

Abstract
Ca2+ influx through voltage-gated Ca2+ channels (CaVs) at the plasma membrane is the major pathway responsible for the elevation of the intracellular Ca2+ concentration ([Ca2+]i), which activates various physiological activities. Calmodulin (CaM) is known to be involved in the Ca2+-dependent inactivation (CDI) of several types of CaVs; however, little is known about how CaM modulates CaV2.2. Here, we expressed CaV2.2 with CaM or CaM mutants with a Ca2+-binding deficiency in HEK293T cells and measured the currents to characterize the CDI. The results showed that CaV2.2 displayed a fast inactivation with Ca2+ but not Ba2+ as the charge carrier; when CaV2.2 was co-expressed with CaM mutants with a Ca2+-binding deficiency, the level of inactivation decreased. Using glutathione S-transferase-tagged CaM or CaM mutants as the bait, we found that CaM could interact with the intracellular C-terminal fragment of CaV2.2 in the presence or absence of Ca2+. However, CaM and its mutants could not interact with this fragment when mutations were generated in the conserved amino acid residues of the CaM-binding site. CaV2.2 with mutations in the CaM-binding site showed a greatly reduced current that could be rescued by CaM12 (Ca2+-binding deficiency at the N-lobe) overexpression; in addition, CaM12 enhanced the total expression level of CaV2.2, but the ratio of CaV2.2 present in the membrane to the total fraction remained unchanged. Together, our data suggest that CaM, with different Ca2+-binding abilities, modulates not only the inactivation of CaV2.2 but also its expression to regulate Ca2+-related physiological activities.
Keywords: Biotinylation, Ca2+-dependent inactivation, Calmodulin, CaV2.2, Voltage-gated Ca2+ channels
Highlights
-
•
CaM binds to the conserved IQ motif of CaV2.2 and triggers inactivation.
-
•
Mutations in the conserved IQ motif decrease the current density.
-
•
The IQ motif is the main CaM binding site.
-
•
CaM with Ca2+-binding deficiency at the N-lobe increases the CaV2.2 level.
1. Introduction
Intracellular Ca2+ concentration ([Ca2+]i) changes are an important signal for a wide spectrum of cell activities from short-term neurotransmitter release to long-term control of gene expression (Berridge, 2014, Lian and Zheng, 2016, Mintz et al., 1995, Simms and Zamponi, 2014, Soderling and Derkach, 2000, Yagami et al., 2012). The voltage-gated Ca2+ channels (CaVs) at the plasma membrane are encoded by various isoforms in different cells and are the main pathway for Ca2+ influx. One of the CaV isoforms, CaV2, is widely expressed in the central and peripheral nervous system (Coppola et al., 1994, Fujita et al., 1993, Mills et al., 1994, Westenbroek et al., 1992, Westenbroek et al., 1998) and is involved in synaptic transmission in most neurons (Dunlap et al., 1995, Olivera et al., 1994).
The opening of the CaVs elevates the [Ca2+]i to a μM level upon membrane depolarization from a resting level of approximately 50 nM (Simons, 1988). Both the CaV1s and CaV2s show Ca2+-dependent inactivation (CDI), and calmodulin (CaM), which has 4 EF-hand Ca2+ binding motifs, may play a role in this inactivation (Johny et al., 2013, Peterson et al., 1999, Soong et al., 2002, Tadross et al., 2008). CaM binds to the IQ motifs located at the C-terminals of several subtypes of voltage-gated Na+ and Ca2+ channels to regulate the channel activity in a Ca2+-dependent manner (Ben-Johny et al., 2015). CDI is a typical feedback inhibition for Ca2+ homeostasis (Eckert and Chad, 1984, Eckert and Tillotson, 1981, Tillotson, 1979, Zweifach and Lewis, 1995); in contrast, Ca2+ signaling also induces facilitation (Ca2+-dependent facilitation, CDF) in several types of CaVs (Chaudhuri et al., 2007, Lee et al., 2000).
CaV2.2 shows CDI and voltage-dependent inactivation (VDI), but no CDF has been reported (Ben-Johny and Yue, 2014). Similar to other CaVs, the CDI is mediated by CaM, but the detailed interaction is not clear. In this report, we verified that bovine CaV2.2 showed an apparent CDI, and CaM interacted with the intracellular C-terminal of CaV2.2 (CaV2.2-CT). Mutating the IF residues in the IQ motif to AF (CaV2.2IF/AF) or AA (CaV2.2IF/AA) not only decreased the current amplitude but also abolished the interaction with CaM. However, the co-expression of CaM12 (Ca2+-binding deficiency at the N-lobe), which has a Ca2+-binding deficiency in the N-lobe, enhanced the currents of CaV2.2 and its mutants. Thus, these results reveal that the C- and N-terminals of CaM have differential effects on binding to CaV2.2 and regulating the channel activities.
2. Material and methods
2.1. Chemicals
Lipofectamine 2000, mouse monoclonal antibodies against the CaM and glutathione S-transferase (GST), Dulbecco's modified Eagle's medium, and other chemicals for cell culture were obtained from Invitrogen Inc. (Carlsbad, CA, USA). Mouse monoclonal antibodies against the Flag epitope and all additional chemicals, unless otherwise indicated, were purchased from Sigma-Aldrich Inc. (St. Louis, MO, USA).
2.2. Plasmid preparation
The plasmids for bovine CaV2.2 and their accessory subunits were generously provided by Dr. Aaron P. Fox (University of Chicago) (Currie and Fox, 2002). The protocol used to prepare rat brain cDNA and mutants of CaM was described previously using Pfu Ultra AD polymerase (Agilent Technologies, USA) (Lin et al., 2013, Shih et al., 2009). The primers for the AF mutant were (Forward) 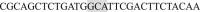 and (Reverse)
and (Reverse) 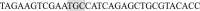 ; the primers for the AA mutant were (Forward)
; the primers for the AA mutant were (Forward) 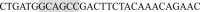 and (Reverse)
and (Reverse) 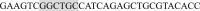 (shaded nucleotides indicate the mutated IF sequence). The triple-Flag-tagged CaV2.2s (T- CaV2.2s) were constructed by digesting a synthetic dsDNA (TCTAGACTTAAGACCGGTGCCACCATGGATTACAAGGATGACGACGATAAGGACTATAAGGACGATGATGACAAGGAC/TACAAAGATGATGACGATAAAG AATTCAAGCTTACCGGTATGGTCCGCTTCGGGGACGAGCTGGGCGGCCGCGGATCC, gene synthesized by Thermo Fisher Scientific, the underline indicates the triple-Flag sequence) with AflII and NotI sites and inserting the fragment at the 5′-terminal of CaV2.2. The clones with the intracellular C-terminal segment of T-CaV2.2s were digested with EcoRI from T-CaV2.2s (aa 1710-2332, gene number: NM_174632.2).
(shaded nucleotides indicate the mutated IF sequence). The triple-Flag-tagged CaV2.2s (T- CaV2.2s) were constructed by digesting a synthetic dsDNA (TCTAGACTTAAGACCGGTGCCACCATGGATTACAAGGATGACGACGATAAGGACTATAAGGACGATGATGACAAGGAC/TACAAAGATGATGACGATAAAG AATTCAAGCTTACCGGTATGGTCCGCTTCGGGGACGAGCTGGGCGGCCGCGGATCC, gene synthesized by Thermo Fisher Scientific, the underline indicates the triple-Flag sequence) with AflII and NotI sites and inserting the fragment at the 5′-terminal of CaV2.2. The clones with the intracellular C-terminal segment of T-CaV2.2s were digested with EcoRI from T-CaV2.2s (aa 1710-2332, gene number: NM_174632.2).
The primers for cloning the CaM from the rat brain were (Forward) 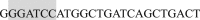 (BamHI) and (Reverse)
(BamHI) and (Reverse)  (XbaI) (shaded nucleotides indicated the restriction enzyme site); the construct was then subcloned into pcDNA3.1 plasmid. To synthesize the Ca2+-binding deficiency mutation, we mutated the last a.a., glutamate, of each EF-hand motif to glutamine using the following primers: EF1 Forward
(XbaI) (shaded nucleotides indicated the restriction enzyme site); the construct was then subcloned into pcDNA3.1 plasmid. To synthesize the Ca2+-binding deficiency mutation, we mutated the last a.a., glutamate, of each EF-hand motif to glutamine using the following primers: EF1 Forward 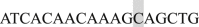 and Reverse
and Reverse 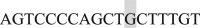 ; EF2 Forward
; EF2 Forward 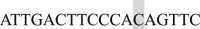 and Reverse
and Reverse 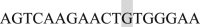 ; EF3 Forward
; EF3 Forward 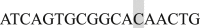 and Reverse
and Reverse 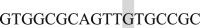 ; EF4 Forward
; EF4 Forward 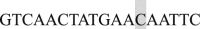 , and Reverse
, and Reverse 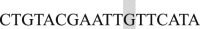 (shaded nucleotides indicate the mutated ones). All of the primers listed are in the 5′ to 3′ direction.
(shaded nucleotides indicate the mutated ones). All of the primers listed are in the 5′ to 3′ direction.
2.3. Transfection of HEK293T cells
For transient expression of the genes in HEK293T cells grown in a 35-mm dish, we mixed α1B, β2a, and α2δ (1 μg total at a ratio of 1:1:1 and 0.1 μg of a green fluorescence protein (GFP) plasmid) with Lipofectamine 2000 according to the manufacturer's instructions. We used GFP fluorescence to identify transfected cells and performed experiments 24–36 h after transfection.
2.4. Protein extraction
HEK293T cells were dissolved in a lysis buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 7.5, Bioman Inc., Taiwan) containing a protease inhibitor cocktail (Set V, 1:100 dilution, Calbiochem, La Jolla, California, USA). The lysates were centrifuged at 1000 ×g for 30 min, and the supernatant was collected as the total lysate.
2.5. GST pulldown assays
We purified the GST-fused CaM, CaM12, CaM34 or CaM1234 (has no Ca2+-binding ability at both the N- and C-lobes) expressed in E. coli as described previously (Chou et al., 2015) and used a Bradford-based protein assay kit (Bio-Rad, USA) to determine the protein concentration. To pull down interacting proteins, we incubated the GST-fused protein or GST with GSH-Sepharose 4B beads (GF Healthcare, USA) following the protocol suggested by the manufacturer. We mixed the beads with cell lysate at room temperature for 1 h or 4 °C overnight in a lysis buffer. The proteins that bound to the beads were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting.
2.6. Electrophysiological recording
The recording procedure was described previously (Shih et al., 2009). The recordings were performed at room temperature with an EPC-10 amplifier and were controlled by the Pulse program (HEKA Elektronik, Lambrecht/Pfalz, Germany). In brief, a cell was incubated in NMG buffer (in mM, 130 NMG, 20 glucose, 10 HEPES, 1 MgCl2·6H2O, 2 KCl, 10 CaCl2·2H2O, pH 7.2 with KOH, 300–305 mOsm) and patched in whole-cell mode. For the Ba2+ current measurement, Ca2+ was replaced with 10 mM Ba2+. The membrane potential was held at −70 mV and depolarized to various potentials for channel activation. The pipette solution consisted of (in mM) 120 aspartic acid, 5 MgCl2, 40 HEPES, 0.1 EGTA, 2 ATP, and 0.3 GTP, pH 7.3 with CsOH (310 mOsm/kg). The R250 ratio was measured by holding at −70 mV and depolarizing for 250 ms from −50 to +120 mV. The activation curves were created from the tail currents obtained by a 10-ms depolarization to various potentials and used for the analysis of V1/2 and slope.
2.7. Biotinylation
The HEK293T cells were seeded on poly-L-lysine-coated 35-mm dishes and transfected with different plasmids as previously described. The cells were incubated on ice and washed with D-PBS buffer supplemented with 0.5 mM CaCl2 and 2 mM MgCl2, followed by 1 mg/mL sulfo-NHS-LC-biotin (Thermo Fisher Scientific) in 1 mL of D-PBS on ice with gentle rocking for 1 h. After being washed with 100 mM of glycine in PBS (in mM, 142 NaCl, 2 KCl, 8 Na2HPO4·7H2O, and 1.5 NaH2PO4·H2O, pH 7.2) two times and TBS (in mM, 20 Tris-HCl, and 150 NaCl, pH 7.4) one time, solubilization was performed using 400 μL of lysis buffer (in mM, 150 NaCl, 5 EDTA, and 50 Tris-HCl, pH 7.6 and 1% Triton-X 100 with dithiothreitol, 1 phenylmethylsulfonyl fluoride and protease inhibitors). The cell lysates were incubated with 30 μL streptavidin-agarose beads (Thermo Scientific) overnight at 4 °C. The beads were washed once in lysis buffer, followed by another wash in high salt buffer (in mM, 500 NaCl, 5 EDTA, and 50 Tris-HCl, pH 7.6 and 0.1% Triton-X 100) and low salt buffer (in mM, 2 EDTA, and 10 Tris-HCl, pH 7.6 and 0.1% Triton-X 100). Then, Laemmli sample buffer (containing 6% SDS) was added to the samples, and the samples were boiled at 70 °C for 10 min and prepared for Western blot analysis.
2.8. Statistical analysis
Data are presented as the mean ± SEM from at least three different batches of cells and were analyzed by one-way ANOVA with Fisher's post hoc test. Differences were considered significant when the p value was less than 0.05.
3. Results
3.1. Bovine CaV2.2 shows Ca2+-dependent inactivation
To examine the inactivation of bovine CaV2.2 currents, we expressed bovine α1B and accessory subunits in HEK293T cells and measured the whole-cell currents in buffers containing Ca2+ or Ba2+ as the charge carrier (Fig. 1). When depolarized to various potentials for 250 ms from a holding potential of −70 mV, the representative current traces of cells incubated in buffer containing Ca2+ but not Ba2+ showed a fast inactivation after reaching the maxima (Fig. 1A). The current-voltage relationship showed that the depolarization potential for the maximum peak inward current was +30 mV for CaV2.2 using either Ca2+ or Ba2+ as the charge carrier (Fig. 1B). The residual currents at the end of the depolarization (R250) of a representative cell were 27.8 or 77.5% of the peak current using Ca2+ (R250/Ca2+) or Ba2+ (R250/Ba2+), respectively, as the charge carrier at a depolarization potential of +30 mV (Fig. 1C). Fig. 1D shows that the average R250/Ca2+ and R250/Ba2+ were 17.4 ± 2.3 (n = 31) and 79.0 ± 2.5% (n = 15), respectively. These results demonstrate that Ca2+ is responsible for the majority of the inactivation of bovine CaV2.2 currents.
Fig. 1.

Bovine CaV2.2 shows Ca2+-dependent inactivation. HEK293T cells transfected with CaV2.2 were whole-cell patched in voltage-clamp mode and depolarized to various potentials from a holding potential of −70 mV for 250 ms. A. The current traces of a representative cell at different potentials using Ca2+ (middle panel) or Ba2+ (bottom panel) as the charge carrier. The black line indicates the trace obtained at +30 mV. B. The normalized current-voltage relationship. The peak inward current measured using Ca2+ (n = 45) or Ba2+ (n = 9) as the charge carrier at different potentials was normalized to the respective maxima of each single cell and then averaged. C. The current evoked by a step depolarization from −70 to +30 mV for 250 ms was recorded using Ca2+ (red) or Ba2+ (blue) as the charge carrier. The residual current at the end of the depolarization was normalized to the peak current as the R250/Ca2+ or R250/Ba2+. D. The average R250/Ca2+ and R250/Ba2+ of CaV2.2. Data are presented as the mean ± SEM. **, p < 0.01 compared to the Ca2+ group using Student's t-test.
3.2. Both lobes of CaM modulate the CDI effect
CaM has 4 Ca2+ binding sites with 2 at each of the N- or C-lobes, and each lobe has different effects in modulating various CaVs (Ben-Johny et al., 2015, Yang et al., 2006). To verify the effect of each lobe of CaM in regulating CaV2.2, we measured the CaV2.2 currents using Ca2+ or Ba2+ as the charge carrier with co-expression of CaM or Ca2+-binding-deficient mutants in HEK293T cells (Fig. 2 & Table 1). The results indicated that the current traces in cells expressing CaM showed an apparent inactivation when using Ca2+ as the charge carrier; in contrast, the inactivation level was decreased in cells expressing CaM12, CaM34, and CaM1234 with Ca2+-binding deficiencies at the N-lobe, C-lobe, and both, respectively. The results (Fig. 2D & Table 1) showed that CaM mutants, CaM12, CaM34, and CaM1234, all showed a significant increase in R250/Ca2+. In contrast, CaM and CaM12 significantly lowered the R250/Ba2+; CaM34 and CaM1234 had no effect on the R250/Ba2+. However, CaM and CaM12 significantly decreased and increased, respectively, the peak inward current at +30 mV (I30/Ca2+) when using Ca2+ as the charge carrier; CaM34 and CaM1234 had no effect when compared with that of the CaV2.2 alone. However, using Ba2+ as the charge carrier enhanced the current density (I30/Ba2+) except in the co-expression of CaM12 (Table SI). These results suggest that the N- and C-lobes of CaM have differential effects in modulating the current amplitude and inactivation.
Fig. 2.

Ca2+-binding-deficient CaM mutants increase the R250/Ca2+. We co-expressed CaV2.2 with CaM and CaM mutants with binding deficiencies at the N-, C-, and both lobes (CaM12, CaM34, and CaM1234, respectively) in HEK293T cells. The currents were recorded at +30 mV using Ca2+ or Ba2+ as the charge carrier, and the R250/Ca2+ and R250/Ba2+ were analyzed. A. Schematic representation of the CaM and CaM mutants. B. Representative normalized current traces (each trace was normalized to the inward peak current) from cells expressing CaM or mutants. C. Average R250/Ca2+ and R250/Ba2+. The numbers on each column refer to the number of cells used in each group. Data are presented as the mean ± SEM and were analyzed by one-way ANOVA with Fisher's post hoc test. *: p < 0.05, **: p < 0.01, and ***: p < 0.001 compared to the same expression group using Ca2+ as the charge carrier. #: p < 0.05 and ##: p < 0.001 compared to the R250/Ba2+ of the CaM group. D. Plot of R250/Ca2+ against I30/Ca2+. The R250/Ca2+ of each cell in Fig. 2C was plotted against the I30/Ca2+. The lines indicated the linear regression of each group.
Table 1.
The basic properties of CaV2.2 with mutations in the IQ motif.
| Co-expression | V1/2 (mV) | Slope (mV) | I30/Ca2+ (pA/pF) | R250/Ca2+ (%) | R250/Ba2+ (%) | |
|---|---|---|---|---|---|---|
| CaV2.2 | Mock | 18.7 ± 2.7 n = 20 |
6.2 ± 0.8 n = 20 |
−71.1 ± 8.4 n = 37 |
17.4 ± 2.3 n = 31 |
79.0 ± 2.5 n = 13 |
| CaM | 13.5 ± 2.7 n = 8 |
4.6 ± 1.0 n = 8 |
−29.9 ± 11.7* n = 10 |
23.8 ± 5.1 n = 11 |
60.7 ± 5.2* n = 10 |
|
| CaM12 | 24.7 ± 2.4 n = 4 |
7.1 ± 0.6 n = 4 |
−170.3 ± 25.7** n = 6 |
45.1 ± 4.2* n = 11 |
68.7 ± 4.3* n = 8 |
|
| CaM34 | 25.5 ± 3.2 n = 5 |
7.7 ± 1.3 n = 5 |
−78.0 ± 16.5 n = 8 |
45.0 ± 4.3* n = 13 |
82.5 ± 3.8 n = 6 |
|
| CaM1234 | 21.9 ± 2.9 n = 16 |
5.9 ± 0.8 n = 16 |
−51.8 ± 11.3* n = 11 |
41.0 ± 4.1* n = 17 |
82.1 ± 4.8 n = 5 |
|
| CaV2.2IF/AF | Mock | 31.6 ± 1.9 n = 7 |
8.8 ± 1.2 n = 7 |
−38.5 ± 9.4 n = 17 |
21.5 ± 4.0 n = 17 |
68.5 ± 3.9 n = 4 |
| CaM | 26.2 ± 3.4 n = 5 |
8.6 ± 1.9 n = 5 |
−52.8 ± 15.0 n = 9 |
24.9 ± 3.5 n = 8 |
64.7 ± 6.0 n = 11 |
|
| CaM12 | 15.4 ± 5.7 n = 5 |
6.8 ± 1.6 n = 5 |
−109.5 ± 40.9 n = 7 |
45.2 ± 2.8 n = 8 |
61.5 ± 5.5 n = 5 |
|
| CaM34 | 37.7 ± 2.5 n = 5 |
14.2 ± 3.2 n = 5 |
−23.7 ± 7.9 n = 12 |
25.0 ± 4.1 n = 12 |
67.6 ± 5.5 n = 6 |
|
| CaM1234 | 30.7 ± 2.8 n = 5 |
9.1 ± 0.6 n = 5 |
−33.3 ± 9.3 n = 16 |
33.5 ± 7.6 n = 7 |
69.5 ± 8.6 n = 5 |
|
| CaV2.2IF/AA | Mock | 41.4 ± 2.0 n = 6 |
13.2 ± 1.1 n = 6 |
−13.4 ± 3.3 n = 9 |
19.5 ± 2.6 n = 6 |
57.7 ± 4.8 n = 6 |
| CaM | 30.0 ± 4.9 n = 6 |
7.9 ± 2.1 n = 6 |
−60.5 ± 19.9 n = 9 |
25.2 ± 5.7 n = 7 |
63.4 ± 6.8 n = 4 |
|
| CaM12 | 10.6 ± 13.1 n = 3 |
6.4 ± 2.1 n = 3 |
−118.8 ± 41.3 n = 4 |
37.8 ± 5.6 n = 6 |
82.6 ± 4.3 n = 4 |
|
| CaM34 | 7.6 ± 10.1 n = 3 |
0.8 ± 0.5 n = 3 |
−63.1 ± 20.3 n = 7 |
22.5 ± 2.6 n = 5 |
63.4 ± 6.8 n = 3 |
|
| CaM1234 | 25.0 ± 6.4 n = 4 |
8.1 ± 2.5 n = 4 |
−20.0 ± 4.4 n = 9 |
16.0 ± 8.1 n = 8 |
42.7 ± 3.1 n = 3 |
|
Data are presented as the mean ± SEM and were analyzed by one-way ANOVA with Fisher’s post hoc test. *: p < 0.05 and **: p < 0.01 when compared to the Mock group.
3.3. Mutating the IF residues reduces the current amplitude
The IQ motif of CaV1s and CaV2.1 is responsible for CaM binding and the CDI effect (Ben-Johny et al., 2015). To assess the importance of this motif of CaV2.2, we expressed CaV2.2IF/AF or CaV2.2IF/AA in HEK293T cells and measured the currents after depolarization to different potentials for 250 ms (Fig. 3 and Table 1). The normalized current-voltage relationship (Fig. 3B) showed that the voltages of the maximum inward current were positively shifted in these mutants. The activation curves (Fig. 3D and Table 1) showed that the V1/2 values were significantly shifted from 18.7 ± 2.7 mV (n = 20) with wild-type CaV2.2 to 31.6 ± 1.9 mV (n = 7, p < 0.01) and 41.4 ± 2.0 mV (n = 6, p < 0.01) with CaV2.2IF/AF and CaV2.2IF/AA, respectively. The I30/Ca2+ of CaV2.2IF/AF and CaV2.2IF/AA was significantly reduced to −38.5 ± 9.4 pA/pF (n = 17, p < 0.05) and −13.4 ± 3.3 pA/pF (n = 9, p < 0.001), respectively, from that of the wild-type, with a value of −71.1 ± 8.4 pA/pF (n = 37). Therefore, the conserved IF residues are important in maintaining the current amplitude and current-voltage relationship.
Fig. 3.

Mutations in the IF residues of CaV2.2 reduce the current. CaV2.2 with mutations in the IF residues (CaV2.2IF/AF and CaV2.2IF/AA) was expressed in the HEK293T cells. A. Schematic representation of CaV2.2 and the constructs. B. The current-voltage relationship. The cell was whole-cell patched and depolarized for 250 ms from a holding potential of −70 mV to various potentials using Ca2+ as the charge carrier. The peak current recorded at each potential was normalized to the inward maxima of each cell. C. The activation curves. Cells were depolarized for 10 ms from a holding potential of −70 mV to various potentials, and the tail inward current obtained at each potential was normalized to the maxima. D. The I30/Ca2+ of CaV2.2 and the mutants. The current was evoked by a depolarization to +30 mV for 250 ms, and the peak inward current was measured. Data are presented as the mean ± SEM and were analyzed by one-way ANOVA with Fisher's post hoc test. *: p < 0.05 and ***: p < 0.001 compared to the wild-type.
3.4. The IF amino acid residues are important for current inactivation
To further assess the importance of the conserved IF residues of CaV2.2 in current inactivation, we co-expressed CaV2.2IF/AF or CaV2.2IF/AA with CaM and measured the currents (Fig. 4 and Table 1). Representative traces (Fig. 4A and B) of CaV2.2IF/AF and CaV2.2IF/AA showed a higher level of inactivation when using Ca2+ as the charge carrier than Ba2+. Fig. 4C and D shows that the average R250/Ca2+ of CaV2.2IF/AF and CaV2.2IF/AA was similar to that of CaV2.2; however, the R250/Ba2+ was 68.5 ± 3.9% (n = 4, p < 0.05) and 57.7 ± 4.8% (n = 6, p < 0.01), respectively, which were both significantly lower than that of the wild-type form (79.0 ± 2.5%, n = 13). CaM12 significantly increased the R250/Ca2+ of CaV2.2IF/AF to 45.2 ± 2.8% (n = 8, p < 0.05) compared to that of the CaM group, but CaM34 and CaM1234 had little effect on the R250/Ca2+. In contrast, the R250/Ba2+ values for CaV2.2IF/AF with different CaM constructs were approximately the same in a range between 60 and 70%. For CaV2.2IF/AA, the co-expression of CaM and mutants did not have any significant effect on the R250/Ca2+ and R250/Ba2+. The activation curves of the wild-type and mutated channels co-expressing CaM and CaM mutants are shown in Fig. S1; the calculated V1/2 and slope are listed in Table 1. These results suggest that mutations in the IF residues blocked the effects of the CaM mutants, except CaM12, in suppressing the current inactivation.
Fig. 4.

CaM12 increases the R250/Ca2+ of mutated CaV2.2. The conserved IF residues of CaV2.2 were mutated to AF (CaV2.2IF/AF) or AA (CaV2.2IF/AF) and expressed in HEK293T cells. A & B. The representative current traces from cells expressing CaV2.2IF/AF and CaV2.2IF/AA co-expressing CaM and CaM mutants. The patched cell was depolarized to +30 mV from a holding potential of −70 mV for 250 ms using Ca2+ (red traces) or Ba2+ (blue traces) as the charge carrier. C & D. Average R250 of CaV2.2IF/AF and CaV2.2IF/AF, respectively. Data are presented as the mean ± SEM and were analyzed by one-way ANOVA with Fisher's post hoc test. *: p < 0.05 and ***: p < 0.001 compared to the same expression group or as indicated.
3.5. CaM12 enhances the attenuated current density
In addition to the effects on current inactivation, the mutations in the IF residues greatly reduced the current amplitude (Table 1). Compared to the group expressing CaV2.2IF/AF and CaM, which had an I30/Ca2+ of −52.8 ± 15.0 pA/pF (n = 9), CaM12 substantially enhanced the I30/Ca2+ to −109.5 ± 40.9 pA/pF (n = 7, p < 0.05); in contrast, CaM34 and CaM1234 slightly decreased the I30/Ca2+ but not significantly. For CaV2.2IF/AA, CaM, CaM12, and CaM34 could slightly but not significantly enhance the I30/Ca2+. By plotting the R250/Ca2+ against the I30/Ca2+ (Fig. 2D) from cells expressing CaV2.2 and CaM/mutants, the linear regression curves showed an inverse relationship, except for the CaM12 group, which had a R250/Ca2+ above 40% regardless of the current density. In addition, the I30/Ba2+ were larger than those I30/Ca2+ of the same groups except the CaM12 group (Table SI). For CaV2.2 wild type and mutants, the I30/Ca2+ and I30/Ba2+ were about the same in the co-expression of CaM12. These results indicated that the conserved IF residues and each lobe of CaM are important in regulating the current density.
3.6. Differential binding of CaM mutants to the C-terminal fragment of CaV2.2
To confirm that CaM interacts with CaV2.2 at the C-terminal, we used GST-tagged CaM or Ca2+-binding-deficient mutants as the bait to pull down CaV2.2-CT expressed in HEK293T cells (Fig. 5A–D). A representative Western blot shows that GST-CaM pulled down a protein with a MW similar to the expected size of CaV2.2-CT (∼72 kD) in the presence of Ca2+ (100 μM), EGTA (5 μM), or no addition; however, GST-CaM12 and -CaM1234 could only interact with this fragment in the presence of Ca2+ but with a lower capacity than that of CaM. CaM34 did not show any interaction with the CaV2.2-CT. We then mutated the conserved IF residues to AA (CaV2.2-CTIF/AA) for pulldown assays to characterize the importance of the IF residues in this interaction. Western blot analysis (Fig. 5E) showed that little CaV2.2-CTIF/AA was observed regardless of the presence of CaM or mutants with or without Ca2+. These results illustrate the importance of the IF residues in the binding of CaM to CaV2.2 and indicate that each lobe of CaM has differential contributions to this interaction.
Fig. 5.

CaM interacts with the C-tail fragment of CaV2.2. The lysates from HEK293T cells expressing the intracellular C-tail fragment of the wild-type (CT, 72 kDa) (A–D) or mutant CaV2.2 (CTIF/AA) (E) were used for pulldown assays with GST-tagged CaM and mutants (43 kDa) as the baits. The reaction buffer for the assays had no extra addition (None) or contained 100 μM of Ca2+ (Ca) or 5 μM of EGTA (EGTA). The pulldown fraction was then analyzed by Western blotting using a monoclonal antibody against the C-terminal of CaV2.2 (CT) or CaM.
3.7. CaM12 enhances the expression level of CaV2.2
Because CaM and the mutants affected the I30/Ca2+, we examined the total expression level and the fraction of CaV2.2 localized at the cell membrane (Fig. 6). The lysates from cells transfected with CaV2.2 showed a protein band with a MW greater than 250 kD that was not observed in the control group without CaV2.2 expression (Fig. 6A). With co-expression of CaM and the mutants, the lysate containing CaM12 showed an increased band intensity compared to that of the other groups. The average total CaV2.2 level from cells co-expressing CaM12 was 1.6 ± 0.1 (n = 3, p < 0.05) when normalized to the group with CaM overexpression, which was significantly higher than that with CaM overexpression (Fig. 6B). CaM34 (1.1 ± 0.3) resulted in a similar level of CaV2.2 expression to that of the CaM group; CaM1234 (0.7 ± 0.3, n = 3, p = 0.06) slightly reduced the expression level but not significantly. Because the number of channels at the plasma membrane determines the current density, we used biotinylation to label the proteins at the plasma membrane and analyzed the ratio of CaV2.2 at the membrane to the total lysate (Fig. 6C–D). Western blot analysis showed that the antibody against CaV2.2 could recognize a protein in the biotinylation fraction with a MW similar to that in the total lysate. After analyzing the intensities of these bands, the CaV2.2 located at the membrane surface was 40–50% of the total CaV2.2 in all cell groups. These results indicate that CaM12 increases the total amount of CaV2.2 in the cells and plasma membrane.
Fig. 6.

CaM12 enhances the total expression level of CaV2.2. A. Representative Western blot of CaV2.2 in total lysates. Cell lysates isolated from HEK293T cells with no transfection (None), transfected with T-CaV2.2 only (T-CaV2.2, Mock), or transfected with co-expression of CaM, CaM12, CaM34, and CaM1234 were used for Western blot analysis using antibodies against FLAG and β-actin. B. Normalized total CaV2.2 level. The level of T-CaV2.2 was normalized to the level of β-actin in each sample; the values were then normalized to the value of the CaM group in each independent experiment. C. Representative staining of CaV2.2 after being biotinylated. Membrane proteins were first labeled with biotin and then isolated with avidin. The T-CaV2.2 in the total lysate (Total) and avidin-purified fraction (Biotinylated) was analyzed by an antibody against FLAG. D. The ratio of CaV2.2 in the membrane fraction to the total lysate. Data are presented as the mean ± SEM from at least 3 independent experiments and were analyzed by one-way ANOVA with Fisher's post hoc test. *: p < 0.05 compared to the CaM group.
4. Discussion
Ca2+ influx through the CaVs is the major elicitor of exocytosis and many other Ca2+-related activities in excitable cells; thus, regulating the kinetics of CaVs could be an effective way to modulate different cellular functions (Dubel et al., 1992, Williams et al., 1992). Our results demonstrated that CaM binds to the conserved IQ motif of CaV2.2 at the intracellular C-terminal to trigger CDI. In addition, CaM12 enhanced the current amplitude and the total expression level of CaV2.2 at the cell membrane. Therefore, the N- and C-lobes of CaM have differential effects in regulating CaV2.2 activity.
For CaV1s, CaV2.1, and CaV2.3, binding of CaM to the C-terminal IQ motif determines the CDI (DeMaria et al., 2001, Yang et al., 2006). Several reports utilizing a gene-shuffled chimeric C-terminal of CaV2.2 and CaV1s have also suggested that CaM binds to the C-terminal end and promotes CDI of the chimeric channels (Kim et al., 2008, Mori et al., 2008). The IQ segments of CaV2.1-3 interact with CaM as determined by X-ray crystallography and isothermal titration calorimetry (Fallon et al., 2009, Kim et al., 2008, Wang et al., 2014). The X-ray structure analysis shows that the IQ-helix peptides of CaV1.2 and CaV2s bind the pocket of CaM in the opposite orientation. The N-lobe of CaM interacts more with the a. a. residues located at the N-terminal portion of the CaV1.2 IQ peptide relative to the conserved IQ residues but the C-terminal portion of the IQ peptide of the CaV2s and vice versa. Even so, the CDI is determined by the interaction between the CaM N-lobe and the C-terminal portion of the IQ motif; therefore, mutations at the N-lobe (CaM12 and CaM1234) lose the ability to induce CDI (Liang et al., 2003). However, our results showed that losing the Ca2+-binding ability at either the N- or C-lobes of CaM decreases the CDI to a similar level, suggesting that either interaction is necessary and sufficient to maintain the CDI. We mutated the last a. a., E, in each EF-hand motif of CaM to Q to reduce the Ca2+-binding capability, while Liang et al. (2003) converted the first residues, D, of each motif to A. This discrepancy may differentially affect the functions of CaM and explain the difference of the results.
An amino-terminal Ca2+/CaM binding segment (NSCaTE, N-terminal Spatial Ca2+ Transforming Element) of CaV1.3 is known to interact with the N-lobe of CaM (Liu and Vogel, 2012). In the N-portion of the C-terminus of CaV1s and CaV2s, the pre-IQ domain and IQ domain are also CaM binding sites (Ben-Johny et al., 2015, Johny et al., 2013). The NSCaTE element can interact with Ca2+/CaM prebound to an IQ domain peptide, suggesting the possible bridging of the channel amino- and carboxyl-termini; however, CaV2.2 does not contain the NSCaTE element (Taiakina et al., 2013). Therefore, CaM may interact with the CaV2.2 mostly through the intracellular C-terminal.
Here, using pulldown assays and current measurement, we further demonstrated that the binding of CaM to CaV2.2 is not Ca2+-dependent, suggesting that CaM could interact with CaV2.2 at rest; in addition, the over-expression of CaM did not enhance the CDI, indicating that most CaV2.2 molecules are bound with CaM at rest, and a global elevation of [Ca2+]i would activate the CDI (Ben-Johny and Yue, 2014, Dick et al., 2008, Dick et al., 2016, Few et al., 2012, Liang et al., 2003). The plot of the R250/Ca2+ against the I30/Ca2+ from cells expressing CaV2.2 shows an inverse relationship, supporting the global Ca2+ effect on current inactivation. The expression of CaM and the mutants showed an inverse relationship as well, except for CaM12, which not only enhanced the I30/Ca2+ but also maintained the R250/Ca2+ above 40%. However, using Ba2+ as the charge carrier did not enhance the current amplitude in the co-expression of CaM12. It is possible that CaM12 blocks the CDI even when the current density is high or increases the VDI portion of the evoked current. The mechanism by which CaM12 modulates the current inactivation needs to be investigated further.
In contrast to the wild-type CaM, the binding of CaM12 to CaV2.2-CT requires Ca2+; therefore, when [Ca2+]i is elevated, CaM12 competes with the endogenous bound CaM and increases the R250/Ca2+. Although CaM34 is incapable of binding to CaV2.2-CT, CaM34 may interfere with the interaction between endogenous CaM and CaV2.2 to increase the R250/Ca2+. The CaV2.2-CT we constructed includes a Ca2+-binding motif (Delcour et al., 1993, Johny et al., 2013, Yang et al., 2006); upon Ca2+ binding, this motif may prepare CaV2.2-CT for CaM binding. As CaM can have different conformations when free or bound with Ca2+ (Fallon et al., 2009, Hultschig et al., 2004), CaM with a mutated N-lobe (CaM12 and CaM1234) may have a conformation that allows Ca2+-dependent binding to CaV2.2.
The binding of CaM to the IQ motif of CaV2.2 is important in maintaining the current amplitude as mutations in the conserved IF residues greatly reduced the I30/Ca2+, and the overexpression of CaM, especially CaM12, rescued the current amplitude of these mutants. The pulldown assays showed that CaM and the mutants did not bind to CaV2.2-CTIF/AA, and CaM12 overexpression increased the total amount of CaV2.2 expressed in HEK293T cells but did not affect the fraction of CaV2.2 at the plasma membrane (Fig. 6). This may partly explain how CaM12 overexpression enhances the current. However, it is not clear how CaM12 modulates the R250/Ca2+ level. It is possible that most of the current either enhanced by CaM12 or decreased by the channel mutations was due to CDI, but this needs to be further characterized.
The β and α2δ subunits of CaVs are involved in modulating the kinetics and amplitude of the currents, as well as targeting the channels to the plasma membrane (Brice and Durward, 1997, Singer et al., 1991). The transient over-expression of α2δ-1, α2δ-2 and α2δ-3 subunits in cultured hippocampal neurons increases not only the presynaptic abundance of P/Q-type channels but the probability of vesicular release in response to a single action potential (Hoppa et al., 2012). The cytosolic β subunits have a chaperone-like effect in promoting the functional expression of the subunits of CaV2s at the plasma membrane (Bichet et al., 2000, Brice and Durward, 1997, Raghib et al., 2001). In addition, β subunits control the gating properties of the CaVs and hyperpolarize the voltage-dependence of activation, as well as increase the maximum open probability resulting in increasing the macroscopic current density (Matsuyama et al., 1999, Meir et al., 2000, Neely et al., 1993). Our results showed that CaM12 could enhance the total expression level of CaV2.2 but not the ratio of the membrane localization. It is not clear how CaM regulates the membrane targeting of CaV2.2 and needs to be further characterized.
5. Conclusion
Our findings suggest that CaM binds to CaV2.2 via the conserved IQ motif to modulate the expression level and current density at rest or low [Ca2+]i; when [Ca2+]i elevates, the bound CaM enhances CDI. Because CaM12 and CaM34 show different effects in modulating these above-mentioned activities, the N- and C-lobes of CaM work differentially in modulating CaV2.2. In addition to providing various binding sites for activity regulation, the C-terminus of CaV1.2 and CaV2.1 could be cleaved by a protease, such as calpain, and the dissociated distal fragments may either interact with the proximal C-terminus to inhibit the channel activity or translocate to the nucleus, acting as a transcription factor (Abele and Yang, 2012, Hell et al., 1996). These results reveal that the versatile pathways for regulating channel activity via the C-terminus of CaVs and CaM, with different Ca2+-binding abilities, are an immediate regulatory factor for various cellular functions (Ben-Johny et al., 2015).
Acknowledgments and conflict of interest disclosure
This work was supported by the Ministry of Science and Technology of Taiwan under grant Nos. of MOST 104-2627-M-002-003 and 103-2320-B-002-060-MY3. Technical support from Technology Commons, College of Life Science, National Taiwan University (Taiwan) is also acknowledged.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ibror.2017.03.002.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- Abele K., Yang J. Regulation of voltage-gated calcium channels by proteolysis. Sheng Li Xue Bao. 2012;64:504–514. [PMC free article] [PubMed] [Google Scholar]
- Ben-Johny M., Dick I.E., Sang L., Limpitikul W.B., Kang P.W., Niu J., Banerjee R., Yang W., Babich J.S., Issa J.B. Towards a unified theory of calmodulin regulation (calmodulation) of voltage-gated calcium and sodium channels. Curr. Mol. Pharmacol. 2015;8:188–205. doi: 10.2174/1874467208666150507110359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Johny M., Yue D.T. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol. 2014;143:679–692. doi: 10.1085/jgp.201311153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M.J. Calcium signalling and psychiatric disease: bipolar disorder and schizophrenia. Cell Tissue Res. 2014;357:477–492. doi: 10.1007/s00441-014-1806-z. [DOI] [PubMed] [Google Scholar]
- Bichet P., Mollat P., Capdevila C., Sarubbi E. Endogenous glutathione-binding proteins of insect cell lines: characterization and removal from glutathione S-transferase (GST) fusion proteins. Protein Expr. Purif. 2000;19:197–201. doi: 10.1006/prep.2000.1239. [DOI] [PubMed] [Google Scholar]
- Brice E., Durward H. Multidisciplinary records: a step in the right direction? Paediatr. Nurs. 1997;9:26–27. doi: 10.7748/paed.9.10.26.s25. [DOI] [PubMed] [Google Scholar]
- Chaudhuri D., Issa J.B., Yue D.T. Elementary mechanisms producing facilitation of CaV2.1 (P/Q-type) channels. J. Gen. Physiol. 2007;129:385–401. doi: 10.1085/jgp.200709749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou A.C., Ju Y.T., Pan C.Y. Calmodulin interacts with the sodium/calcium exchanger NCX1 to regulate activity. PLoS One. 2015;10:e0138856. doi: 10.1371/journal.pone.0138856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola T., Waldmann R., Borsotto M., Heurteaux C., Romey G., Mattei M.G., Lazdunski M. Molecular cloning of a murine N-type calcium channel alpha 1 subunit. Evidence for isoforms, brain distribution, and chromosomal localization. FEBS Lett. 1994;338:1–5. doi: 10.1016/0014-5793(94)80105-3. [DOI] [PubMed] [Google Scholar]
- Currie K.P., Fox A.P. Differential facilitation of N- and P/Q-type calcium channels during trains of action potential-like waveforms. J. Physiol. 2002;539:419–431. doi: 10.1113/jphysiol.2001.013206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcour A.H., Lipscombe D., Tsien R.W. Multiple modes of N-type calcium channel activity distinguished by differences in gating kinetics. J. Neurosci. 1993;13:181–194. doi: 10.1523/JNEUROSCI.13-01-00181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria C.D., Soong T.W., Alseikhan B.A., Alvania R.S., Yue D.T. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Dick I.E., Limpitikul W.B., Niu J., Banerjee R., Issa J.B., Ben-Johny M., Adams P.J., Kang P.W., Lee S.R., Sang L. A rendezvous with the queen of ion channels: three decades of ion channel research by David T Yue and his calcium signals laboratory. Channels (Austin) 2016;10:20–32. doi: 10.1080/19336950.2015.1051272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick I.E., Tadross M.R., Liang H., Tay L.H., Yang W., Yue D.T. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubel S.J., Starr T.V., Hell J., Ahlijanian M.K., Enyeart J.J., Catterall W.A., Snutch T.P. Molecular cloning of the alpha-1 subunit of an omega-conotoxin-sensitive calcium channel. Proc. Natl. Acad. Sci. U. S. A. 1992;89:5058–5062. doi: 10.1073/pnas.89.11.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K., Luebke J.I., Turner T.J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- Eckert R., Chad J.E. Inactivation of Ca2+ channels. Prog. Biophys. Mol. Biol. 1984;44:215–267. doi: 10.1016/0079-6107(84)90009-9. [DOI] [PubMed] [Google Scholar]
- Eckert R., Tillotson D.L. Calcium-mediated inactivation of the calcium conductance in caesium-loaded giant neurones of Aplysia californica. J. Physiol. 1981;314:265–280. doi: 10.1113/jphysiol.1981.sp013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon J.L., Baker M.R., Xiong L., Loy R.E., Yang G., Dirksen R.T., Hamilton S.L., Quiocho F.A. Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+* calmodulins. Proc. Natl. Acad. Sci. U. S. A. 2009;106:5135–5140. doi: 10.1073/pnas.0807487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Few A.P., Nanou E., Watari H., Sullivan J.M., Scheuer T., Catterall W.A. Asynchronous Ca2+ current conducted by voltage-gated CaV2.1 and CaV2.2 channels and its implications for asynchronous neurotransmitter release. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E452–E460. doi: 10.1073/pnas.1121103109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y., Mynlieff M., Dirksen R.T., Kim M.S., Niidome T., Nakai J., Friedrich T., Iwabe N., Miyata T., Furuichi T. Primary structure and functional expression of the omega-conotoxin-sensitive N-type calcium channel from rabbit brain. Neuron. 1993;10:585–598. doi: 10.1016/0896-6273(93)90162-k. [DOI] [PubMed] [Google Scholar]
- Hell J.W., Westenbroek R.E., Breeze L.J., Wang K.K., Chavkin C., Catterall W.A. N-methyl-D-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc. Natl. Acad. Sci. U. S. A. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa M.B., Lana B., Margas W., Dolphin A.C., Ryan T.A. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–125. doi: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultschig C., Hecht H.J., Frank R. Systematic delineation of a calmodulin peptide interaction. J. Mol. Biol. 2004;343:559–568. doi: 10.1016/j.jmb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Johny M.B., Yang P.S., Bazzazi H., Yue D.T. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat. Commun. 2013;4:1717. doi: 10.1038/ncomms2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.Y., Rumpf C.H., Fujiwara Y., Cooley E.S., Van Petegem F., Minor D.L., Jr. Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure. 2008;16:1455–1467. doi: 10.1016/j.str.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A., Scheuer T., Catterall W.A. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J. Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H., Zheng H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer's disease. J. Neurochem. 2016;136:475–491. doi: 10.1111/jnc.13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H., DeMaria C.D., Erickson M.G., Mori M.X., Alseikhan B.A., Yue D.T. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Lin T.Y., Li B.R., Tsai S.T., Chen C.W., Chen C.H., Chen Y.T., Pan C.Y. Improved silicon nanowire field-effect transistors for fast protein-protein interaction screening. Lab. Chip. 2013;13:676–684. doi: 10.1039/c2lc40772h. [DOI] [PubMed] [Google Scholar]
- Liu Z., Vogel H.J. Structural basis for the regulation of L-type voltage-gated calcium channels: interactions between the N-terminal cytoplasmic domain and Ca2+-calmodulin. Front. Mol. Neurosci. 2012;5:38. doi: 10.3389/fnmol.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama Z., Wakamori M., Mori Y., Kawakami H., Nakamura S., Imoto K. Direct alteration of the P/Q-type Ca2+ channel property by polyglutamine expansion in spinocerebellar ataxia 6. J. Neurosci. 1999;19:RC14. doi: 10.1523/JNEUROSCI.19-12-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir A., Bell D.C., Stephens G.J., Page K.M., Dolphin A.C. Calcium channel beta subunit promotes voltage-dependent modulation of α1B by Gβγ. Biophys. J. 2000;79:731–746. doi: 10.1016/S0006-3495(00)76331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills L.R., Niesen C.E., So A.P., Carlen P.L., Spigelman I., Jones O.T. N-type Ca2+ channels are located on somata, dendrites, and a subpopulation of dendritic spines on live hippocampal pyramidal neurons. J. Neurosci. 1994;14:6815–6824. doi: 10.1523/JNEUROSCI.14-11-06815.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz I.M., Sabatini B.L., Regehr W.G. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Mori M.X., Vander Kooi C.W., Leahy D.J., Yue D.T. Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high-resolution mechanistic implications for channel regulation by Ca2+ Structure. 2008;16:607–620. doi: 10.1016/j.str.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely A., Wei X., Olcese R., Birnbaumer L., Stefani E. Potentiation by the beta subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- Olivera B.M., Miljanich G.P., Ramachandran J., Adams M.E. Calcium channel diversity and neurotransmitter release: the omega-conotoxins and omega-agatoxins. Annu. Rev. Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- Peterson B.Z., DeMaria C.D., Adelman J.P., Yue D.T. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Raghib A., Bertaso F., Davies A., Page K.M., Meir A., Bogdanov Y., Dolphin A.C. Dominant-negative synthesis suppression of voltage-gated calcium channel Cav2.2 induced by truncated constructs. J. Neurosci. 2001;21:8495–8504. doi: 10.1523/JNEUROSCI.21-21-08495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih P.Y., Lin C.L., Cheng P.W., Liao J.H., Pan C.Y. Calneuron I inhibits Ca2+ channel activity in bovine chromaffin cells. Biochem. Biophys. Res. Commun. 2009;388:549–553. doi: 10.1016/j.bbrc.2009.08.046. [DOI] [PubMed] [Google Scholar]
- Simms B.A., Zamponi G.W. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron. 2014;82:24–45. doi: 10.1016/j.neuron.2014.03.016. [DOI] [PubMed] [Google Scholar]
- Simons T.J. Calcium and neuronal function. Neurosurg. Rev. 1988;11:119–129. doi: 10.1007/BF01794675. [DOI] [PubMed] [Google Scholar]
- Singer D., Biel M., Lotan I., Flockerzi V., Hofmann F., Dascal N. The roles of the subunits in the function of the calcium channel. Science. 1991;253:1553–1557. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- Soderling T.R., Derkach V.A. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- Soong T.W., DeMaria C.D., Alvania R.S., Zweifel L.S., Liang M.C., Mittman S., Agnew W.S., Yue D.T. Systematic identification of splice variants in human P/Q-type channel alpha1(2.1) subunits: implications for current density and Ca2+-dependent inactivation. J. Neurosci. 2002;22:10142–10152. doi: 10.1523/JNEUROSCI.22-23-10142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross M.R., Dick I.E., Yue D.T. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell. 2008;133:1228–1240. doi: 10.1016/j.cell.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiakina V., Boone A.N., Fux J., Senatore A., Weber-Adrian D., Guillemette J.G., Spafford J.D. The calmodulin-binding, short linear motif, NSCaTE is conserved in L-type channel ancestors of vertebrate CaV1.2 and CaV1.3 channels. PLoS One. 2013;8:e61765. doi: 10.1371/journal.pone.0061765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillotson D. Inactivation of Ca conductance dependent on entry of Ca ions in molluscan neurons. Proc. Natl. Acad. Sci. U. S. A. 1979;76:1497–1500. doi: 10.1073/pnas.76.3.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Chung B.C., Yan H., Wang H.G., Lee S.Y., Pitt G.S. Structural analyses of Ca2+/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat. Commun. 2014;5:4896. doi: 10.1038/ncomms5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek R.E., Hell J.W., Warner C., Dubel S.J., Snutch T.P., Catterall W.A. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- Westenbroek R.E., Hoskins L., Catterall W.A. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J. Neurosci. 1998;18:6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.E., Brust P.F., Feldman D.H., Patthi S., Simerson S., Maroufi A., McCue A.F., Velicelebi G., Ellis S.B., Harpold M.M. Structure and functional expression of an omega-conotoxin-sensitive human N-type calcium channel. Science. 1992;257:389–395. doi: 10.1126/science.1321501. [DOI] [PubMed] [Google Scholar]
- Yagami T., Kohma H., Yamamoto Y. L-type voltage-dependent calcium channels as therapeutic targets for neurodegenerative diseases. Curr. Med. Chem. 2012;19:4816–4827. doi: 10.2174/092986712803341430. [DOI] [PubMed] [Google Scholar]
- Yang P.S., Alseikhan B.A., Hiel H., Grant L., Mori M.X., Yang W., Fuchs P.A., Yue D.T. Switching of Ca2+-dependent inactivation of CaV1.3 channels by calcium binding proteins of auditory hair cells. J. Neurosci. 2006;26:10677–10689. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A., Lewis R.S. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. J. Biol. Chem. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.