org/responders/). This database provides experimental linear MS response curves of peptides and fragment ions (transitions) at a proteome-wide scale. It includes over 19 815 proteins, 121 318 unique peptides and 2 647 773 fragment ions with 1% FDR at protein level, representing a dataset with quantitative linear information with the deepest proteome coverage. In the database, the linear curve of each identified peptide and its transitions are plotted against the actual amount of protein-loading. For selected proteins and peptides, an overview plot or check-selection plot of the corresponding peptides and transitions are also available. A 5-level hierarchy, including pathway, gene, protein, peptide and transition, is integrated and presented. Users can either browse peptide/transition response curves in the ‘View’ panel, or search for interested Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways or genes using the search panel. Searching for pathways or genes in a batch is supported. There is also a filter for further selection, and result can also be downloaded (Supplementary Fig. S3).
3.3 Evaluation of quantification accuracy and versatility
In order to evaluate the quantification accuracy based on the BR method, we compared proteome quantification by the BR method and the iBAQ method. We performed MS analyses of whole cell extracts of HeLa in triplicate, then employing iBAQ or BR method to quantify proteins. In the BR approach, protein abundances were calculated by BR peptides indexed in the SCRIPT-MAP database. Correlation coefficients for the repeated triplicates were compared between these two quantification approaches. The R2 values based on the top 1 to top 3 best-responder peptides were significantly higher than those obtained using the iBAQ, top peptides only based on the slope, or slope*R2 (Fig. 3B). Significantly lower CV values were also observed using the BR approach (Fig. 3A), demonstrating that quantification using BRs indexed in SCRIPT-MAP database resulted in the best reproducibility and accuracy among these methods. We calculated CV values for peptides and proteins and ranked them in the order of their scores indexed in SCRIPT-MAP. Higher-ranked responders displayed lower CV (Supplementary Fig. S4).
Fig. 3.

Comparison of the best-responder peptide approach with iBAQ. (A) Relatively lower CVs were obtained using the BR peptide approach. Peptides were scored and ranked, then the CV was calculated for the top one, top two, top three and lowest ranked peptides. Random1 indicates a random selection of one of the ranked peptides. Furthermore, CV of top ranked peptides by slope, R2, as well as slope*R2 were also calculated. The box plot combined with the violin plot depicts the density distribution of CVs of three repeat experiments. (B) R2 values for proteome quantification based on top one to three peptides by the BR approach in three repeats were significantly higher than those obtained using the iBAQ algorithm, and better than the results by the top peptides only based on slope, R2, or slope*R2
We then evaluated the accuracy of quantification using SCRIPT-MAP by measuring the abundance of Universal Proteome Standard (UPS2)—a proteomics dynamic range standard set (obtained from SIGMA®) containing 48 proteins covering 6 orders of magnitude in abundance. We chose the first ranked BR of the UPS2 proteins for quantification. If there were cleavages or modifications for the first ranked BR, then the second ranked BR without cleavages or modifications was chosen. We also chose the first ranked peptide calculated from the method PREGO (Searle et al., 2015) for comparison of quantification. The result showed that the correlation between the calculated amount and the actual amount based on SCRIPT-MAP (R2 = 0.908) was higher than those calculated by the MaxQuant iBAQ (R2 = 0.886), or the PREGO (R2 = 0.857) (Supplementary Fig. S5), demonstrating that the BR method is more accurate.
As the BRs were obtained by measuring HeLa proteins, we tested whether the method can be used to quantify 293T proteins. Better quantification results were obtained using the BR method than the iBAQ method when quantifying the 293T proteome using BR peptides generated from the HeLa proteins (Fig. 4A). For low abundant proteins, the BR method was also superior to the iBAQ (Fig. 4B).
Fig. 4.
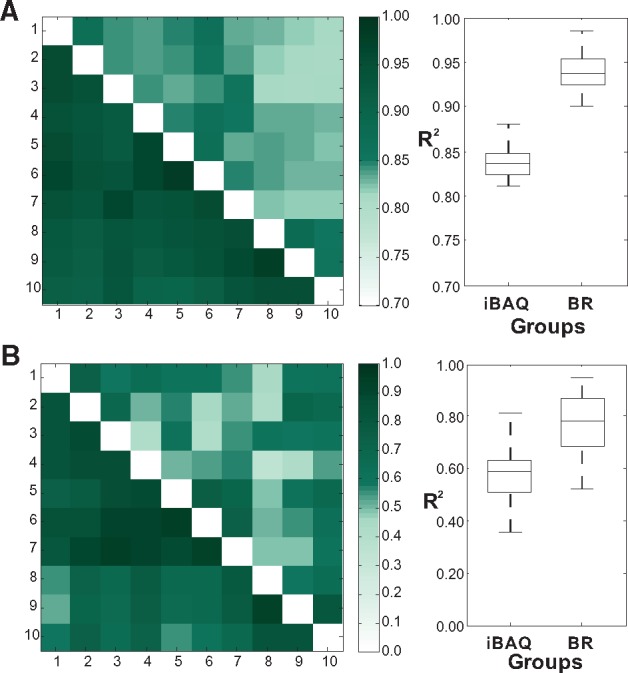
Comparison of quantification consistency of best-responder peptides with iBAQ using 293T proteins. (A) Heatmap illustrates the pairwise correlation coefficient (R2) of quantifications of all proteins between each pair of experiments. The upper triangular part of heatmap is the coefficient by iBAQ, and the lower triangular part is the coefficient by best responder peptides method. Darker colours reflect higher coefficient of experiments. Box plot illustrates the distribution between the two groups. (B) Heatmap illustrate the pairwise correlation coefficient (R2) of quantifications of lowest-abundance proteins between each pair of experiments. Box plot illustrates the distribution between the two groups
As we obtained the BR peptides using the QTOF type of instrument, we tested whether BR peptides are instrument dependent. We carried out a set of 293T dilution experiments using the Velos Orbitrap instrument and scored the peptides by the Fquan_293T equation. Then we matched the identified peptides with the SCRIPT-MAP database and tracked back Fquan scores indexed in the SCRIPT-MAP database to evaluate the consistency of SCRIPT-MAP database and 293T dilution experiments. The result showed that 76.6% of the highest ranked peptides by score based on the SCRIPT-MAP database in 293T dilution experiments were ranked as top three peptides based on calculated (level 1); 6.8% were ranked as top 50% but not in top 3 (level 2). The rest of peptides (16.6%) have relatively poor rank (level 3). And the 2nd and 3rd ranked peptides by scores of 293T dilution experiments have a total of 75 and 69% good ranked peptides (level 1 + 2) based on calculated (Supplementary Fig. S6A). According to the scores, ∼84% of the top ranked peptides have excellent linearity (R ≥ 0.9) in the 293T dilution experiments; and over 90% of the top ranked peptides have good linearity (R ≥ 0.8) (Supplementary Fig. S6B). The evaluations suggested that the BR approach indexed in the SCRIPT-MAP is independent of protein source and mass spectrometer type, and has higher reliability, repeatability and accuracy in proteome-scale quantification than the iBAQ method.
3.4 An example of quantification of selected metabolic pathway proteins
We used proteins in metabolic pathways as a proof-of-principle experiment for protein quantification using the BR method. We selected BR peptides and synthesized a QconCAT protein covering 32 metabolic proteins (Supplementary Table S1), including proteins from the TCA cycle, lipid metabolism and glucose metabolism. The QconCAT protein was expressed and labelled in SILAC medium. Comparison of the QconCAT protein with a purified recombinant protein ZSCAN21 showed that AUCs of the QconCAT peptides displayed a 3-fold variation at most, while peptides from ZSCAN21 were dispersed over 4 orders of magnitude (Supplementary Table S2, Supplementary Fig. S7), demonstrating BR peptides from different proteins displayed similar MS response.
As the QconCAT proteins were isotope labelled, we used them to determine the stoichiometry of enzymes in metabolic pathways and to compare protein abundances in human heart, liver, lung and stomach (Fig. 5 and Supplementary Table S3). Interestingly, profound difference in abundance was observed in these organs, with 18 out of the 32 proteins exhibiting significant differences between organs (P < 0.05, n = 3). Even in the highly conserved TCA cycle, five out of the seven proteins varied considerably across the four organs. As a major organ for lipid metabolism, lipid catalysis enzymes such as FASN, CPT1A, ACSS2 and ACAT2 were more abundant in liver than in other organs, while in the heart, proteins in the glycolysis pathway (PFKP, ALDOA) and TCA (DLST, OGDH and CS) were more abundant, reflecting the requirement for intensive glucose consumption and energy usage in the heart. Despite of being generally considered as abundant proteins, several key enzymes were expressed at low levels in some tissues. For instance, FASN was expressed at very low level in the heart; pyruvate kinase (PKM) was much less abundant in the liver than in other tissues. Interestingly, the abundance of the TCA cycle enzymes were different: MDH2, CS, FH and ACO1 were more abundant than others (IDH3A, OGDH and DLST) (Fig. 5). For comparison, we also obtained protein expression levels of the 32 metabolic proteins from The Human Protein Atlas (Uhlen et al., 2015) (https://www.proteinatlas.org/), and marked them in the colourmap in Figure 5. In general, the quantitative results obtained by the QconCAT method were consistent with those of the Human Protein Atlas database. Low-expression proteins detected by QconCAT method, such as PFKFB2 and BPGM, were generally marked as low or not detected in Human Protein Atlas database. Both methods showed GPAM was relatively higher in liver, ENO1 was relatively lower in heart and PFKP was relatively lower in liver. Our stoichiometric measurement revealed selective enhancement of different metabolic pathways in each of the organs.
Fig. 5.

Using QconCAT to determine stoichiometry in metabolic pathways in human liver, heart, stomach and lung. Expression intensities for organs and conditions are colour-coded
4 Discussion
As the specific mass spectrometry signal response of different tryptic peptides from the same protein can differ by as much as 100-fold in intensity (Picotti et al., 2007) and a similar situation also exists in the signal response of transitions from the same peptide in MRM measurements, the consensus in the field of proteome quantification has been that the selection of suitable quantification peptides strongly influences or even entirely determines the accuracy of the quantification (Picotti and Aebersold, 2012), In our previous study (Ding et al., 2013a,b), we reported that the errors in quantification came from at least two major sources: (i) a false discovery rate (FDR) in identification that cannot be avoided, and (ii) inaccuracy in quantification that may be reduced by selecting suitable quantification peptides and transitions. We therefore constructed an experimental database entitled SCRIPT-MAP to facilitate accurate MS1 and MS2 based quantification. SCRIPT-MAP (http://www.firmiana.org/responders) enables the exploration of MS response curves for precursor and fragment ion. Comprehensive quantification scores for precursor and fragment ions have been compiled, which provides a reference library of BR peptides/transitions in proteome measurement. We ranked the peptides of proteins using our algorithm and calculated the CV of the responders. The order of the CV is consistent with the ranking score of the peptides, demonstrating the practicality of our procedure. This method empirically identify the peptides that response well in MS as well as show linear response as a function of peptide concentration to achieve more accurate proteome quantification than the commonly used iBAQ method.
We show that BR peptides in the SCRIPT-MAP database are protein source independent and largely MS instrument independent, this will allow the widest use of the method independent of MS platforms. Using the BR peptides will achieve better relative quantification than the iBAQ method. Combining with the QconCAT approach may allow for absolute quantification and even protein stoichiometry measurement as we demonstrated in the paper, in which we shown that enzymes in the major metabolic pathways in heart, liver, lung and stomach have extensive variations in stoichiometry in the four organs, which may reflect the differences in functions. Furthermore, the hard choice how to pick peptides in the QconCAT design has been solved by our BR peptides in the SCRIPT-MAP database. The BR peptides tend to be excellent choice for the QconCAT design.
While the current version of SCRIPT-MAP database covers quantification peptides and transitions of over 10 000 gene products, it is still a work in progress. Linear MS response curves for global peptides and transitions from more cell lines and species are being incorporated. As additional data are generated, the dataset will undergo the same quality control procedures as described in this study and the SCRIPT-MAP database will receive continuous original data support and updates. Our goal is to expand the repertoire of SCRIPT-MAP database to the limit of the proteome coverage, and eventually develop an accurate proteome quantification method.
Supplementary Material
Acknowledgements
We thank Dr. Jie Ma for advice on protein quantification, and Dr. Tao Chen for advice on web server development.
Funding
This work has been supported by International Science & Technology Cooperation Program of China (2014DFB30010; 2014DFA33160; 2012DFB30080); National Program on Key Basic Research Project (973 Program, 2014CBA02000; 2012CB910300); National Key Research and Development Program of China (2017YFA0505102 and 2017YFC0908404); National High-tech R&D Program of China (863 program, 2012AA020201); National Natural Science Foundation of China (31200582; 31170779; 31200992; 31270822; 31770886 and 31700682); Beijing Natural Science Foundation (5132012; Z131100005213003) and Shanghai Municipal Science and Technology Major Project (2017SHZDZX01).
Conflict of Interest: none declared.
References
- Addona J.Q. et al. (2019) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol., 27, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L., Hunter C.L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics, 5, 573–588. [DOI] [PubMed] [Google Scholar]
- Bensimon A. et al. (2012) Mass spectrometry-based proteomics and network biology. Annu. Rev. Biochem., 81, 379–405. [DOI] [PubMed] [Google Scholar]
- Beynon R.J. et al. (2005) Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods, 2, 587–589. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol., 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Deutsch E.W. et al. (2008) PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep., 9, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C. et al. (2013a) Proteome-wide profiling of activated transcription factors with a concatenated tandem array of transcription factor response elements. Proc. Natl. Acad. Sci. USA, 110, 6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C. et al. (2013b) A fast workflow for identification and quantification of proteomes. Mol. Cell. Proteomics, 12: 2370–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C. et al. (2011) Quantitative analysis of cohesin complex stoichiometry and SMC3 modification-dependent protein interactions. J. Proteome Res., 10, 3652–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet L.C. et al. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics, 11, O111 016717.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange V. et al. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol., 4, 222.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M. (2006) Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell. Biol., 7, 952–958. [DOI] [PubMed] [Google Scholar]
- Martin D.B. et al. (2008) MRMer, an interactive open source and cross-platform system for data extraction and visualization of multiple reaction monitoring experiments. Mol. Cell. Proteomics, 7, 2270–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead J.A. et al. (2009) MRMaid, the web-based tool for designing multiple reaction monitoring (MRM) transitions. Mol. Cell. Proteomics, 8, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller L.N. et al. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res., 7, 51–61. [DOI] [PubMed] [Google Scholar]
- Ong S.E. et al. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Picotti P., Aebersold R. (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods, 9, 555–566. [DOI] [PubMed] [Google Scholar]
- Picotti P. et al. (2007) The implications of proteolytic background for shotgun proteomics. Mol. Cell. Proteomics, 6, 1589–1598. [DOI] [PubMed] [Google Scholar]
- Picotti P. et al. (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell, 138, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle B.C. et al. (2015) Using data independent acquisition (DIA) to model high-responding peptides for targeted proteomics experiments. Mol. Cell. Proteomics, 14, 2331–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood C.A. et al. (2009) MaRiMba: a software application for spectral library-based MRM transition list assembly. J. Proteome Res., 8, 4396–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou C.C. et al. (2010) IDEAL-Q, an automated tool for label-free quantitation analysis using an efficient peptide alignment approach and spectral data validation. Mol. Cell. Proteomics, 9, 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M. et al. (2015) Proteomics. Tissue-based map of the human proteome. Science (New York, N.Y.), 347, 1274.. [DOI] [PubMed] [Google Scholar]
- Whiteaker J.R. et al. (2010) An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics, 9, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F. et al. (2011) Liquid chromatography-mass spectrometry-based quantitative proteomics. J. Biol. Chem., 286, 25443–25449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. et al. (2004) DBParser: web-based software for shotgun proteomic data analyses. J. Proteome Res., 3, 1002–1008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.