

Summary
Loss of the CDKN2A tumor suppressor is associated with melanoma metastasis, but the mechanisms connecting the phenomena are unknown. Using CRISPR-Cas9 to engineer a cellular model of melanoma initiation from primary human melanocytes, we discovered that a lineage restricted transcription factor, BRN2, is downstream of CDKN2A and directly regulated by E2F1. In a cohort of melanocytic tumors that capture distinct progression stages, we observed that CDKN2A loss coincides with both the onset of invasive behavior and increased BRN2 expression. Loss of the CDKN2A protein product p16INK4A permitted metastatic dissemination of human melanoma lines in mice, a phenotype rescued by inhibition of BRN2. These results demonstrate a mechanism by which CDKN2A suppresses the initiation of melanoma invasion through inhibition of BRN2.
In Brief
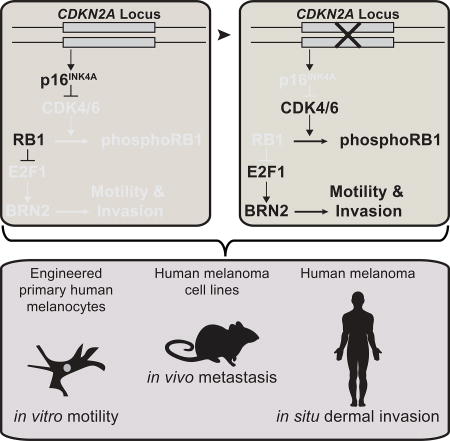
Zeng et al. find that complete CDKN2A loss coincides with the onset of invasiveness in melanocytic tumors at distinct progression stages. pl6INK4A, encoded by CDKN2A, inhibits E2F1-mediated transcriptional activation of BRN2, a transcription factor that has been associated with melanocytic invasive programs.
Introduction
Melanoma is a skin cancer that initiates from pigment-producing cells called melanocytes and progresses in a step-wise fashion(Bastian, 2014). Early lesions are curable through excision, but once melanomas become invasive, the 5-year patient survival rate drops precipitously from over 95% for patients with early stages of invasive melanoma to less than 30% for patients with metastatic disease(Gershenwald et al., 2017; Kunwor et al., 2017). The acquisition of invasive behavior therefore marks a critical transition during melanoma progression. The most common acquired genetic change distinguishing precursor lesions, such as melanocytic nevi or melanoma in situ (MIS), from invasive melanomas is loss of the CDKN2A locus(Shain et al., 2015). Over 75% of melanoma metastases have lost one or both alleles of this tumor suppressor (TCGA Research Network). Likewise, the most common somatic mutations associated with familial melanoma also disrupt the CDKN2A locus(Eliason et al., 2006; FitzGerald et al., 1996; Hussussian et al., 1994). In genetic mouse models, disruption of CDKN2A in conjunction with other oncogenic mutations promotes melanoma formation and metastases(Ackermann et al., 2005; Damsky et al., 2015; Dhomen et al., 2009; Krimpenfort et al., 2001). Despite this abundance of genetic data supporting a role for CDKN2A in suppressing melanoma initiation, the mechanism connecting loss of this locus to invasive behavior is uncharacterized.
The CDKN2A locus encodes two gene products - p14ARF and p16INK4A – each under transcriptional regulation by independent promoters and each with distinct tumor suppressive functions(Sharpless and DePinho, 1999). Studies in human genetics and mouse modeling have demonstrated that p16INK4A is the predominant suppressor of melanoma progression(Bennett, 2016; Goldstein et al., 2006; Hussussian et al., 1994; Kamb et al., 1994; Krimpenfort et al., 2001; Sviderskaya et al., 2003). The p16INK4A protein is a cyclin-dependent kinase (CDK) inhibitor, which binds to CDK4 and CDK6 and prevents the phosphorylation of retinoblastoma protein (RB1)(Rubin, 2013; Serrano et al., 1993). In its hypophosphorylated state RB1 directly binds to and inhibits E2F family transcription factors, prohibiting cell cycle progression(Chellappan et al., 1991; van den Heuvel and Dyson, 2008).
Due to its known function in cell cycle inhibition, p16INK4A is hypothesized to suppress melanoma progression by inducing cellular senescence downstream of BRAFV600E, the oncogene most commonly associated with nevi and melanoma(Bennett, 2016; Michaloglou et al., 2005; Zhao et al., 2016). However, several lines of evidence are inconsistent with this model. In vitro, small interfering (si)RNA-mediated knock-down of p16INK4A fails to rescue BRAFV600E-induced growth arrest(Haferkamp et al., 2009; Halaban et al., 1998; McNeal et al., 2015; Michaloglou et al., 2005). In vivo, mice engineered to express BRAFV600E in melanocytes still form nevus-like lesions even on a p16INK4A- or CDKN2A- null background(Damsky et al., 2015; Dhomen et al., 2009). In situ, immuno-histochemical analyses have consistently demonstrated that expression of p16INK4A is retained in proliferating MIS melanocytes, and that germline CDKN2A mutations do not alter the proliferation rate of nevi(Florell et al., 2002; Funk et al., 1998; Keller-Melchior et al., 1998; Reed et al., 1995; Sparrow et al., 1998; Talve et al., 1997; Wang et al., 1996). In contrast, the CDKN2A locus is most commonly lost concurrently with the transition into invasive melanoma(Reed et al., 1995; Shain et al., 2015). In aggregate, these observations suggest there is additional complexity governing transformation of nevi and the mechanisms by which CDKN2A and p16INK4A inhibit melanoma initiation.
Here, we sought to determine the oncogenic behaviors induced by CDKN2A loss in both BRAF wild-type and BRAFV600E human melanocytes and to characterize the downstream mechanism by which CDKN2A loss contributes to melanoma invasion.
Results
Targeted engineering of primary human melanocytes
Recent advances in genome editing using CRISPR/Cas9-mediated homology directed repair (HDR) have made possible the precise engineering of primary human cells (Hsu et al., 2014). To explore the consequence of CDKN2A loss in a model that retains both the cell type and genome representative of the early stages of melanoma progression, we harnessed CRISPR-Cas9-mediated HDR to edit the endogenous loci of primary human melanocytes. In human melanomas, focal deletions and point mutations in CDKN2A most commonly affect exon 2(Sviderskaya et al., 2003). We therefore designed a strategy to engineer a focal replacement of CDKN2A exon 2 with a CMV-promoter driven EGFP (Figure 1A). Candidate sgRNAs for targeting exon 2 were designed and introduced in combination with Cas9 into normal human melanocytes (NHMs) (Figure S1A) using an optimized electroporation protocol. The Cas9 cut the expected genome sequence with high efficiency and without notable effects on the top predicted off-target sites (Figure S1B). We used a double nickase (dCas9) cutting strategy to further reduce off-target cutting(Chen et al., 2013; Ran et al., 2013). We introduced plasmids expressing each sgRNA and dCas9 in combination with a double-stranded template for HDR containing 2 kb homology arms. Three weeks after transfection 8–17% of NHMs remained EGFP-positive, whereas control cells that received only the HDR template were EGFP-negative (Figure 1B). EGFP-expressing NHMs were isolated via flow cytometric cell sorting (FACS) and compared to sibling NHMs with an intact CDKN2A locus (Figure 1C). Polyclonal EGFP-expressing NHMs exhibited the correct genotype for successful knock-in (Figure S1C) and the corresponding loss of p16INK4A and p14ARF expression, whereas expression of p15INK4B from the CDKN2B locus was retained (Figure 1D–E). To further validate the specificity of our editing, we expanded three single cell clones and performed targeted genome sequencing of over 500 cancer genes. In each clone, there was only one fully-clonal genetic alteration, a bi-allelic CDKN2A deletion (Figure 1F). Other alterations were present in only small fractions of the clonal populations, implicating CDKN2A editing as the only genetic change induced by the engineering event. These data demonstrate that our optimized strategy for engineering locus-specific changes in NHMs is both tractable and precise.
Figure 1. Engineering of CDKN2A null melanocytes.

(A) Strategy for introducing focal knock-in of CMV-EGFP to replace exon 2 of CDKN2A.
(B) Percentage of EGFP-positive NHMs three weeks after editing the CDKN2A locus. Data represent mean, 25th and 75th percentiles, minimum and maximum values of eight CRISPR reactions on three preparations of NHMs.
(C) Strategy for isolating CDKN2A null NHMs for comparison to wild-type siblings cells. Directly after transfection, both Cas9− and Cas9+ conditions contain EGFP+ cells (top). Over time, Cas9− cells become uniformly EGFP−, and the remaining EGFP+ cells in the Cas9+ population are isolated (middle). Isolated cells are stably EGFP+ and compared to matched EGFP- cells (bottom).
(D) Western blot detection of both CDKN2A protein products (p16INK4A and p14ARF, left) and CDKN2B protein product (p15INK4B, right) in isolated EGFP+ and EGFP− NHM populations.
(E) Immunofluorescent visualization of p16INK4A expression in EGFP+ and matched EGFP− NHMs.
(F) Targeted sequencing of three clonal EGFP+ populations to monitor for off-target Cas9 cutting. Graphed are the mutant allele frequencies of deletions (Del) and point mutations (PM) in intergenic, coding and intronic regions.
NHM1–3 refers to independent primary melanocyte derivations and engineering events.
See also Figure S1.
p16INK4A suppresses melanocyte motility and invasion
We next sought to characterize the oncogenic contributions of CDKN2A loss in primary NHMs. CDKN2A deletions were engineered into low-passage neonatal NHMs, as above, and compared to CDKN2A wild-type sibling cells. To compare cell behavior in the two sorted populations, we utilized a platform of digital holographic cytometry that permits simultaneous quantification of cell division, death, senescence, and motility of adherent cells over long-term culture (Figure 2A)(Hejna et al., 2017). Compared to wild-type sibling cells, CDKN2A null NHMs proliferated more rapidly and exhibited increased longevity in culture, consistent with previous observations (Figure S2A–B)(Obata et al., 1997; Sviderskaya et al., 2003; Sviderskaya et al., 2002). However, despite increased longevity, CDKN2A null NHMs were not immortalized and possessed reduced telomere length before undergoing growth-arrest after 60–70 doublings (Figure S2B–C). Growth-arrest could be rapidly induced through transduction of BRAFV600E cDNA as previously reported (Figure S2D)(Haferkamp et al., 2009; Kumar et al., 2014; Lu et al., 2016; Michaloglou et al., 2005). Interestingly, the percentages of cells that underwent BRAFV600E-induced growth-arrest were comparable in both CDKN2A null and wild-type cells suggesting BRAFV600E overexpression can induce growth arrest in a CDKN2A independent manner. Neither population presented notable occurrences of cell death or markers of pre-apoptosis (Figure S2E).
Figure 2. CDKN2A loss promotes melanocyte motility and invasion.

(A) Schematic of experimental set-up. For each experiment, NHMs are derived from donated tissue and engineered for CDKN2A loss as in Figure 1. After isolation, CDKN2A null and wild-type sibling cells are monitored via digital holographic cytometry for 72 hr. The rate of cell division, motility, morphology, growth arrest and detachment are quantified. Representative holographic phase shift images (bottom left) show colored comet tails tracking cells.
(B) Single cell motility analysis of CDKN2A null NHMs compared to wild-type siblings. Box and whisker plot represents mean, 10th, 25th,75th and 90th percentiles of at least fifty cells each from three CRISPR reactions of three preparations of NHMs.
(C) Quantification as in (B) but excluding cells that divided during the 72 hr time period surrounding 24 hr of analysis.
(D and E) Time-lapsed holographic phase shift images (D) and quantification (E) of NHMs migrating into scratched region. Yellow line indicates scratch limit at first time point. Error bars represent standard deviation of the mean of three CRISPR reactions of three preparations of NHMs.
(F) Quantification of transwell invasion assays where CDKN2A null NHMs and wild-type siblings were required to migrate through high-density basement membrane extract. Box and whisker plots represent mean, 25th and 75th percentiles, minimum and maximum values of five CRISPR reactions of three preparations of NHMs.
(G) Single cell motility analysis of CDKN2A null NHMs transduced with and sorted for the indicated vectors. Box and whisker plot represents mean, 10th, 25th, 75th and 90th percentiles of at least fifty cells each from three transductions.
Asterisks indicate p value of * <0.05 to ***** <0.000005 from unpaired t-test. N.S. indicates no significant difference.
See also Figure S2.
In addition to the expected effect of CDKN2A loss on cell proliferation, another interesting phenotype emerged from our digital holographic analysis. CDKN2A null NHMs were significantly more motile compared to wild-type sibling cells (Figure 2B). To account for movement associated with cell division, actively dividing cells were excluded from the analysis, and the increased motility remained significant (Figure 2C). Corroborating these observations, CDKN2A null NHMs also exhibited enhanced migration in live-imaged scratch assays (Figure 2D–E) and enhanced invasion across high-density basement membrane extracts in transwell assays (Figure 2F). The increased motility of CDKN2A null NHMs was independent of the expression level, intracellular location or identity of the fluorescent protein used in the engineering (Figure S2F–G). To verify the phenotype was the consequence of loss of a CDKN2A gene product, we transduced CDKN2A null NHMs with either p14ARF or p16INK4A cDNA. Expression of p16INK4A reverted the motility phenotype, whereas expression of p14ARF did not (Figure 2G). These data demonstrate that the p16INK4A product encoded by the CDKN2A locus inhibits cellular motility in primary NHMs.
To determine whether CDKN2A loss alters melanocyte motility in the presence of melanoma driver mutations, we first engineered wild-type or CDKN2A null NHMs to harbor the BRAFV600E point mutation. We transfected ribonucleoprotein (RNP) complexes containing Cas9 and sgRNA targeting exon 15 of the BRAF locus (Figure 3A & Figure S3A–B). We designed a single-stranded (ss)DNA HDR template to introduce the point mutation encoding V600E along with three silent mutations that suppress subsequent re-binding of the sgRNA (Figure 3A). To detect mutant BRAF, we developed a digital droplet quantitative (dd-q)PCR assay with 100 percent specificity for the V600E allele and sensitivity down to 0.1 percent mutant allele frequency (MAF) (Figure S3C–E). Three weeks after co-transfection of the RNP with the HDR template into NHMs, 5–9% of the BRAF sequences in the culture were mutant (Figure S3F). The MAF of the engineered NHMs increased rapidly in standard culture conditions over two months (Figure S3F). The resulting populations were ~80% heterozygous for BRAFV600E (Figure S3G), expressed the BRAFV600E protein, and demonstrated the concomitant increase in MAPK signaling (Figure S3H). The selective growth advantage of cells harboring the mutant allele was reverted by treatment with the BRAFV600E-specific inhibitor vemurafenib, indicating dependence on expression of the mutant protein (Figure S3F).
Figure 3. p16INK4A regulates motility independent of driver mutation.

(A) Strategy for introducing BRAFV600E point mutation. Center of HDR template is shown (blue) with point mutations causing intended codon change (orange) or silent mutations disrupting sgRNA binding (green) highlighted. sgRNA sequence shown in purple.
(B) Western blot detection of BRAFV600E and p16INK4A expression and ERK phosphorylation in engineered NHMs.
(C) Population motility analysis of NHMs engineered with BRAFV600E and/or CDKN2A deletion. Error bars represent standard deviation of the mean of three experiments.
(D) Comparison of motility in ten melanoma lines with different genetic backgrounds. Lines were classified by activating driver mutation (BRAF, NRAS, CKIT or Wild-type) and CDKN2A status. CDKN2A status was designated as either wildtype and expressed (Wild-type), mono-allelic disruption and expressed (Heterozygous), bi-allelic disruption (Homozygous), or wild-type and silenced (Silenced). Box and whisker plot represents mean, 10th, 25th, 75th and 90th percentiles of forty cells per line.
(E) Effect of EF1a-driven p16INK4A expression on motility in CDKN2A null or silenced melanoma lines (left). Effect of shRNA-mediated p16INK4A knockdown on motility in p16INK4A expressing melanoma lines (right). Error bars represent standard deviation of the mean of forty cells per condition.
(F) Comparison of the effect of p16INK4A loss on proliferation and motility in ten melanoma lines. Red dots represent comparison of shRNA-mediated p16INK4A knock-down normalized to vector controls in p16INK4A expressing melanoma lines. Green dots represent comparison of vector controls normalized to EF1a-driven p16INK4A expression in CDKN2A null or silenced lines. No significant correlation between proliferation and motility was observed.
Asterisks indicate p value of ** <0.005 to **** <0.00005 from unpaired t-test. N.S. indicates no significant difference.
See also Figure S3.
After engineering the BRAFV600E mutation into CDKN2A null and wild-type melanocytes (Figure 3B), we observed that CDKN2A loss induced motility independent of either BRAF status or treatment with vemurafenib (Figure 3C). To further investigate the pervasiveness of p16INK4A-regulated motility, we obtained ten human melanoma cells lines representing different driver mutations and CDKN2A states (Figure 3D & Figure S3I). CDKN2A null or silenced lines were more motile than lines with p16INK4A expression. Similar to the engineered NHMs, the enhanced motilities of lines that did not express p16INK4A were reverted upon ectopic expression of p16INK4A (Figure 3E & Figure S3J). Concordantly, motility was increased in all CDKN2A-expressing lines upon targeted p16INK4A knockdown. In contrast, the manipulation of p16INK4A did not consistently alter proliferation across each line (Figure 3F). These data demonstrate that regulation of cellular motility by p16INK4A occurs independent of the diver mutation and is not constitutively tied to cellular proliferation.
The acquisition of invasive behavior is a critical bottleneck to the series of events that occur during metastatic dissemination of melanoma. Previous reports have connected CDKN2A loss with metastatic dissemination in mouse model systems, but the mechanism has been attributed to indirect effects of p16INK4A on cell cycle regulation or p14ARF on p53-mediated apoptosis(Ackermann et al., 2005; Aguirre et al., 2003; Krimpenfort et al., 2001; Pavey et al., 2002). Since our data connect loss of CDKN2A (specifically p16INK4A) with the acquisition of invasive behavior, we chose two lines from our panel to investigate whether p16INK4A loss could directly affect the outcome of an in vivo assay for metastatic dissemination, independent of its inhibitory role on proliferation.
The WM793 melanoma line was derived from a BRAFV600E – mutant early stage primary melanoma and is immortalized, but does not metastasize when injected subcutaneously into immune-compromised mice(Herlyn et al., 1985). In contrast, its derivative line, 1205Lu, disseminates to the lung(Juhasz et al., 1993). To investigate genetic differences between the non-metastatic WM793 cell line and its metastatic derivative line 1205Lu, we performed targeted sequencing and copy number analysis. Known pathogenic alterations were shared between both cell lines, confirming their common ancestry, but each also harbored private point mutations and copy number alterations, most of which were not considered drivers (Figure 4A–C). However, the metastatic 1205Lu cell line harbored a bi-allelic deletion and associated loss of p16INK4A expression, while the WM793 cell line retained a copy of the CDKN2A locus and expression of p16INK4A (Figure 4B & Figure S4A).
Figure 4. p16INK4A loss induces metastasis in WM793 melanoma line.

(A) A 500 gene panel, comprised of established cancer genes, was used to sequence the WM793 and 1205Lu cell lines. Point mutations are stratified by their mutant allele frequencies (MAFs) in each cell line as shown. Likely pathogenic mutations are highlighted in red. Data points along the diagonal represent mutations shared between the cell lines, and data points along either axis represent private mutations.
(B) Copy number variation between WM793 and 1205Lu cell lines inferred from sequencing depth (top). A focal heterozygous (bottom left, WM793) and homozygous (bottom right, 1205Lu) deletion affecting CDKN2A is highlighted in red.
(C) Phylogenic tree representation of the genetic relationship between the primary WM793 and the derivative metastatic 1205Lu cell lines.
(D) Schematic of experimental setup for metastasis assay. WM793 cells transduced with either pSICOR-mCherry or pSICOR-shp16INK4A-mCherry or 1205Lu cells transduced with either pHIV-zsGreen or pHIV-p16INK4A-IRES-zsGreen were sorted for fluorescent protein expression and injected subcutaneously into NSG mice (n=5 per condition). Primary tumor volume was monitored and lung metastasis was analyzed after five weeks. Representative H&E stained lung section shows metastasized lesion (right). Red scale bars = 100 µm.
(E) Representative lung sections with immunofluorescent stain for human nuclear antigen (HNA). Red scale bars = 100 µm.
(F) Quantification of IF images from (E), showing the number of HNA positive cells in the lung for each condition (each group represents 100 quantified sections over 5 mice). Box and whisker plots represent mean, 10th, 25th, 75th & 90th percentiles.
(G) PCR amplification of genomic DNA from mouse lungs was performed using human (top) or mouse (bottom) specific primers to detect micro-metastases. All lobes were tested and a representative experiment is shown.
(H) Quantification of human specific DNA in murine lung across experiments. Values are absolute concentration of human DNA per sample measured via quantitative PCR. Data represent biopsies from 20 lung lobes over 5 mice per condition. Horizontal bars represent mean.
(I) Volume of primary tumors for all mice. Error bars represent standard deviation of the mean.
Asterisks indicate p value of ** <0.005 and **** <0.00005 from unpaired t-test. N.S. indicates no significant difference.
See also Figure S4.
To assess the effect of p16INK4A expression on metastatic potential, derivative lines of 1205Lu and WM793 that respectively added back and knocked down p16INK4A were developed (Figure S4B–E). Derivative lines were injected subcutaneously into the flanks of immune-comprised mice, and lungs were harvested for analysis after 5 weeks (Figure 4D). Metastasis formation was abolished in the p16INK4A-expressing 1205Lu line, and conversely, WM793 cells acquired the ability to metastasize to the lung upon p16INK4A knockdown (Figure 4E–H & Figure S4F–H). Despite the increase in metastatic potential upon p16INK4A knockdown, the growth rate of the primary tumor at the animals’ flank remained low, and we observed no correlation between in vitro or in vivo proliferation rates and metastasis (Figure 4I & Figure S4I–K). These data demonstrate that metastatic potential can be gained through spontaneous or targeted loss of p16INK4A via a mechanism that is uncoupled from its regulation of proliferation.
p16INK4A loss permits BRN2 transcription
To investigate the mechanism by which p16INK4A regulates melanocyte motility, we transcriptionally profiled three matched CDKN2A null and control NHM populations using RNA sequencing. Among the fifteen most significantly up-regulated genes in the CDKN2A null lines was the transcript, POU3F2 (BRN2), which encodes an established regulator of melanocyte and melanoma invasion (Figure 5A) (Fane et al., 2017; Hoek and Goding, 2010; Pinner et al., 2009). BRN2 protein levels inversely correlated with p16INK4A expression and were associated with higher RB1-phosphorylation across three independently derived and engineered NHM populations (Figure 5B). To determine whether increased BRN2 expression was responsible for the increased motility caused by p16INK4A loss, we monitored CDKN2A null NHMs transfected with siRNA targeting BRN2 and observed reversion of the motility phenotype without altering proliferation (Figure 5C–D). A similar reduction in motility was observed in 1205Lu cells upon BRN2 knockdown (Figure 5E). We also generated 1205Lu cells stably expressing shRNA targeting BRN2 and observed a significant reduction in their potential to metastasize to the lung after subcutaneous injection (Figure 5F). These data demonstrate that p16INK4A loss enhances the motility and invasive potential of melanocytic cells through increased expression of BRN2.
Figure 5. CDKN2A loss induces transcriptional activation of BRN2.

(A) Volcano plot comparing transcriptomes of three independently derived pairs of CDKN2A null NHMs and matched wild-type sibling cells. Differentially expressed transcripts with q-values < 0.05 and log2 fold change > 1.7 are highlighted in aqua. The BRN2 transcript is highlighted in red.
(B) Western blot comparing BRN2, RB1, phospho-RB1, CDK4 and p16INK4A expression in three independently derived sets of CDKN2A null engineered NHMs and wild-type sibling cells.
(C) Motility of CDKN2A null NHMs after transfection with siBRN2 (top) and representative western blot validation of BRN2 knock-down (bottom). At least fifty cells each from three CRISPR reactions of three preparations of NHMs were quantified.
(D) Total cell number of the populations analyzed in (C), three days after transfection, relative to one day after transfection. Error bars represent standard deviation of the mean.
(E) Motility of 1205Lu melanoma cells after transfection with siBRN2 (top) and representative western blot validation of BRN2 knockdown (bottom).
(F) Quantification of human specific DNA in murine lungs comparing 1205Lu cells expressing either pLKO.1-shControl or pLKO.1-shBRN2 (top) and representative western blot validation of BRN2 knockdown (bottom). Values are absolute concentration of human DNA per sample. Data represent biopsies from 25 lung lobes over 5 mice per condition.
All box and whisker plots represent mean, 10th, 25th, 75th & 90th percentiles. Asterisks indicate p value of ** <0.005 and *** <0.0005 from unpaired t-test. N.S. indicates no significant difference.
To determine whether p16INK4A loss and BRN2 expression are associated with acquisition of invasive behavior during the normal course of human melanoma progression, we analyzed a cohort of 82 lesions containing either single stages of melanoma progression, two adjacent regions representing distinct stages of melanoma progression, or in rare cases, three progression stages (Shain et al., 2018). In total 31 nevus regions, 18 MIS regions, and 55 invasive melanoma regions were micro-dissected for genetic analysis. The status of the CDKN2A locus was analyzed in each region and compared to a publically available database of sequenced melanoma metastases (TCGA). We observed that bi-allelic inactivation of CDKN2A most frequently occurred at the transition from MIS to invasive melanoma, consistent with our previous observations (Figure 6A)(Shain et al., 2015). We analyzed mRNA expression in a subset of the transition cases with sufficient material to isolate RNA. BRN2 mRNA levels were significantly increased in invasive melanomas compared to nevi (Figure 6B). Further, there was a correlation between loss of CDKN2A and increased expression of BRN2 mRNA (Figure 6C). In the majority of cases presenting bi-allelic inactivation, at least one allele presented a focal deletion or point mutation affecting exclusively CDKN2A, implicating loss of this gene, and not adjacent genes, in permitting the transition (Figure 6C, hashtags). Interestingly, the fraction of invasive melanomas that possessed bi-allelic CDKN2A loss remained constant when stratified as thin melanoma, thick melanoma, and distal metastases, indicating that loss of the second allele arises early in melanoma invasion and does not undergo further selection in subsequent stages of progression (Figure 6A).
Figure 6. CDKN2A / p16INK4A loss is associated with invasive melanoma and BRN2 gain.

(A) Fraction of nevi or melanomas with mono- or bi-allelic CDKN2A disruption. Melanomas are sub-categorized as melanoma in situ (MIS), thin invasive melanoma (Stage T1), thick invasive melanoma (Stage T2+) and distal metastatic melanoma (obtained from the TCGA public database).
(B) Comparison of BRN2 mRNA levels in regions of nevus (n=12) and melanoma (n=19). Box and whisker plots represent mean, 25th and 75th percentiles, minimum and maximum values.
(C) p16INK4A and BRN2 mRNA expression from next generation sequencing of microdissected transition cases. Cases where at least one allele contained a focal deletion or point mutation that affected exclusively the CDKN2A gene are designated with a hashtag (#).
(D) Representative transition lesion where precursor MIS and descendent invasive components are distinguishable. Dual p16INK4A (brown) and melanocyte marker MelanA (pink) immunohistochemistry (top) and H&E staining (bottom) are shown.
(E and F) Shown are serial sections stained for H&E, MelanA/ p16INK4A, MelanA/ Ki67, and BRN2 from representative MIS from nevus (E) and melanoma from MIS (F) regions. Dotted lines indicate borders of MIS (yellow), nevus (blue) and invasive melanoma (green) regions. Yellow arrowheads highlight MelanA and p16INK4A positive cells in the MIS regions.
(G) Quantification of the percentage of nevus, MIS, and melanoma cases that stained positive for indicated proteins by IHC.
See also Figure S5.
To determine protein expression, we established an independent cohort of melanomas with different progression stages (42 regions) for immunohistochemistry (IHC) (Figure 6D–G). Sixty percent of invasive melanomas had completely lost p16INK4A expression, corroborating our genetic analysis and prior studies (Figure 6F&G)(Funk et al., 1998; Keller-Melchior et al., 1998; Pavey et al., 2002; Reed et al., 1995; Sparrow et al., 1998; Talve et al., 1997; Wang et al., 1996). In contrast, p16INK4A was rarely lost in melanomas in situ, which are nevertheless proliferative, as indicated by Ki67-positive immunostaining (Figure 6D–G). The expression pattern of BRN2 was inverse to that of p16INK4A (Figure 6E–G & Figure S5A and S5B). Invasive melanoma presented BRN2-positive cells in localized mosaic patterns that were rarely observed in the precursor regions (Figure 6F). These tissue analyses thus corroborate our RNA expression analysis, NHM engineering, and in vivo metastasis assay and validate that p16INK4A loss and increased BRN2 expression accompany the transition into invasive melanoma.
BRN2 is a direct target of E2F1
To determine how p16INK4A loss is mechanistically linked to increased expression of BRN2 mRNA, we analyzed the transcriptional changes that occurred upon CDKN2A loss, using both our engineered in vitro system and our database of sequenced progression stages. We specifically searched for changes in known transcription factor target networks. We identified E2F1 targets as one of the most consistently enriched networks (Figure 7A). E2F1 activation occurs downstream of p16INK4A loss through hyperphosphorylation of RB1 by CDK4/6(Hara et al., 1996). To investigate the regulation of BRN2 by the CDK4/6-RB1-E2F1 pathway, we engineered a luciferase construct to report POU3F2 promoter activity in NHMs. We demonstrated that CDKN2A loss increases transcriptional activity of the POU3F2 promoter and BRN2 mRNA levels, and that this activity was reversed in a dose-dependent manner by a small molecule inhibitor of CDK4/6 (Figure 7B–C). Similarly, direct targeting of E2F1 using siRNA also reduced luciferase expression and BRN2 mRNA levels (Figure 7D & Figure S6A). Consistent with these observations, treatment with either a small molecule inhibitor of CDK4/6 or siE2F1 resulted in loss of BRN2 protein expression and reversion of the enhanced motility phenotype (Figure 7E–F & Figure S6B). We observed a similar relationship between CDK4/6 or E2F1 inhibition and reduction in BRN2 protein in most CDKN2A null melanoma cell lines (Figure S6C–E). Overall, these data demonstrate that BRN2 transcriptional activation is downstream of both CDK4/6 and E2F1.
Figure 7. p16INK4A loss transcriptionally activates BRN2 through an E2F1-mediated pathway.

(A) Gene set enrichment analysis of three CDKN2A null NHM lines compared to three CDKN2A wild-type NHM lines (Engineered NHMs) or CDKN2A wild-type regions compared to CDKN2A null regions of the clinical cohort (Transition case cohort). # indicates both the NOM p value and FDR q-values for the enrichment score < 0.0005.
(B) Expression of firefly luciferase under the control of the POU3F2 promoter normalized to constitutively expressed renilla luciferase in CDKN2A null NHMs (n=10) and wild-type sibling cells (n=6) treated with PBS or indicated concentrations of the CDK4/6 inhibitor PD 0332991 for 24 hr. Box and whisker plots represent mean, 25th and 75th percentiles, minimum and maximum values.
(C) RT-qPCR quantification of BRN2 mRNA levels in CDKN2A null NHMs (n=9) and wild-type sibling cells (n=6) treated with PBS and CDK4/6 inhibitor for 24 hr. Box and whisker plots represent mean, 25th and 75th percentiles, minimum and maximum values.
(D) Expression of firefly luciferase under the control of the POU3F2 promoter normalized to constitutively expressed renilla luciferase in CDKN2A null NHMs and wild-type sibling cells transfected with control siRNA (siCon) or E2F1 siRNA. Box and whisker plots represent mean, 25th and 75th percentiles, minimum and maximum values of eleven experiments.
(E) Motility (top) and western blot validation (bottom) of CDKN2A null NHMs after treatment with PBS or 10 µM of CDK4/6 inhibitor PD 0332991 for 24 hr. At least fifty cells each from three CRISPR reactions of three preparations of NHMs were quantified. Box and whisker plots represent mean, 10th, 25th, 75th & 90th percentiles.
(F) Motility (top) and western blot validation (bottom) of CDKN2A null NHMs after transfection with siE2F1. At least fifty cells each from three CRISPR reactions of three preparations of NHMs were quantified. Box and whisker plots represent mean, 10th, 25th, 75th & 90th percentiles.
(G) Schematic of BRN2 transcriptional start site (TSS, red) and 0 to 500 bp upstream sequence. Design of PCR products for ChIP experiments (blue horizontal lines) and predicted E2F1 binding sites (orange vertical lines) are indicated.
(H) ChIP-qPCR detection of E2F1 binding to the POU3F2 promoter region. Relative amplification of primer sets indicated in (G) precipitated by either anti-E2F1 or anti-IgG antibodies are plotted as percent of input. Error bars represent standard deviation of the mean of four experiments.
(I) Representative images of co-immunofluorescent visualization of p-RB1 and BRN2 in invasive melanoma tissue.
(J) Single cell quantification of nuclear p-RB1 intensity in eight cases stained as in (I). Cells are stratified as either BRN2 positive (n=103) or negative (n=3877). Box and whisker plots represent mean, 25th and 75th percentiles, minimum and maximum values.
Asterisks indicate p value of * <0.05 to **** <0.00005 from unpaired t-test (panels B–H) or Mann-Whitney test (panel J) See also Figure S6.
We identified four predicted E2F1 binding motifs within the POU3F2 promoter and performed chromatin immunoprecipitation with an anti-E2F1 antibody to determine whether the transcription factor directly bound to the POU3F2 promoter (Figure 7G). Endogenous E2F1 precipitated with the genomic POU3F2 promoter sequence surrounding both predicted binding sites, specifically in human melanoma cell lines, but interestingly not in the MCF10A human breast cell line (Figure 7H). These data demonstrate that E2F1 directly binds the POU3F2 promoter in human melanoma cells.
To confirm whether the induction of BRN2 expression by the CDK4/6-RB1-E2F1 pathway can be observed during the progression of melanoma, we performed co-immunofluorescence visualization of S807/811 phosphorylated RB1 and BRN2. Consistent with IHC, nuclear BRN2 expression was found in invasive melanomas, in a mosaic pattern (Figure 7I). Phosphorylated RB1 presented the expected punctate sub-nuclear localization pattern(Szekely et al., 1991). There was a significant positive correlation between RB1 hyperphosphorylation and BRN2 expression (Figure 7J & Figure S6F). All together, these data demonstrate that BRN2 is transcriptionally activated by p16INK4A loss through direct binding by E2F1, as a consequence of CDK4/6-mediated hyperphosphorylation of RB1.
Discussion
Here, we report a previously uncharacterized mechanism by which CDKN2A loss contributes to melanoma progression. Utilizing precision engineering and digital holographic cytometry, we monitored the direct effect of genetic CDKN2A disruption on low-passage primary human melanocytes. We observed that loss of p16INK4A promotes melanocyte motility and the invasive and metastatic capacity of melanoma cells through the transcriptional activation of BRN2, a transcription factor previously associated with melanocytic invasive programs during both development and disease (Besch and Berking, 2014; Fane et al., 2017; Hoek and Goding, 2010; Pinner et al., 2009; Reed et al., 1995). These observations complement previous studies linking p16INK4A to cellular motility via cytoskeletal and metabolic intermediates (Fahraeus and Lane, 1999; Tyagi et al., 2017). Thus the critical role of CDKN2A as a melanoma tumor suppressor gene involves not only the canonical mechanism of 16INK4A preventing cell cycle entry and that of p14ARF inducing the p53 pathway, but additionally includes the suppression of melanocyte migration. Loss of CDKN2A, and consequently p16INK4A, occurs in approximately half of invasive melanomas, implicating CDKN2A loss with induction of BRN2 as a common, but not ubiquitous, mechanism for melanoma initiation.
The regulation of BRN2 transcription is complex. The MAPK, PI3K, and WNT signaling pathways are known to converge on the POU3F2 promoter (Cook and Sturm, 2008). It is therefore likely that melanomas can evolve distinct mechanisms for deregulating this critical gene. Indeed, the expression of BRN2 in melanoma cell lines, which are representative of more advanced disease, is more variable than in normal melanocytes. Nonetheless, our study demonstrates that in primary human melanocytes BRN2 expression is directly repressed by the p16INK4A-RB1 pathway. These data suggest that regulation of the POU3F2 promoter can oscillate with the cell cycle. Consistent with this hypothesis of cyclical expression, BRN2 IHC staining in invasive melanomas exhibits a mosaic pattern. We also observed that E2F1 binding to the POU3F2 promoter was specific to the melanocyte lineage, suggesting that either gain or loss of a second co-factor or a specific epigenetic state is required for promoter activation. RB1 has also been shown to bind both MITF and TBX2, in addition to E2F1, indicative of a complex link between the cell cycle and lineage-restricted transcriptional networks (Carreira et al., 2005; Halaban, 2005; Vance et al., 2010).
Loss of the CDKN2A locus has been previously demonstrated to be a common genetic alteration in melanoma progression. The locus is corrupted, primarily by homozygous deletions, in the majority of melanoma cell lines and melanomas (Curtin et al., 2005; Fountain et al., 1992; Reed et al., 1995). In genetically engineered mouse models, the in vivo introduction of BRAFV600E into mouse melanocytes induces the formation of intradermal melanocytic lesions that are stably growth-arrested, similar to nevi in humans(Dankort et al., 2009; Dhomen et al., 2009). When crossed onto a CDKN2A- or p16INK4A-null background these mice develop melanomas at increased rates, although most of the nevi remain growth arrested, consistent with our observations that CDKN2A suppresses melanoma initiation but does not mediate BRAFV600E-induced growth arrest(Damsky et al., 2015; Dankort et al., 2009). Interestingly, unlike most human melanomas, which typically go through a melanoma in situ stage confined to the epidermis, the neoplastic transformation in these mouse models occurs in the dermis. These models thereby implicate other mechanisms of CDKN2A-driven tumor suppression in addition to the inhibition of dermal invasion.
In this study, we link CDKN2A loss to the propensity of neoplastic BRAF-mutant melanocytes to invade from the epidermis and colonize the dermis. We provide direct evidence that this ability is promoted by increased cellular motility that results from CDKN2A loss. However, the invasive phenotype not only requires migration of neoplastic melanocytes from the epidermis into the subjacent dermis, but also requires their ability to survive in this environment. Future investigations should focus on whether additional tumor suppressive functions associated with CDKN2A loss beyond motility contribute to all aspects of invasion and subsequent survival and proliferation in the dermis, or whether other factors are involved. Nonetheless, here we observed that the timing of CDKN2A loss coincides with the step-wise transformation from melanoma in situ to invasive melanoma and that the enhanced invasive potential is explained in part by BRN2-mediated melanocyte motility.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
- EXPERIMENTAL MODEL AND SUBJECT DETAILS
-
◦Mouse model
-
◦Engineered primary normal human melanocytes
-
◦Human melanoma cell lines o Human melanocytic neoplasia (FFPE)
-
◦
- METHOD DETAILS
-
◦Analysis of clinical specimens and sequence
-
■Histopathologic Assessment
-
■Genomic Analysis
-
■Transcriptomic analysis
-
■
-
◦Cell culture and analysis
-
■Primary melanocyte isolation
-
■Lentivirus transduction
-
■Transfection
-
■Flow cytometric analysis
-
■Holographic imaging and microscopy
-
■Invasion analyses
-
■Mycoplasma detection
-
■
-
◦NHM engineering
-
■Introduction of BRAFV600E point mutation
-
■Introduction of CDKN2A knock-in
-
■
-
◦Lung metastasis assay
-
◦Molecular Biology
-
■Western blot analysis
-
■RT-qPCR
-
■Human genomic DNA qPCR
-
■ChIP
-
■Digital droplet qPCR
-
■Telomere length assay
-
■Plasmid construction
-
■
-
◦
QUANTIFICATION AND STATISTICAL ANALYSIS
- DATA AND SOFTWARE AVAILABILITY
-
◦Data Resources
-
◦
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents maybe directed to, and will be fulfilled by the corresponding author Robert L. Judson (robert.judson@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse model
All protocols described in this and other sections regarding animal studies were approved by the UCSF Institutional Animal Care and Use Committee. Ethical endpoint for tumor transplantation experiments was reached when a tumor was 2.5 cm or more in any single dimension. Transduced WM793 and 1205Lu cells were sorted and subcutaneously injected (8 × 106 cells) into immuno-deficient female mice (NOD.Cg-Prkdcscid I12rgtmlWjl/SzJ) aged 6~8 weeks by a technician blinded to the identity of the cells. Each week the mice were monitored for tumor size using a digital caliper and recorded with the guidance of the Helen Diller Comprehensive Cancer Center Preclinical Core (UCSF). 5 weeks post injection the lungs were perfused with 4% PFA, and mice were humanely sacrificed. A piece of each lobe was harvested for obtaining genomic DNA and the remaining lobes were incubated in sucrose overnight to be used for immunofluorescence and H&E staining.
Engineered primary normal human melanocytes
This study was approved by the UCSF human research protection program, and all tissues were collected in accordance with the institutional review board with regard to informed consent. Primary melanocytes isolated from normal human foreskin were engineered with either endogenous BRAFV600E mutations, CDKN2A deletion, or a combination of both alterations.
Human melanoma cell lines
The WM793 and 1205Lu melanoma lines were gifted by the Herlyn lab; WM1366, WM3670 and SK-MEL2 and were gifted by the Ortiz Lab. WM793 and 1205Lu were grown in DMEM H-16 supplemented with 10% FBS (Corning, 35-010-CV), 0.1 mg/mL Pen-Strep, and 0.002 mg/mL L-Glutamine at 37°C with 5% C02. M230, M368, WM1366, 501Mel, SK-MEL5, A375, WM3670 and SK-MEL2 were grown in RPMI 1640 supplemented with 10% FBS, 0.1 mg/mL Pen-Strep, and 0.002 mg/mL L-Glutamine. 293T and MCF10A cells were grown in DMEM H-21 supplemented with 10% FBS and 0.1 mg/mL Pen-Strep.
Human melanocytic neoplasia (FFPE)
This study was approved by the UCSF human research protection program, and all tissues were collected in accordance with the institutional review board with regard to informed consent. Two cohorts of transitional lesions were analyzed in this study. A transitional lesion is a lesion with two or more histopathologically distinct areas – for example, a portion of benign nevus and a portion of invasive melanoma; a portion of melanoma in situ and a portion of melanoma; or a portion of melanoma in situ and a portion of nevus. Transitional lesions include nevus, melanoma in situ, invasive thin melanoma (Tl melanoma) and invasive thick melanoma (T2 melanoma or higher). By analyzing transitional lesions we could perform inter- and intra- tumoral comparisons in order to identify the stage at which CDKN2A loss occurred both within individual tumors and across many tumors. In addition, transitional lesions permitted comparison of relative protein expression between progression stages using IHC.
The first cohort of transitional lesions comprised 82 cases for which we performed DNA sequencing to determine the timing of CDKN2A somatic alterations during melanoma progression (related to Figure 6A and discussed in more detail by Shain, et al (Shain et al., 2018). This cohort spanned a broad spectrum of histopathologically distinct areas, including 31 nevus areas, 18 melanoma in situ areas, 23 thin invasive melanoma areas (stage Tl), and 32 thick invasive melanoma areas (stage T2+). The second cohort of transitional lesions was comprised of 23 cases for which we utilized Melan-A, BRN2, Ki67 and pl6INK4A immunostaining in order to confirm the timing of pl6INK4A loss at the protein level and its relationship to other BRN2 and Ki67 (related to Figure 6D–G, Figure 7I–J and Figure S5). This cohort had a more focused spectrum of histopathologic areas, encompassing nevi associated with melanoma in situ or melanoma in situ associated with invasive melanoma. Cases from both cohorts were retrieved from the UCSF Dermatopathology archive as formalin-fixed paraffin-embedded tissue (FFPE) blocks.
METHOD DETAILS
Analysis of clinical specimens and sequence
Histopathologic Assessment
Protein staining was performed by the UCSF Dermatopathology and Oral Pathology Service and the UCSF Diller Cancer Center Biospecimen and Biostatistics Core. Protein levels were classified as either “expressed” or “not expressed” based upon the presence or absence of visual IHC signal (related to Figure 6 and Figure S5). In adjacent regions of transitional lesions, relative pl6INK4A levels were further classified depending on whether IHC signal was visible in both regions but uniformly less in one (“Partial loss”) or visible in one region but uniformly absent in the other (“Complete loss”) (related to Figure S5).
Genomic Analysis
For genomic analysis, distinct areas were manually microdissected with a scalpel and dissection scope from a series of 10–20 µm unstained sections following the guidance of a pathologist. In a subset of 31 of these transitional lesions with sufficient material to isolate both DNA and RNA, we also performed RNA-sequencing to identify the relationship between CDKN2A loss and pl6INK4A or BRN2 expression (related to Figure 6B–C). Sequencing was performed by the UCSF Clinical Cancer Genomics Laboratory. Genomic DNA was sequenced as previously described(Shain et al., 2015). Briefly, 25–250 ng of genomic DNA was prepared for sequencing using KAPA Biosystems Illumina compatible library preparation kit (KAPA Hyper Prep Kit, Cat# KK8504). Target enrichment was performed using a custom gene panel of approximately 500 genes and including the TERT promoter (SeqCap EZdeveloper library 24 reactions, Ref #: 06471706001, gmi.ucsf.edu/testing/). 100bp paired-end sequencing was performed on an Illumina HiSeq 2500 instrument. For clinical samples, only CDKN2A gene status was analyzed in this study. Read alignment, mutational analysis, and copy number analysis was performed as previously described(Shain et al., 2015; Talevich et al., 2016). CDKN2A deletions, loss of chromosomal regions containing CDKN2A, or point mutations known to disrupt protein function were considered inactivating (related to Figure 6A). Thirty-seven transitional lesions were previously sequenced and reanalyzed here(Shain et al., 2015). Results in Figure 6A are also in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
For comparison of WM793 and 1205Lu cell lines, after sequencing as described above, their genomic landscapes were compared, as previously described(Shain et al., 2015), to delineate the shared (trunk) and private (branch) mutations in each cell line (see Figure 4C). There was no source of normal tissue associated with these cell lines, and therefore somatic point mutations were inferred by subtracting known SNPs (IK Genome Project) from variants. It is likely that a small number of private, germline SNPs are present in the trunk of the phylogenetic tree, however, this did not hinder the goal of this analysis, which was to identify differences between the two cell lines.
Transcriptomic analysis
Matched RNA-sequencing was performed on a subset of 31 transitional lesions. For this subset of cases, every other unstained section was microdissected and used instead for RNA isolation (Ambion 1975). 100 ng of total RNA was prepared for sequencing using KAPA stranded RNA-Seq Library Preparation Kit (KR0934). This kit uses random priming for cDNA synthesis, and thus target enrichment was performed using xGen Lockdown Reagents (Cat# 1072281) and Nimblegen Capture Baits (Roche, customized), thereby depleting ribosomal RNA prior to sequencing. 100bp paired-end sequencing was performed on an Illumina HiSeq 2500 instrument. BRN2 and pl6INK4A transcript status were analyzed in this study. Sequencing reads were aligned using STAR(Dobin et al., 2013) and transcript abundance was estimated using RSEM(Li and Dewey, 2011).
For melanocytes, RNA sequencing was performed on 3 sets of wild-type and CDKN2A ex2::CMV-EGFP melanocytes preparations. Library construction was performed using TruSeq Stranded mRNA LT (Illumina) followed by single end 75-bp sequencing on the NextSeq 500 (Illumina). QC was performed using FastQC followed by trimming using Trimmomatic(Bolger et al., 2014). Sequencing reads were aligned using Tophat with differential analysis performed using CuffDiff (Trapnell et al., 2012).
Cell culture and analysis
Primary melanocyte isolation
Foreskins were provided by Dr. Thea Mauro in accordance with UCSF Human Research Protection Program protocol. After PBS washing, the skin was cut into 1 cm × 1 cm squares and cleared of fat prior to incubation with Dispase for 24 hr at 4°C with the epidermis side facing upwards. After washing, the epidermis was isolated, sectioned into small pieces, and trypsinized at 37°C for 5 min. After quenching and low-speed centrifugation, cell pellets were plated in NHM media (Medium 254 (ThermoFisher M254500) supplemented with HMGS1 (ThermoFisher S0025) and Pen-Strep) and incubated at 37°C overnight. Fresh media containing G418 (1 µg/ml) was added 24 hr later until a pure NHM population was observed. Once established in culture, NHMs were verified as pigmented, possessing melanocyte morphology, and expressing SoxlO and Melan-A (Figure S1A) as follows: cells were washed with PBST (PBS with 0.05% TritonX), blocked with PBST with 3% BSA and 0.01% Donkey serum for 1 hr, incubated with primary antibody in blocking buffer overnight at 4°C, washed, incubated with secondary antibody (anti-rabbit-Cy5, anti-mouse 488 and anti-goat 594 (Life Technologies)) for 2 hr, washed with DAPI-containing PBST, and imaged. After verification, NHMs were maintained in NHM media. Medium was refreshed twice a week and cells were passaged upon reaching 80% confluence with 0.05% Trypsin quenched with lml 0.5 mg/ml soybean trypsin inhibitor (Life technologies 17075029).
Lentivirus transduction
293T cells were co-transfected with 3rd generation lentivirus packaging vectors and expression construct (pSico-mcherry, pSico-shINK4A-mCherry, MISSION pLKO.1-puro shRNA Control or shBRN2, pHIV-EFla-IRES-ZsGreen, pHIV-EFla-IRES-INK4A-ZsGreen, pHIV-EFla-IRES-INK4A-mOrange2 or pHIV-EFla-IRES-ARF-mOrange2) using jetPRIME transfection reagent (Polyplus 114-15). Viral supernatants were harvested 72 hr post transfection, filtered through 0.45 pm filter and 10 µg/ml of Polybrene (SIGMA H9268) was added prior to plating. Media was switched to standard culture media either 24 hr (cell lines) or 6 hr (NHM) post infection. 72 hr post infection the cells were trypsinized, washed in PBS, filtered and sorted for mCherry, mOrange2 or ZsGreen positive cells.
Transfection
NHMs were transfected using the Neon Electroporation system (ThermoFisher), as described in the manufacture’s protocol. Briefly, NHMs were trypsinized, washed, and resuspended in R buffer at a final concentration of 2.0 × 107 cells/mL. Cells were mixed with either 150 pmol siRNA, 2 µg expression or reporter plasmid or CRISPR/Cas9-HDR reagents (see NHM engineering) and electroporated with two 20 ms 1500 V pulses in a 10 µL Neon transfection tip (ThermoFisher MPK1096), then plated into 500 µL pre-warmed media in a 24-well tissue culture treated plate.
Cell lines were transfected using either jetPRIME transfection reagent (Polyplus 114-15) for plasmid or Lipofectamine RNAiMax (ThermoFisher 13778150) for siRNA. The concentration of siRNA, plasmid and transfection reagent followed the manufacture’s protocol.
Flow cytometric analysis
Primary melanocytes, 1205Lu and WM796 melanoma cells were either CRISPR engineered to be EGFP or mOrange2 positive, or transduced to be mCherry, mOrange2 or ZsGreen positive. Fluorescent protein expressing cells were sorted out for pure populations. The cells were trypsinized, centrifuged and resuspended in PBS, following which they were analyzed and sorted on the FACSAria III (BD biosciences) on the FITC channel (ZsGreen+ and EGFP+ cells) or PE channel (mOrange2+ and mCherry+ cells). Further analysis and quantification was performed using Flowjo (Treestar).
For cell cycle analysis, primary melanocytes and melanoma lines were harvested and fixed with cold 70% ethanol in 4°C overnight. Cells were washed with PBS with 0.1% FBS twice, centrifuged at 850 g for 5 min, and resuspended in 500 µl PBS with 0.1% FBS followed by incubation with 1 µl of 200 µM FxCycle Far Red Stain (ThermoFisher F10348) or 25 µl of 0.5 µg/ml propidium iodide (BioLegend 421301) along with 100 µg/ml of RNAase for 30 min. At least l×104 cells per sample were analyzed through FACSAria III (BD biosciences) according to their individual emission spectra.
For apoptosis analysis, 1×106 cells were harvested and washed twice with cold PBS, resuspended in 100 µl binding buffer, then incubated with 10 µl of 0.5 mg/ml propidium iodide (BioLegend 421301) and 5 µl of APC-labelled antibody against ANNEXIN V (BioLegend 640920) for 30min. At least l×104 cells per sample were analyzed through FACSAria III (BD biosciences).
Holographic imaging and microscopy
Holographic imaging was conducted as previously described(Hejna et al., 2017). Briefly, imaging was conducted with a HoloMonitor M4 imaging cytometer with high precision automated stage (Phase Holographic Imaging, Lund, Sweden) installed in a standard tissue culture incubator. Cells were plated in 6-well standard tissue culture plates, plated at 10,000 – 100,000 cells per well depending on cell type and experiment and covered with a Hololid (PHI). For motility analyses, holograms were generated every hr for indicated time and analyzed using the Hstudio software package (PHI). For growth arrest analyses, cells were classified based upon quantitative optical metrics as previously described(Hejna et al., 2017), where cells were defined as growth arrested if they stopped dividing while adopting a morphology indistinguishable from NHMs treated with CDK4/6 inhibitor(Fry et al., 2004). For cell migration analysis, cells were plated in two nearby migration chambers (Ibidi 80241). Each chamber was grown to confluence and migration was monitored every hr for 24 hr by holographic imaging after removal of the chamber.
Fluorescent images were captured with an EVOS FL inverted microscope (ThermoFisher), colored images were captured with a Lieca Dmil light microscope, and high quality scans of clinical specimens were scanned with a ScanScope XT scanner (Aperio).
Invasion analyses
In vitro invasion assays were conducted using transwell cassettes coated with high-density basement membrane extract as described in manufacture’s protocol (Trevigen, 3483-096-K)
Mycoplasma detection
All cell lines and primary cells in the laboratory, including those used during these studies, were verified as mycoplasma negative using the ATCC Mycoplasma Detection Kit (30-1012-K) first upon arrival or isolation, and monthly thereafter for cells in active culture.
NHM engineering
Introduction of BRAFV600E point mutation
crRNA (KEY RESOURCES TABLE, IDT) and tracrRNA (IDT 1072533) were resuspended in nuclease-free IDTE buffer (IDT 11-01-02-02) and combined for a final concentration of 100 µM each and assembled into sgRNA via heating at 95°C for 5 min then cooled to room temperature. Cas9:sgRNA RNPs were formed by combining 120 pmol sgRNA and 100 pmol recombinant Cas9 protein (qb3 Berkeley), heating at 37°C for 5 min then cooling to room temperature. 120 pmol of crRNA:tracrRNA complexes was mixed with 100 pmol BRAF ssOligo HDR template, then electroporated into low-passage NHMs (passage 2–5 after isolation) as described above. sgRNA and ssOligo HDR template, without recombinant Cas9, were transfected for no Cas9 controls. After electroporation, NHMs were plated directly into media containing 5 µM L755507 for enhancement of HDR recombination(Yu et al., 2015). After 3 days, genomic DNA was harvested for indel analysis via the TIDE approach(Brinkman et al., 2014).
Introduction of CDKN2A knock-in
Low-passage NHMs (passage 2–5 after isolation) were electroporated as described above with 800 ng each of pHDR-CDKN2A-Ex2-CMV-EGFP (Addgene #110734), pEN35-CDKN2A-Ex2-L (Addgene #110736) and pEN35-CDKN2A-Ex2-R (Addgene #110737) and plated directly into media containing 5 µM L755507 for enhancement of HDR recombination (Yu et al., 2015). pHDR plasmids alone were transfected as no Cas9 controls. Transfected NHMs were maintained and expanded in normal conditions until fluorescent signal was no longer observed in no Cas9 controls (~3 weeks). EGFP positive and EGFP negative cells were then sorted from the Cas9 and no Cas9 populations, respectively to generate CDKN2A null and wild-type sibling NHMs. For Figure S2F, engineering was conducted identically but with pHDR-CDKN2A-Ex2-EFla-H2B-mOrange2 as the template.
Lung metastasis assay
Immuno-deficient mice were subcutaneously injected with human melanoma cells as described in EXPERIMENTAL MODELS AND SUBJECT DETAILS. Presence of lung metastasis was determined by staining of human nuclei specific primary antibody (HNA). Primary tumor and lung were sectioned on a cryostat and incubated at room temperature for 20 min. The sections were washed with PBST, blocked with 3% BSA PBST, incubated with primary antibody 1:50 in blocking buffer overnight, washed 2 times with PBST, and incubated with secondary antibody anti-Cy5/488 (Life Technologies) for 2 hr. After washing, the slides were coated with 20 µl of mounting medium with DAPI (Life Technologies) and HNA positive cells were counted by an observer blinded to the identity of each sample.
Molecular Biology
Western blot analysis
Protein extracts were mixed with NuPAGE LDS Sample Buffer (ThermoFisher NP0007), NuPAGE Sample Reducing Agent (ThermoFisher NP0004) and heated for 10min at 70°C. Total protein extracts (10 µg/lane) were resolved on NuPAGE Novex 4–12% or 16% Bis-Tris Protein Gels, 1.0 mm, 15-well (ThermoFisher NP0323BOX) and transferred using a Mini-Trans-Blot Cell Trans-Blot semi-Dry transfer cell (BioRad) onto 0.22 µm PVDF membranes (ThermoFisher 88520). Membranes were blocked with Membrane Blocking Solution (ThermoFisher 000105) for 1 hr at room temperature (22°C). The membranes were then incubated overnight at 4°C with primary antibodies. The membranes were washed 3 times for 10 min with TBS and 0.1% Tween20 (TBST), incubated with horseradish peroxidase-conjugated secondary antibody (GE Healthcare NA934) 1:5000 for 1 hr at room temperature, washed four times with TBST and visualized with Lumina Forte Western HRP substrate (Millipore WBLUF0500) or Clarity™ ECL Western Blotting Substrate (BioRad 1705061).
RT-qPCR
RNA from cell lines was isolated with TRI reagent according to the manufacturer’s protocol (Sigma T9424). RT-qPCR was performed following the instructions for SensiFAST cDNA Synthesis Kit and SensiFAST SYBR No-ROX Kit (Bioline BIO-65053 & BIO-98020) with CFX96 Real-Time PCR Detection System (BioRad). Relative expression level was calculated using 2−Δ ΔCT method. RPL37A was used as a housekeeping gene for normalization. The sequences of primers used for qPCR analysis are listed in the KEY RESOURCES TABLE.
Human genomic DNA qPCR
Genomic DNA was isolated from mouse lung biopsies using ISOLATE II Genomic DNA Kit (Bioline BIO-52067). Quantitative PCR was performed to detect human DNA using SensiFAST SYBR No-ROX Kit (Bioline BIO-98020) on the CFX96 Real-Time PCR Detection System (BioRad). Standard curves using known concentrations of pure human genomic DNA were generated to calculate absolute value. The sequences of primers used for qPCR analysis are listed in the KEY RESOURCES TABLE.
ChIP
ChIP experiments were performed using SimpleChIP Enzymatic Chromatin IP Kit (#9003, Cell Signaling Technology) according to the procedures provided by the manufacturer starting with 5×106 cells and using normal mouse IgG (Santa Cruz sc-2025) or anti-E2F1 (Millipore, 05-379). Precipitates were then used as templates in qPCR reactions. The primers for detecting E2F1 binding to POU3F2 promoter region are listed in the KEY RESOURCES TABLE.
Digital droplet qPCR
FAM- and HEX- tagged probes that specifically bound BRAF V600 and E600 alleles, respectively, were designed as non-binding sense strands to the ddOligo HDR template. A PCR primer was designed to span the probe-binding region. Probes and primers were synthesized by Bio-Rad. PCR reactions were assembled using premixtures of each probe (5 µM) and primer (18 µM), 150 ng genomic DNA, FAM and HEX probes and ddPCR Supermix (Bio-Rad) according to manufacture’s protocol. Droplet generation with a QX100 Droplet Generator was performed according to the manufacturer’s instructions (Bio-Rad), and the reaction was transferred into a 96-well PCR plate for standard PCR on a C1000 Thermal Cycler (Bio-Rad). The thermal cycling program conducted was: step 1, 95 °C 10 min; step 2, 94 °C 30 s; step 3, 60 °C 1 min; repeat steps 2 and 3 39 times; step 4, 98 °C 10 min. After the PCR was completed, the droplets were analyzed using a QX100 Droplet Reader (Bio-Rad). BRAFV600E mutation percentage was calculated based on the copy number of BRAFV600E per pi (x) and the copy number of BRAFWT per µl (y) with the formula: percentage of BRAFV600E (%)= X/(X+Y) *100.
Telomere length assay
The telomere length measurement assay is adapted from the published original method by Cawthon(Cawthon, 2002; Lin et al., 2010). The telomere thermal cycling profile consists of:
Cycling for T(telomic) PCR: 96°C for 1 min; denature at 96°C for 1 s, anneal/extend at 54°C for 60 s, with fluorescence data collection, 30 cycles.
Cycling for S (single copy gene) PCR: PCR: 96°C for 1 min; denature at 95°C for 15 s, anneal at 58°C for 1 s, extend at 72°C for 20 s, 8 cycles; followed by denature at 96°C for 1 s, anneal at 58°C for 1 s, extend at 72°C for 20 s, hold at 83°C for 5 s with data collection, 35 cycles.
The primers for the telomere PCR are tellb [5'-CGGTTT(GTTTGG)5GTT-3'], used at a final concentration of 100 nM, and tel2b [5'-GGCTTG(CCTTAC)5CCT-3'], used at a final concentration of 900 nM. The primers for the single-copy gene (human beta-globin) PCR are hbgl [5' GCTTCTGACACAACTGTGTTCACTAGC-3'], used at a final concentration of 300 nM, and hbg2 [5'-CACCAACTTCATCCACGTTCACC-3'], used at a final concentration of 700 nM. The final reaction mix contains 20 mM Tris-HCl, pH 8.4; 50 mM KC1; 200 µM each dNTP; 1% DMSO; 0.4× Syber Green I; 22 ng E. coli DNA per reaction; 0.4 Units of Platinum Taq DNA polymerase (Invitrogen) per 11 microliter reaction; ~6 ng of genomic DNA. Tubes containing 26, 8.75, 2.9, 0.97, 0.324 and 0.108 ng of a reference DNA (human genomic DNA from buffy coat, Roche 11691112001) are included in each PCR run so that the quantity of targeted templates in each research sample can be determined relative to the reference DNA sample by the standard curve method. The same reference DNA is used for all PCR runs.
To control for inter-assay variability, 8 control DNA samples are included in each run, and the T/S ratio is calculated. If the T/S ratio for duplicates varies by more than 7%, the sample is re-run. The average CV for this study was 1.6%.
Plasmid construction
The lentiviral expression construct pHIV-ZsGreen was a gift from Bryan Welm and Zena Werb (Addgene plasmid # 18121)(Welm et al., 2008). pHIV-mOrange2 was constructed by digesting pHIV-zsgreen with Baul51 and inserting the mOrange2 coding sequence. MISSION pLKO.1-puro Non-Mammalian shRNA Control Plasmid (SHC002) and shPOU3F2 MISSION shRNA Bacterial Glycerol Stock (TRCN0000218221) were purchased from Sigma. pHIV-INK4A-zsGreen (Addgene #110728), pHIV-ARF-zsGreen (Addgene #110729), pHIV-ARF-mOrange2 (Addgene #110731), pHIV-INK4A-mOrange2 (Addgene #110730), and pHIV-BRAFV600E-mOrange2 (Addgene #110732) were all generated by inserting the indicated coding sequence into the Ecorl and Xbal sites of the indicated parental pHIV constructs. pSicoR-Efla-mCh-Puro was a gift from Bruce Conklin (Addgene plasmid # 31845)(Salomonis et al., 2010). pGL410-BRN2p (Addgene #110733, BRN2 promoter luciferase reporter) was generated by placing 2kb of sequence upstream of the BRN2 transcriptional start site into the Bgl2 and Xhol sites. pHDR-CDKN2A-Ex2-CMV-EGFP (Addgene #110734) was generated by amplifying 2kb left and right arms from human genomic DNA and using Cold Fusion (System Bio) to combine with the CMV promoter, EGFP ORF, and an Amp/Ori cassette. pHDR-CDKN2A-Ex2-EFla-H2B-mOrange2 (Addgene #110735) was constructed similarly but with a EFla promoter and mOrange2 ORF fused to H2B. For conducting double nickase CRISPR-Cas9, we used pEN35, generously provided by Elphege Nora. pEN35 was constructed by introducing the (F&E) modification into the sgRNA backbone of px335(Chen et al., 2013; Ran et al., 2013).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unpaired t-tests were used for Gaussian distributions and Mann-Whitney tests for non-Gaussian distributions or for samples of unequal variance. Exact p values were calculated as reported by Prism 6.
DATA AND SOFTWARE AVAILABILITY
Data Resources
Sequencing data is deposited in dbGaP (Accession Number phs001550.vl.pl).
Supplementary Material
Significance.
We report the development of CRISPR/Cas9-mediated genetic engineering for precision editing of primary human melanocytes. This approach provides a flexible cellular model for assessing the acute effects of pathogenic mutations commonly associated with melanoma initiation. Bi-allelic deletion of the CDKN2A tumor suppressor did not influence BRAFV600E-induced growth arrest as expected, but rather resulted in hyper-motile and invasive melanocyte behavior. Further in vivo and in situ analyses revealed a mechanism by which p16INK4A loss contributes to melanoma invasion and metastasis through transcriptional activation of the lineage-restricted transcription factor BRN2. Overall, this study describes an approach for functionally dissecting the earliest stages of human melanoma initiation and uncovers the release of p16INK4A – mediated BRN2 inhibition as a major contributor to melanoma invasion.
Highlights.
Engineering of human melanocytes is a tractable model for melanoma initiation.
The CDKN2A locus suppresses melanocyte migration and melanoma invasion.
p16INK4A loss drives melanoma invasion via BRN2 activation.
BRN2 is a direct transcriptional target of E2F1.
Acknowledgments
This research was supported in part by NIH DP5OD019787 and the Sandler Foundation and Program for Breakthrough Biomedical Research to R.L.J., the UCSF Resource Allocation Program to R.L.J. and B.C.B., the Melanoma Research Alliance to B.C.B., I.Y. and R.L.J., NIH 5T32 CA177555 to A.H.S, and the NCI Outstanding Investigatory Award (1R35CA220481) to B.C.B. We thank M. McMahon for significant intellectual contribution and providing kinase inhibitors; V.K. Mishra, J. Phillips, and the Helen Diller Cancer Center Core Facilities for technical and experimental design assistance; and G. Panagiotakos, E. Van Veen, and M. Foth for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
H.Z. conducted engineering, cell and molecular assays in Figures 3,5,7, S1–S4 & S6. A.J. conducted cell, molecular, and mouse studies in Figures 3D&3E, 4, 5E, & S4. A.H.S conducted genome and RNA analyses in Figures 1F, 4A–C & 6A. U.E.L. contributed to case identification and analyses in Figures 6, 7I–J, S5 & S6. R.T. conducted RNA analysis in Figure 5A & 7A. J.L. conducted telomere length assay in Figure S2B. Y.Z, T.B., A.S.M., M.D., I.N.B. and R.Y. assisted in molecular, mouse, and computational work. J.P.N., L.P., and B.S.R. provided histopathology and diagnosis for cases in Figure 6. N.M.J. provided histopathology, diagnosis, and microdissection for cases in Figure 6. I.Y. and B.C.B. contributed to case identification, histopathology, diagnosis, and analyses for cases in Figure 6 and provided significant intellectual contribution. R.L.J. conceived of and directed all experiments and conducted engineering, cell and molecular assays in Figures 1, 2, 7, S1, S2 & S6. R.L.J., B.C.B., H.Z., T.B. and A.H.S wrote the manuscript.
Declaration of Interests: B.C.B. has served as a consultant for Lilly Inc.
References
- Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005;65:4005–4011. doi: 10.1158/0008-5472.CAN-04-2970. [DOI] [PubMed] [Google Scholar]
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol. 2014;9:239–271. doi: 10.1146/annurev-pathol-012513-104658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DC. Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res. 2016;29:122–140. doi: 10.1111/pcmr.12422. [DOI] [PubMed] [Google Scholar]
- Besch R, Berking C. POU transcription factors in melanocytes and melanoma. Eur J Cell Biol. 2014;93:55–60. doi: 10.1016/j.ejcb.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, Larue L, Goding CR. Mitf cooperates with Rbl and activates p21Cipl expression to regulate cell cycle progression. Nature. 2005;433:764–769. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2f Transcription Factor Is a Cellular Target for the Rb Protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AL, Sturm RA. POU domain transcription factors: BRN2 as a regulator of melanocytic growth and tumourigenesis. Pigment Cell Melanoma Res. 2008;21:611–626. doi: 10.1111/j.1755-148X.2008.00510.x. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, et al. Distinct sets of genetic alterations in melanoma. New Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Damsky W, Micevic G, Meeth K, Muthusamy V, Curley DP, Santhanakrishnan M, Erdelyi I, Platt JT, Huang L, Theodosakis N, et al. mTORCl Activation Blocks Braf(V600E)-Induced Growth Arrest but Is Insufficient for Melanoma Formation. Cancer Cell. 2015;27:41–56. doi: 10.1016/j.ccell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;75:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliason MJ, Larson AA, Florell SR, Zone JJ, Cannon-Albright LA, Samlowski WE, Leachman SA. Population-based prevalence of CDKN2A mutations in Utah melanoma families. J Invest Dermatol. 2006;126:660–666. doi: 10.1038/sj.jid.5700094. [DOI] [PubMed] [Google Scholar]
- Fahraeus R, Lane DP. The pl6(INK4a) tumour suppressor protein inhibits alpha(v)beta(3) integrin-mediated cell spreading on vitronectin by blocking PKC-dependent localization of alpha(v)beta(3) to focal contacts. Embo Journal. 1999;18:2106–2118. doi: 10.1093/emboj/18.8.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fane ME, Chhabra Y, Hollingsworth DEJ, Simmons JL, Spoerri L, Oh TG, Chauhan J, Chin T, Harris L, Harvey TJ, et al. NFIB Mediates BRN2 Driven Melanoma Cell Migration and Invasion Through Regulation of EZH2 and MITF. EBioMedicine. 2017;16:63–75. doi: 10.1016/j.ebiom.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzGerald MG, Harkin DP, Silva-Arrieta S, MacDonald DJ, Lucchina LC, Unsal H, O'Neill E, Koh J, Finkelstein DM, Isselbacher KJ, et al. Prevalence of germ-line mutations in pl6, pl9ARF, and CDK4 in familial melanoma: analysis of a clinic-based population. Proc Natl Acad Sci U S A. 1996;93:8541–8545. doi: 10.1073/pnas.93.16.8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florell SR, Boucher KM, Holden JA, Meyer LJ, Samlowski WE, Cannon-Albright LA, Zone JJ, Leachman SA. Failure to detect differences in proliferation status of nevi from CDKN2A mutation carriers and non-carriers. Journal of Investigative Dermatology. 2002;118:386–387. doi: 10.1046/j.1523-1747.2002.01659.x. [DOI] [PubMed] [Google Scholar]
- Fountain JW, Karayiorgou M, Ernstoff MS, Kirkwood JM, Vlock DR, Titusernstoff L, Bouchard B, Vijayasaradhi S, Houghton AN, Lahti J, et al. Homozygous Deletions within Human-Chromosome Band-9p21 in Melanoma. P Natl Acad Sci USA. 1992;89:10557–10561. doi: 10.1073/pnas.89.21.10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade MA, Trachet E, Albassam M, Zheng XX, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–1437. [PubMed] [Google Scholar]
- Funk JO, Schiller PI, Barrett MT, Wong DJ, Kind P, Sander CA. pl6INK4a expression is frequently decreased and associated with 9p21 loss of heterozygosity in sporadic melanoma. J Cutan Pathol. 1998;25:291–296. doi: 10.1111/j.1600-0560.1998.tb01748.x. [DOI] [PubMed] [Google Scholar]
- Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, Ross MI, Lazar AJ, Faries MB, Kirkwood JM, McArthur GA, et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin. 2017;67:472–492. doi: 10.3322/caac.21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF, Azizi E, Bianchi-Scarra G, Bishop DT, Bressac-de Paillerets B, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66:9818–9828. doi: 10.1158/0008-5472.CAN-06-0494. [DOI] [PubMed] [Google Scholar]
- Haferkamp S, Scurr LL, Becker TM, Frausto M, Kefford RF, Rizos H. Oncogene-induced senescence does not require the pl6(INK4a) or pl4ARF melanoma tumor suppressors. J Invest Dermatol. 2009;129:1983–1991. doi: 10.1038/jid.2009.5. [DOI] [PubMed] [Google Scholar]
- Halaban R. Rb/E2F: a two-edged sword in the melanocytic system. Cancer Metastasis Rev. 2005;24:339–356. doi: 10.1007/s10555-005-1582-z. [DOI] [PubMed] [Google Scholar]
- Halaban R, Cheng E, Zhang Y, Mandigo CE, Miglarese MR. Release of cell cycle constraints in mouse melanocytes by overexpressed mutant E2F1(E132), but not by deletion of pl6(INK4A) or p21(WAF/CIPl) Oncogene. 1998;16:2489–2501. doi: 10.1038/sj.onc.1201773. [DOI] [PubMed] [Google Scholar]
- Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of pl6CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejna M, Jorapur A, Song JS, Judson RL. High accuracy label-free classification of single-cell kinetic states from holographic cytometry of human melanoma cells. Sci Rep. 2017;7:11943. doi: 10.1038/s41598-017-12165-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlyn M, Thurin J, Balaban G, Bennicelli JL, Herlyn D, Elder DE, Bondi E, Guerry D, Nowell P, Clark WH, et al. Characteristics of cultured human melanocytes isolated from different stages of tumor progression. Cancer Res. 1985;45:5670–5676. [PubMed] [Google Scholar]
- Hoek KS, Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigm Cell Melanoma R. 2010;23:746–759. doi: 10.1111/j.1755-148X.2010.00757.x. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, Clark WH, Jr, Tucker MA, Dracopoli NC. Germline pl6 mutations in familial melanoma. Nat Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- Juhasz I, Albelda SM, Elder DE, Murphy GF, Adachi K, Herlyn D, Valyi-Nagy IT, Herlyn M. Growth and invasion of human melanomas in human skin grafted to immunodeficient mice. Am J Pathol. 1993;143:528–537. [PMC free article] [PubMed] [Google Scholar]
- Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, Ding W, Hussey C, Tran T, Miki Y, Weaver-Feldhaus J, et al. Analysis of the pl6 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8:23–26. doi: 10.1038/ng0994-22. [DOI] [PubMed] [Google Scholar]
- Keller-Melchior R, Schmidt R, Piepkorn M. Expression of the tumor suppressor gene product pl6INK4 in benign and malignant melanocytic lesions. J Invest Dermatol. 1998;110:932–938. doi: 10.1046/j.1523-1747.1998.00211.x. [DOI] [PubMed] [Google Scholar]
- Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of pl6Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- Kumar SM, Dai J, Li S, Yang R, Yu H, Nathanson KL, Liu S, Zhou H, Guo J, Xu X. Human skin neural crest progenitor cells are susceptible to BRAF(V600E)-induced transformation. Oncogene. 2014;33:832–841. doi: 10.1038/onc.2012.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunwor R, Nepal M, Ho D, Ghimire KB. Survival trends among patients with metastatic melanoma in the United States: A population based study. Journal of Clinical Oncology. 2017;35 [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinformatics. 2011;12 doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Epel E, Cheon J, Kroenke C, Sinclair E, Bigos M, Wolkowitz O, Mellon S, Blackburn E. Analyses and comparisons of telomerase activity and telomere length in human T and B cells: Insights for epidemiology of telomere maintenance. J Immunol Methods. 2010;352:71–80. doi: 10.1016/j.jim.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HZ, Liu SJ, Zhang G, Kwong LN, Zhu YY, Miller JP, Hu Y, Zhong WQ, Zeng JW, Wu L, et al. Oncogenic BRAF-Mediated Melanoma Cell Invasion. Cell Rep. 2016;15:2012–2024. doi: 10.1016/j.celrep.2016.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeal AS, Liu KV, Nakhate V, Natale CA, Duperret EK, Capell BC, Dentchev T, Berger SL, Herlyn M, Seykora JT, Ridky TW. CDKN2B Loss Promotes Progression from Benign Melanocytic Nevus to Melanoma. Cancer Discov. 2015;5:1072–1085. doi: 10.1158/2159-8290.CD-15-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Obata M, Lee GH, Kanda H, Kitagawa T, Ogawa K. Loss of heterozygosity at loci on chromosome 4, a common genetic event during the spontaneous immortalization of mouse embryonic fibroblasts. Mol Carcinogen. 1997;19:17–24. [PubMed] [Google Scholar]
- Pavey SJ, Cummings MC, Whiteman DC, Castellano M, Walsh MD, Gabrielli BG, Green A, Hayward NK. Loss of pl6 expression is associated with histological features of melanoma invasion. Melanoma Res. 2002;12:539–547. doi: 10.1097/00008390-200212000-00003. [DOI] [PubMed] [Google Scholar]
- Pinner S, Jordan P, Sharrock K, Bazley L, Collinson L, Marais R, Bonvin E, Goding C, Sahai E. Intravital Imaging Reveals Transient Changes in Pigment Production and Brn2 Expression during Metastatic Melanoma Dissemination. Cancer Research. 2009;69:7969–7977. doi: 10.1158/0008-5472.CAN-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JA, Loganzo F, Jr, Shea CR, Walker GJ, Flores JF, Glendening JM, Bogdany JK, Shiel MJ, Haluska FG, Fountain JW, et al. Loss of expression of the pl6/cyclin-dependent kinase inhibitor 2 tumor suppressor gene in melanocytic lesions correlates with invasive stage of tumor progression. Cancer Res. 1995;55:2713–2718. [PubMed] [Google Scholar]
- Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38:12–19. doi: 10.1016/j.tibs.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomonis N, Schlieve CR, Pereira L, Wahlquist C, Colas A, Zambon AC, Vranizan K, Spindler MJ, Pico AR, Cline MS, et al. Alternative splicing regulates mouse embryonic stem cell pluripotency and differentiation. Proc Natl Acad Sci U S A. 2010;107:10514–10519. doi: 10.1073/pnas.0912260107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, Ruben B, North J, Pincus L, Yeh I, et al. Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell. 2018 doi: 10.1016/j.ccell.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, et al. The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med. 2015;373:1926–1936. doi: 10.1056/NEJMoa1502583. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999;9:22–30. doi: 10.1016/s0959-437x(99)80004-5. [DOI] [PubMed] [Google Scholar]
- Sparrow LE, Eldon MJ, English DR, Heenan PJ. pl6 and p21WAFl protein expression in melanocytic tumors by immunohistochemistry. Am J Dermatopathol. 1998;20:255–261. doi: 10.1097/00000372-199806000-00006. [DOI] [PubMed] [Google Scholar]
- Sviderskaya EV, Gray-Schopfer VC, Hill SP, Smit NP, Evans-Whipp TJ, Bond J, Hill L, Bataille V, Peters G, Kipling D, et al. pl6/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: possible implications for melanoma progression. J Natl Cancer Inst. 2003;95:723–732. doi: 10.1093/jnci/95.10.723. [DOI] [PubMed] [Google Scholar]
- Sviderskaya EV, Hill SP, EvansWhipp TJ, Chin L, Orlow SJ, Easty DJ, Cheong SC, Beach D, DePinho RA, Bennett DC. pl6 (Ink4a) in melanocyte senescence and differentiation. J Natl Cancer I. 2002;94:866–866. doi: 10.1093/jnci/94.6.446. [DOI] [PubMed] [Google Scholar]
- Szekely L, Uzvolgyi E, Jiang WQ, Durko M, Wiman KG, Klein G, Sumegi J. Subcellular localization of the retinoblastoma protein. Cell Growth Differ. 1991;2:287–295. [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talve L, Sauroja I, Collan Y, Punnonen K, Ekfors T. Loss of expression of the pl6INK4/CDKN2 gene in cutaneous malignant melanoma correlates with tumor cell proliferation and invasive stage. Int J Cancer. 1997;74:255–259. doi: 10.1002/(sici)1097-0215(19970620)74:3<255::aid-ijc4>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi E, Liu B, Li C, Liu T, Rutter J, Grossman D. Loss of pl6(INK4A) stimulates aberrant mitochondrial biogenesis through a CDK4/Rb-independent pathway. Oncotarget. 2017;8:55848–55862. doi: 10.18632/oncotarget.19862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol. 2008;9:713–724. doi: 10.1038/nrm2469. [DOI] [PubMed] [Google Scholar]
- Vance KW, Shaw HM, Rodriguez M, Ott S, Goding CR. The retinoblastoma protein modulates Tbx2 functional specificity. Mol Biol Cell. 2010;21:2770–2779. doi: 10.1091/mbc.E09-12-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YL, Uhara H, Yamazaki Y, Nikaido T, Saida T. Immunohistochemical detection of CDK4 and pl6INK4 proteins in cutaneous malignant melanoma. Br J Dermatol. 1996;134:269–275. [PubMed] [Google Scholar]
- Welm BE, Dijkgraaf GJ, Bledau AS, Welm AL, Werb Z. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell. 2008;2:90–102. doi: 10.1016/j.stem.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y, Liu H, La Russa M, Xie M, Ding S, Qi LS. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell. 2015;16:142–147. doi: 10.1016/j.stem.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Choi BY, Lee MH, Bode AM, Dong Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (pl6(INK4a)) in Cancer. EBioMedicine. 2016;8:30–39. doi: 10.1016/j.ebiom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.