

Abstract
Objective
Individuals with Parkinson disease are more likely to develop melanoma, and melanoma patients are reciprocally at higher risk of developing Parkinson disease. Melanoma is strongly tied to red hair/fair skin, a phenotype of loss-of-function polymorphisms in the MC1R (melanocortin 1 receptor) gene. Loss-of-function variants of MC1R have also been linked to increased risk of Parkinson disease. The present study is to investigate the role of MC1R in dopaminergic neurons in vivo.
Methods
Genetic and pharmacological approaches were employed to manipulate MC1R, and nigrostriatal dopaminergic integrity was determined by comprehensive behavioral, neurochemical, and neuropathological measures.
Results
MC1Re/e mice, which carry an inactivating mutation of MC1R and mimic the human redhead phenotype, have compromised nigrostriatal dopaminergic neuronal integrity, and they are more susceptible to dopaminergic neuron toxins 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Furthermore, a selective MC1R agonist protects against MPTP-induced dopaminergic neurotoxicity.
Interpretation
Our findings reveal a protective role of MC1R in the nigrostriatal dopaminergic system, and they provide a rationale for MC1R as a potential therapeutic target for Parkinson disease. Together with its established role in melanoma, MC1R may represent a common pathogenic pathway for melanoma and Parkinson disease.
Patients with Parkinson disease (PD), one of the most common neurodegenerative diseases, generally have reduced risk of developing almost all types of cancer, with one notable exception—melanoma, a malignant tumor of melanin-producing cells in skin.1,2 A large number of epidemiological studies have reported that occurrence of melanoma is higher than expected among subjects with PD, and melanoma patients are reciprocally more likely to develop PD.1,2 Although well documented since the 1970s, mechanisms underlying this association between PD and melanoma remain largely unknown. Recent epidemiological studies further demonstrated the positive, bidirectional link not only in the patients themselves but also in their relatives,3,4 suggesting a possible genetic basis for the association between the two seemingly distinct conditions.
A cyclic adenosine monophosphate–stimulating G-protein– coupled receptor (GPCR), melanocortin 1 receptor (MC1R) contributes to the regulation of skin physiology through the melanin synthetic pathway as well as pigmentation-independent mechanisms.5,6 Loss-of-function variants of MC1R in humans are associated with red hair and fair skin and increased risk of developing melanoma.7,8 In mice, an inactivating mutation of MC1R (MC1R extension, e/e) with a phenotype analogous to red hair/fair skin in humans,9 results in impaired skin protection and is sufficient to enhance melanoma formation.10,11 Furthermore, MC1Re/e mice display greater oxidative damage in skin, suggesting an oxidative stress–mediated mechanism in carcinogenesis.11 In addition to skin melanocytes, other tissue and cell types that express MC1R include adrenal tissue, immunocytes, endothelial cells,12 and possibly human astrocytes13 and periaqueductal gray neurons,14 suggesting functions of MC1R extending beyond those in skin. In an analysis of 2 large prospective cohorts led by our epidemiologist collaborators, we found that red hair individuals and individuals homozygous for the red hair–associated Arg151Cys allele of Cys were associated with an increased risk for PD.15 The association was substantiated by another study linking PD with an alternative red hair MC1R variant in an independent cohort.16 Although other studies17–20 did not replicate a significant MC1R– PD association, which may be attributable to technical or population differences across studies,21 MC1R has nevertheless emerged as having a potential role at the interface of melanoma and PD.
To explore such a role of MC1R in dopaminergic neurons and therefore a biological basis for the PD–melanoma link, we first assessed expression of MC1R in dopaminergic neurons of the substantia nigra (SN) in the mouse brain. Redhead MC1Re/e mice were then employed to investigate effects of MC1R inactivation on the nigrostriatal dopaminergic system under basal conditions and in well-established 6-hydroxydopamine (6- OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) models of PD.22 Furthermore, the neuroprotective potential of MC1R stimulation in an MPTP model was explored using a potent, specific MC1R agonist 1-(1-[3- methyl-L-histidyl-O-methyl-D-tyrosyl]-4-phenyl-4-piperidinyl)- 1-butanone (BMS-470539).23
Materials and Methods
Animals
First reported as an extension in 1993,9 MC1Re/e mice carry a frameshift mutation due to a deletion of a single nucleotide at position 549. The mutation produces a prematurely terminated, nonfunctioning protein and results in a yellow/red coat in homozygous mice despite being on a C57BL/6J background. 9–11 Heterozygous (MC1Re/+) mice, conversely, do not display melanoma-related skin phenotypes. MC1Re/e mice were backcrossed with C57BL/6J mice from the Jackson Laboratory (Bar Harbor, ME). Homozygous MC1Re/e and littermate wildtype (WT) mice were used for experiments.
For effects of MC1R agonist BMS-470539, adult (3 months old) male C57Bl/6J mice were purchased from the Jackson Laboratory.
Animals were maintained in home cages at constant temperature with a 12-hour light/dark cycle and free access to food and water. All experiments were performed in accordance with a protocol approved by the Massachusetts General Hospital Animal Care and Use Committee.
Measurement of Locomotion
Locomotion was assessed using an automated open field recording system (San Diego Instruments, San Diego, CA). Horizontal locomotor activity during the first 60 minutes (7–8 PM) of the dark phase of the light cycle was recorded and analyzed as described.24 Ambulation was quantified as sequential breaks in adjacent beams (55mm apart).
MC1R Immunohistochemistry
Naive adult male C57Bl/6J mice were perfused, and brains were postfixed. Following cryoprotection, brains were sectioned. The sections were heated for antigen retrieval25 and then treated with H2O2 and blocking milk containing 0.3% Triton X-100. Primary anti-MC1R (Santa Cruz Biotechnology, Santa Cruz, CA; #SC-19485 at 1:50 dilution) was applied overnight at 4 °C. For a peptide blocking control, the primary antibody was preincubated with immunizing peptide (#SC-19485P, 1:25 dilution) overnight. For a control without primary antibody, it was replaced by phosphate-buffered saline only. Sections were Nissl counterstained.
Immunofluorescence Double Staining
For MC1R and tyrosine hydroxylase (TH) double labeling, mice were perfused and their brains were sectioned. Tyramide signal amplification (Life Technologies, Carlsbad, CA; #MP20911) was employed per the manufacturer’s instructions. Briefly, sections were heated for antigen retrieval and incubated with the above primary MC1R antibody at 1:50 dilution and then stained with tyramide-labeled Alexa Fluor 488. Subsequent TH staining was performed by incubating the sections with anti-TH primary antibody (Sigma, St Louis, MO; #T1299, 1:500 dilution) and Alexa Fluor 546 donkey antimouse secondary antibody.
Immunohistochemistry for TH and Stereological Cell Counting
For TH immunohistochemistry in naive mice, the animals were perfused and the brains were postfixed for 48 hours. For TH immunohistochemistry with tissues from dopaminergic toxin experiments, mice were sacrificed by rapid cervical dislocation. The hinder brain block containing midbrain was immediately dissected and fixed for 48 hours. The brains were then cryoprotected, rapidly frozen by immersion in isopentane on dry ice, and sectioned at 30 μm. Sections were processed free-floating. Primary antibody was mouse monoclonal anti-TH (Sigma, #T1299) at 1:800.26
For TH stereology, a complete set of serial sections were immunostained for TH and counterstained for Nissl. Sections were analyzed stereologically as described.26,27 The number of TH-positive (and TH-negative) neurons in the SN on both sides of each section was counted, and the total number of TH-positive (and TH-negative) neurons on each side (6- OHDA lesion) or both sides of the SN from individual animals was calculated.
Western Blotting
For Western blot analysis of MC1R in total protein, adrenal or brain tissues were obtained. Total protein was extracted and electrophoresed. After transferring, the membrane was blocked in 5% bovine serum albumin and then incubated with the above MC1R antibody at 1:500. β-Tubulin was probed using anti–β-tubulin (Cell Signaling Technology #2146) as loading control.
For Western blot analysis of MC1R in membrane and cytosolic fractions, membrane and cytosolic fractions were obtained with a Membrane Protein Extraction Kit (Thermo Scientific, Waltham, MA; #89842). Na+/K+-adenosine triphosphatase (Cell Signaling Technology #3010) and β-tubulin (Cell Signaling Technology #2146) were probed as loading controls for membrane and cytosolic fractions, respectively.
To validate specificity of the MC1R primary antibody in vitro, B16 mouse melanoma cells were treated with short hairpin RNA (shRNA) construct targeting mouse MC1R (target sequence: 5′-AATGGAGATCAGGAAGGGATG-3′) according to published methods.28 Cells and reagents were kind gifts from Dr Rutao Cui, Boston University School of Medicine. Primary MC1R antibody was diluted at 1:1,000.
Laser Capture Microdissection and Reverse Transcriptase Polymerase Chain Reaction
To detect MC1R mRNA in dopaminergic neurons in the mouse brain, approximately 2,000 nigral TH-positive neurons were laser-captured. RNA was extracted, and reverse transcriptase polymerase chain reaction (RT-PCR) was performed.29 Primer sequences were: forward TGGTAAGTGTCAGCATCGTG and reverse TGATAACGCAGCGCATAGAA.
Determination of Protein Carbonyls
Protein carbonyls in brain tissues were detected using the Oxyblot Protein Oxidation Detection Kit (Millipore, Billerica, MA).26 Band density was analyzed and normalized with Ponceau staining using the ImageJ system.
Determination of Oxidative DNA Damage
Nuclear DNA was digested enzymatically, and N-labeled standard lesions were added. Lesions were separated by high-performance liquid chromatography (HPLC) and analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/ MS/MS) as previously described.11,30
6-OHDA Lesion
Mice were pretreated with desipramine, and a solution of 6- OHDA (5.0 μg/μl) was infused into the left striatum as previously described.26
Rotational Behavior Assessmen
Spontaneous and amphetamine (5mg/kg)-induced rotational behavior was tested at 3 weeks after the 6-OHDA lesion using an automated rotometry system (San Diego Instruments) for 60 minutes as previously described.26
MPTP Treatment Regimens
For MPTP toxicity in MC1Re/e mice, a subacute regimen was employed.31,32 Mice were administered intraperitoneally (i.p.) with MPTP-HCl 20mg/kg or saline once daily for 4 consecutive days and sacrificed 7 days after the last MPTP injection.
For effects of MC1R agonist BMS-470539 on MPTP toxicity, a single dose of MPTP-HCl (40mg/kg)31,32 or saline was injected i.p., and BMS-470539 dihydrochloride (Tocris Bioscience, Bristol, UK) or vehicle (water) was administrated 10 minutes before and 60 minutes after MPTP. Animals were sacrificed 7 days after the injections. The same treatment paradigm was employed to examine whether BMS-470539 changes MPTP metabolism. Mice were sacrificed 90 minutes after MPTP administration.
MC1R Agonist BMS-470539 and Its Pharmacokinetics
Male C57Bl/6J mice were dosed with BMS-470539 (20mg/kg subcutaneously [s.c.])23 and sacrificed at 5 time points (n=3) from 5 to 120 minutes. BMS-470539 was measured in plasma and brain by LC-MS/MS/MS by Cyprotex (Watertwon, MA).
Measurement of MPTP Metabolite
Striatal 1 methyl-4-phenylpyridinium (MPP+) was measured using HPLC-ultraviolet (UV) as previously described.33
Measurement of Dopamine and Metabolites
Dopamine (DA) and metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were determined by standard HPLC with electrochemical detection, as previously described.26
Statistical Analysis
All values are expressed as mean±standard error of the mean. The difference between two groups was analyzed by Student t test. Multiple comparisons among groups were performed by 1- way or 2-way analysis of variance (ANOVA), and Tukey post hoc test was performed when an overall significance was demonstrated with 1-way ANOVA or a main effect of genotype was demonstrated with 2-way ANOVA. p≤0.05 is considered statistically significant.
Results
MC1R Is Expressed in Dopaminergic Neurons of the SN of Adult C57BL Mice
To probe a role of MC1R in dopaminergic neurons of the SN, we first assessed expression of MC1R in naive C57BL/6J mouse brain. Western blot analysis of MC1R immunoreactivity in the striatum and the ventral midbrain revealed a band corresponding to a molecular weight of ~35kDa, the known size of MC1R (Fig 1). Immunohistochemistry demonstrated, in a coronal section of the ventral midbrain, positive staining for MC1R in the SN pars compacta (SNpc), and the staining vanished when antibody peptide was applied or when primary antibody was omitted. Immunofluorescence double labeling indicated colocalization of MC1R and TH, a marker for dopaminergic neurons within SNpc. MC1R appeared to be located on cell surface as well as in cytoplasm, consistent with Western blot results showing the presence of a ~35kDa band in both membrane, although weak, and cytosolic fractions extracted from ventral midbrain and striatal tissues. Specificity of MC1R primary antibody was indirectly verified in vitro by shRNA knockdown of the mouse MC1R in mouse B16 melanoma cells.28 MC1R is known to be expressed in B16 cells.28 Reduced MC1R was demonstrated in cells treated with mouse MC1R shRNA but not scrambled construct or vehicle-treated control. shRNA treatment did not alter β-tubulin band density. The presence of MC1R protein is supported both technically and conceptually by its mRNA expression in the SN, more specifically, in dopaminergic neurons by laser capture microdissection of nigral TH-positive neurons and RT-PCR. A band corresponding to predicted size of MC1R was demonstrated. Despite lack of theoretically definite negative control (eg, MC1R knockout mice), our data using 3 different methods suggest the existence of MC1R in the SN in C57Bl/6J mice.
FIGURE 1.

Expression of MC1R in the substantia nigra (SN) in adult naive C57BL/6J mouse brain. (A) Western blot using anti- MC1R antibody with total tissue lysate (left panel), membrane (m) or cytosolic fraction (c) (middle panel) isolated from ventral midbrain (MB) and the striatum (Str), or B16 mouse melanoma cells treated with shRNA targeting mouse MC1R or scrambled construct (right panel). Adrenal (Adr), where MC1R is known to be expressed, serves as a control.12 β-Tubulin and Na+/K+-adenosine triphosphatase (ATPase) are loading controls for total and cytosolic proteins, and membrane proteins, respectively. (B) Immunostaining of MC1R and Nissl counterstaining in a ventral midbrain coronal section (top panel). Cells in the box in the SN pars compacta are shown at higher magnification on the right. Scale bar=25 μm. Bottom panels show ventral midbrain coronal sections incubated with blocking peptide (left) or no primary antibody (right). (C) Fluorescence double labeling for tyrosine hydroxylase (TH; red) and MC1R (green) in the SN. Scale bars=10 μm. (D) Laser capture microdissection of TH-positive neurons from the SN and reverse transcriptase polymerase chain reaction analysis of MC1R mRNA. Glyceraldehyde-3- phosphate dehydrogenase (GAPDH) serves as internal control.
Impaired Nigrostriatal Dopaminergic Neuronal Integrity in Redhead MC1Re/e Mice
To investigate potential MC1R influence on the nigrostriatal dopaminergic system, we monitored locomotor activity in MC1Re/e mice by open field testing for the first hour of the dark phase in a 12-hour-light/12-hour-dark cycle.24 Despite being generally healthy and fertile, redhead MC1Re/e mice displayed progressive decline in locomotor activity. Compared to littermate WT controls, there was a significant decrease in the number of adjacent photobeam breaks in 8-month-old (p=0.05) and 14- month-old MC1Re/e mice (p=0.01). Young MC1Re/e mice performed normally (Fig 2A).
FIGURE 2.

Compromised nigrostriatal dopaminergic integrity in MC1Re/e mice. (A) Locomotor activity in MC1Re/e mice and littermate wild-type (WT) controls assessed by adjacent beam breaks during the first hour of the dark cycle in an open field chamber (n=15, WT and MC1Re/e 2- and 8-month-old mice; n=14 and 13, WT and MC1Re/e 14-month-old mice). *p=0.05, **p=0.01 versus WT littermates; ##p=0.01 versus 2-month-old self-control MC1Re/e mice by 2-way analysis of variance (ANOVA) followed by Tukey post hoc test. p=0.038 for genotype effect; p=0.0002 for time effect; p=0.11 for interaction. (B) Striatal dopamine (DA) content in MC1Re/e mice and WT littermates determined by high-performance liquid chromatography coupled with electrochemical detection (n=5, WT and MC1Re/e at all 3 age points). *p < 0.05, **p < 0.01 versus WT littermates, 1-way ANOVA followed by Tukey post hoc test. (C) Stereological quantification of tyrosine hydroxylase (TH)-positive and TH-negative (Nissl-positive) neurons in substantia nigra pars compacta in MC1Re/e mice and littermate WT controls (n=5, WT and MC1Re/e 2- and 14-month-old mice; n=6, WT and MC1Re/e 8-month-old mice). **p=0.01 versus WT littermates, 1- way ANOVA followed by Tukey post hoc test. (D) Representative sets of nigral sections from MC1Re/e mice and littermate WT controls stained for TH. Cells in the black boxes at indicated age points are shown at higher magnification in the lower panels. Scale bars=20 μm.
We then assessed neurochemical and morphological measures of dopaminergic nigrostriatal neuron integrity26,27 in MC1Re/e mice as they age. HPLC coupled with electrochemical detection revealed 16% and 26% reduction in DA content in the striatum of 8- and 14-month-old MC1Re/e mice, respectively, significantly lower than their WT littermates (8 months old, p<0.05; 14 months old, p<0.01; see Fig 2). The reduced striatal DA in MC1Re/e mice is likely caused by loss of dopaminergic neurons in the SN, as demonstrated by immunohistochemistry and stereological counting of cells stained positive for TH. MC1Re/e mice at 8 months of age had 24% fewer TH-positive neurons (p<0.01), and at 14 months of age there were 39% fewer nigral dopaminergic neurons compared to WT littermates (p=0.01). Nissl counterstaining and stereological counting of TH-negative neurons in the SNpc revealed no difference between MC1Re/e and WT littermates at any of the ages. Total summed counts of TH-positive and TH-negative cells in the SNpc, which represent its total number of neurons, were significantly lower in MC1Re/e mice at 8 (p=0.01) and 14 months (p<0.01) but not at 2 months (p=0.20) of age as compared to WT littermates. Representative images of TH staining in the SN showed loss of TH-positive cells in 8- and 14-month-old redhead MC1Re/e mice. There is no significant change in the size of either the nigra or individual TH-positive neurons (Fig 2D and quantitative data not shown).
Increased Oxidative Damage in the Ventral Midbrain of Adult MC1Re/e Mice
Our collaborative team previously reported an oxidative stress mechanism in MC1R-mediated, UV-independent carcinogenesis in skin using MC1Re/e mice.11 Oxidative stress has been implicated in neurodegenerative diseases including PD.34 To determine whether disrupted MC1R is associated with changes in oxidative stress in the nigrostriatal system, we assessed protein carbonyls, a marker of oxidative protein damage, by Oxyblot.26 Increased protein carbonyls were detected in the ventral midbrain (p<0.05; Fig 3A) but not the striatum (see Fig 3B) of adult (3–6 months old) MC1Re/e mice compared to littermate WT controls.
FIGURE 3.

MC1Re/e mice have greater oxidative damage in the ventral midbrain. (A, B) Protein carbonyls assessed by Oxyblot in (A) the ventral midbrain and (B) the striatum in adult (3–6-month-old) MC1Re/e mice and littermate wild-type (WT) controls (n=4, WT and MC1Re/e ventral midbrain; n=5, WT and MC1Re/e striatum). *p< 0.05 versus WT by Student t test. (C, D) DNA damage markers 8,5′-cyclo-2′-deoxyadenosine (cdA) and 8,5′-cyclo-2′-deoxyguanosine (cdG) assessed by liquid chromatography–tandem mass spectrometry in (C) the ventral midbrain and (D) the striatum in adult MC1Re/e mice (5–8 months old) and littermate WT controls (n=4, WT and MC1Re/e mice, pooled from 12 animals of each genotype). *p< 0.05 versus WT by Student t test. [Color figure can be viewed at wileyonlinelibrary.com]
Oxidative DNA damage markers 8,5′-cyclo-2′-deoxyadenosine (cdA) and 8,5′-cyclo-2′-deoxyguanosine (cdG) were evaluated in MC1Re/e mice using established LC-MS/ MS/MS.11,30 Increased levels of S-cdA and S-cdG, which lead to A to T and G to A mutations, respectively, were demonstrated in ventral midbrain in adult MC1Re/e mice (5–8 months old) as compared to WT littermates (p=0.036, S-cdA; p=0.046, S-cdG; see Fig 3C). R-cdA did not show a significant difference between the 2 genotypes (13.6±0.8 and 12.4±1.0 lesions per 107 nucleosides, WT and MC1Re/e; p>0.05). There were no significant changes in S-cdA and R-cdA levels in the striatum in adult redhead MC1Re/e mice compared to WT controls (see Fig 3D).
MC1Re/e Mice Are More Susceptible to Dopaminergic Neurotoxin 6-OHDA
In humans, loss-of-function variants of MC1R produce red hair/fair skin phenotype and are linked to higher risk of both melanoma7,8 and PD.15,16 Our collaborative team previously reported the lower threshold for melanoma induction in redhead MC1Re/e mice.11 To investigate whether the MC1R disruption may contribute to greater susceptibility to PD, we assessed dopaminergic phenotype of redhead MC1Re/e mice in a well-established 6-OHDA model of PD.22,26 Adult (6–8 months old) MC1Re/e and >littermate WT mice received unilateral intrastriatal infusion of 10 μg 6-OHDA. This model induces progressive and retrograde degeneration of the nigrostriatal system by a combined effect of reactive oxygen species and quinones. 22 The animals were then assessed for behavioral, neurochemical, and anatomical measures of dopaminergic deficits (Fig 4A).26 Comparing to WT littermates, MC1Re/e mice displayed a statistically significant increase in ipsilateral net rotations after stimulation with a DA-releasing agent amphetamine (p<0.05) despite no difference in spontaneous rotations ipsilateral to the lesion (see Fig 4B).
FIGURE 4.

MC1Re/e mice are more susceptible to dopaminergic toxin 6-hydroxydopamine (6-OHDA). (A) Adult (6–8-month-old) MC1Re/e and littermate wild-type (WT) mice were infused with 10μg 6-OHDA into the left striatum. Rotational behavior was assessed at 3 weeks, and animals were sacrificed at 4 weeks after 6-OHDA lesion. (B) Spontaneous and 5mg/kg amphetamine-induced net ipsilateral rotations in MC1Re/e mice and littermate WT controls (n=12, WT and MC1Re/e). *p< 0.05 versus WT by Student t test. (C) Striatal dopamine (DA) measured by high-performance liquid chromatography (HPLC) and DA turnover rate (homovanillic acid [HVA]/DA ratio, HVA determined by HPLC) on contralateral (Contra) unlesioned side and ipsilateral (Ipsi) lesioned side in MC1Re/e mice and WT littermates (n=12, WT and MC1Re/e). **p < 0.01 versus WT Contra side; ##p< 0.01 versus WT Ipsi side, 1-way analysis of variance (ANOVA) followed by Tukey post hoc test. (D) Disruption of substantia nigra (SN) dopaminergic neurons (stained positive for tyrosine hydroxylase [TH]) on the Ipsi lesioned side of an MC1Re/e mouse after 6-OHDA lesion. (E) Stereological quantification of nigral TH-positive neurons expressed as absolute counts and percentage of Contra side in MC1Re/e mice and WT littermates (n=12,WT and MC1Re/e). **p< 0.01 versus WT Contra side; ##p< 0.01 versus WT Ipsi side, 1-way ANOVA followed by Tukey post hoc test for absolute counts and Student t test for normalized counts. [Color figure can be viewed at wileyonlinelibrary.com]
The greater DA imbalance between ipsilateral and contralateral sides in MC1Re/e mice was confirmed by HPLC analysis of DA content in the striatum. 6- OHDA induced greater depletion of DA and a further increase in ratio of HVA, a DA metabolite, to DA in the striatum in MC1Re/e mice than WT littermates on the lesioned side (p<0.01, striatal DA; p<0.01, HVA/ DA; see Fig 4C). After normalization for the significant difference in DA on the unlesioned side between the 2 groups of animals (p<0.01) by expressing it as percentage of contralateral control value, MC1Re/e mice had 12.8±2.1% residual DA on the lesioned side, significantly lower than the residual in WT mice (31.3±3.0%; p<0.01).
A more substantial loss of nigral dopaminergic neurons induced by 6-OHDA infusion was demonstrated in MC1Re/e mice compared to their WT littermates (see Fig 4D). Stereological cell counts revealed fewer remaining TH-positive neurons on the lesioned side (p<0.01). The loss of dopaminergic neurons was still greater as a proportion of the unlesioned contralateral side in MC1Re/e mice (32.4±4.6%) than in WT (49.8±1.6%; p<0.01; see Fig 4E), despite reduced numbers on the unlesioned side (p<0.01), which confirms the compromised dopaminergic integrity of intact MC1Re/e mice as shown in Figure 2. These results demonstrate higher suceptibility of redhead MC1Re/e mice to the dopaminergic toxin 6-OHDA.
MC1Re/e Mice Are More Susceptible to Dopaminergic Neurotoxin MPTP
Exacerbated dopaminergic neurotoxicity was demonstrated in MC1Re/e mice in another well-established, complementary toxin model of PD.22,31 After systemic administration, MPTP enters the brain and is locally converted to its active metabolite MPP+, which in turn is selectively transported into dopaminergic neurons, where it exerts toxicity by inhibiting complex I of the mitochondrial electron transport chain.22,32 A subacute regimen (20mg/kg MPTP-HCl i.p. once daily for 4 days) induced greater reduction of striatal DA in adult (6–8 months old) MC1Re/e mice than in WT littermates 7 days after the last MPTP administration (p<0.01; Fig 5A). Even with normalization to saline-treated control mice to account for basal differences between the 2 control groups (p=0.01), a greater percentage loss of striatal DA in MC1Re/e mice treated with MPTP (82.2±2.5%) persisted compared to WT MPTP-treated mice (59.6±3.1%; p=0.05). MC1Re/e mice also displayed a 350% increase in DA turnover rate (HVA/DA ratio) after MPTP administration in comparison to a 180% increase in WT littermates (p<0.05).
FIGURE 5.

MC1Re/e mice are more susceptible to dopaminergic toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Adult (6–8-month-old) MC1Re/e and littermate wildtype (WT) mice were injected intraperitoneally (i.p.) with 20mg/kg MPTP-HCl or control vehicle (saline) once daily for 4 days and sacrificed 7 days after the last MPTP administration. (A) Dopamine (DA) in the striatum measured by high-performance liquid chromatography (HPLC) and DA turnover rate (homovanillic acid [HVA]/DA ratio, HVA determined by HPLC) in MPTP or vehicle control (CON) MC1Re/e mice and WT littermates (n=6, WT and MC1Re/e CON; n=8 and 7, WT and MC1Re/e MPTP). *p< 0.05 versus WT CON; #p < 0.05, ##p< 0.01 versus WT MPTP, 1-way analysis of variance (ANOVA) followed by Tukey post hoc test. (B) Substantial loss of TH-positive neurons in the substantia nigra (SN) of an MC1Re/e mouse after MPTP subacute treatment. (C) Stereological quantification of nigral tyrosine hydroxylase (TH)- positive neurons expressed as absolute counts and percentage of CON group in MC1Re/e mice and WT littermates (n=6, WT and MC1Re/e CON; n=8 and 7, WT and MC1Re/e MPTP). **p< 0.01versus WT CON; #p< 0.05, ##p< 0.01 versus WT MPTP, 1-way ANOVA followed by Tukey post hoc test for absolute counts and Student t test for normalized counts. (D) Striatal 1 methyl-4-phenylpyridinium (MPP+) detected by HPLC in MC1Re/e and littermate WT mice following 20mg/kg MPTP-HCl intraperitoneal administration (n=6 and 8, WT and MC1Re/e, both 90 minutes and 6 hours). [Color figure can be viewed at wileyonlinelibrary.com]
Anatomically, TH immunohistochemistry (see Fig 5B) and quantitative stereological analysis indicated an extensive loss of dopaminergic neurons in MPTP-treated MC1Re/e mice, with residual TH-positive SN neuron counts that were 35.5±5.5% of those in saline-treated MC1Re/e littermates, whereas the WT MPTP group had 55.1±5.4% of the nigral TH-positive neurons compared to their saline-treated WT littermates, with the MC1Re/e mice showing significantly greater losses of dopaminergic nigral neurons compared to their controls, either by absolute cell counts (p<0.01) or as a proportion of their vehicle-treated controls’ counts (p<0.05; see Fig 5C).
To evaluate whether exacerbated MPTP toxicity in MC1Re/e mice may be due to greater conversion of MPTP to its toxic metabolite MPP+ (or to slower rates of its clearance), we measured levels of MPP+ in the striatum 33 of MC1Re/e and littermate WT mice 90 minutes and 6 hours after a single i.p. injection of MPTP-HCl 20mg/kg. The 2 time points represent the peak and late phase, respectively, of MPP+ accumulation in brain after i.p. MPTP administration in naive WT mice.32 There was no statistically significant difference in striatal MPP+ levels between MC1Re/e and WT controls at either time point, suggesting MC1R disruption does not alter metabolism of the dopaminergic toxin, and therefore that the exacerbated toxicity in MC1Re/e mice is not due to increased local toxic metabolite after systemic injection (see Fig 5D).
MC1R Agonist Protects against MPTP-Induced Dopaminergic Neurotoxicity
The compromised nigrostriatal integrity in MC1Re/e mice and their high susceptibility to both 6-OHDA and MPTP suggest a protective role of MC1R signaling in the nigrostriatal dopaminergic system. To further probe this role, we investigated whether pharmacological activation of MC1R protects against MPTP toxicity using a commercially available MC1R agonist BMS-470539. BMS-470539 is a selective, potent MC1R agonist used in the laboratory to understand the role of MC1R in immunomodulation and inflammatory responses.23,35 We first determined the pharmacokinetic profile of BMS- 470539 in blood and brain in naive C57Bl/6J mice. After systemic injection (20mg/kg s.c.), the concentration of BMS-470539 in plasma peaked at 15 minutes (1,318±189ng/ml); in brain it peaked at 30 minutes (18±2ng/ml) and remained detectable until at least 120 minutes at ~10ng/ml (n=3 each time point), corresponding to the agonist’s reported median effective concentration of ~10ng/ml.23,35
Based on the pharmacokenetics and metablism of MPTP,32 we injected i.p. adult (3 months old) C57Bl/6J mice with a single dose of MPTP-HCl at 40mg/kg, and BMS-470539 (100mg/kg) was injected s.c. 10 minutes before and 60 minutes after MPTP. Mice were sacrificed 7 days later. The single dose regimen induced 66% reduction in DA in the striatum. BMS-470539 attenuated MPTP-induced DA depletion to 49% of that of the control group, resulting in significantly greater residual DA content compared to MPTP mice treated with vehicle (p<0.01; Fig 6). Measurement of DOPAC showed similar preservation in the DA metabolite in MPTP mice treated with BMS-470539. TH immunohistochemistry and stereological cell counting revealed 65% loss of dopaminergic neurons in the SN in the MPTP group, and treatment with BMS-470539 improved cell survival to 53% of that of the control group (p<0.05). BMS- 470539 itself did not alter striatal DA content or the number of TH-positive neurons. A separate experiment using the same treatment regimen showed BMS-470539 did not change MPTP metabolism. There was no significant difference in MPP+ in the striatum between BMS- 470539–treated and vehicle-treated mice 90 minutes after a single 40mg/kg i.p. injection of MPTP-HCl (14.2±2.3 and 17.6±0.6pmol/mg tissue, n=4 and 5, respectively; p>0.05).
FIGURE 6.
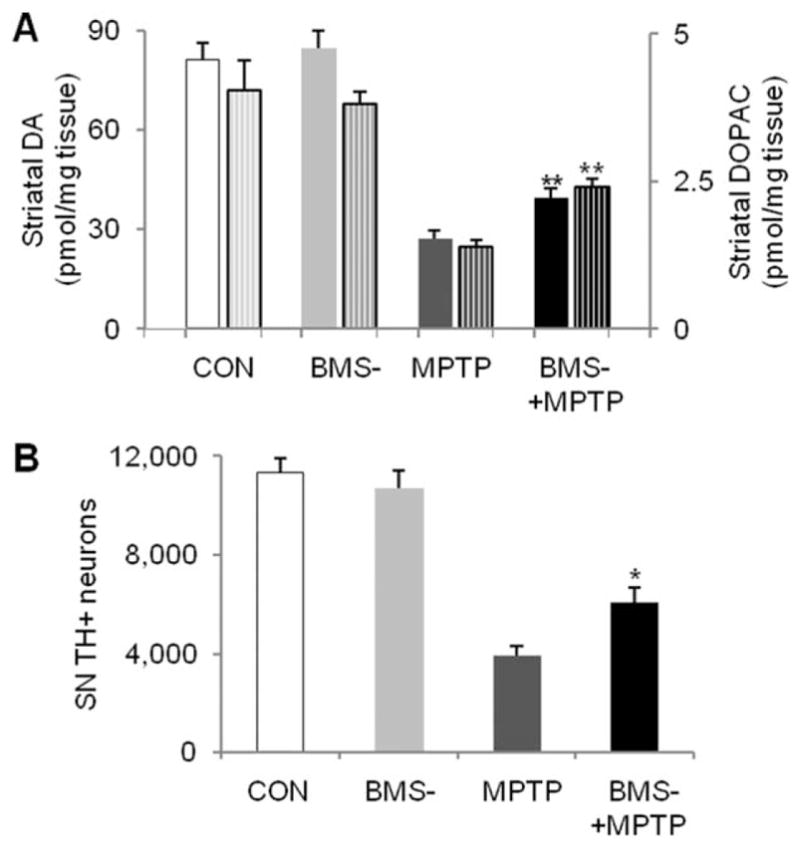
MC1R agonist protects against 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Adult (3-month-old) C57Bl/6 mice were administered with a single dose of MPTP-HCl at 40mg/kg or vehicle (saline) intraperitoneally, and BMS-470539 (BMS-) 100mg/kg or vehicle (water) was injected subcutaneously 10 minutes before and 60 minutes after MPTP. Mice were sacrificed 7 days later. (A) Striatal dopamine (DA) and metabolite 3,4-dihydroxyphenylacetic acid (DOPAC; patterned columns, the right y-axis) were analyzed by high-performance liquid chromatography. (B) Numbers of nigral tyrosine hydroxylase (TH)-positive neurons were counted by stereological method. *p < 0.05, **p < 0.01 versus MPTP group, 1-way analysis of variance followed by Tukey post hoc test (n=5, 4, 6, 7 for control [CON], BMS-, MPTP, and BMS-+MPTP groups, respectively). SN=substantia nigra.
Discussion
Epidemiological studies have demonstrated a reproducible bidirectional association between melanoma and PD.1–4 They also raised the possibility that the MC1R gene may mediate the association.15,16 Built on the previously characterized skin phenotype and melanoma tendency in redhead MC1Re/e mice,10,11 the present interdisciplinary study identifies a novel role of MC1R in the nigrostriatal dopaminergic system. Our findings demonstrate compromised nigrostriatal dopaminergic system in MC1Re/e mice and their greater susceptibility to local and systemic dopaminergic toxins 6-OHDA and MPTP. Furthermore, pharmacological stimulation of MC1R with a selective agonist attenuates MPTP neurotoxicity.
MC1R is expressed in the mouse SN at the protein and mRNA levels. Although juxtaposed to or on the cell surface in some cases, MC1R appears to be located intracellularly, overlapping with cytoplasmic TH fluorescence. Depending on cellular context and cell types, trafficking and localization of GPCRs are tightly regulated and are highly related to their signaling cycle.36 Immunoreactivity of GPCRs detected inside of the cells may reflect not only technical incorporation of fixation and detergent permeabilization but also functional status of the receptors; for example, they are newly synthesized to be transported to the plasma membrane or internalized following agonist stimulation,36,37 both often producing a typical punctate staining pattern in the case of MC1R.38 Relative cell surface expression versus intracellular retention has been reported to correlate with function of MC1R variants.39 The only neuronal demonstration of MC1R was in cytosome of human periaqueductal gray matter neurons.14 Intracellular MC1R undergoes oligomerization, which can significantly affect its signaling.6 It is unclear whether a band at higher molecular weight in both brain tissues and cells that appears to respond modestly to shRNA represents MC1R oligomers. Further characterization of MC1R subcellular distribution and oligomerization would be helpful in deciphering regulation of its function in the SN dopaminergic neurons.
Loss-of-function mutant MC1Re/e mice display loss of nigral dopaminergic neurons, reduced striatal DA content, and impaired locomotor activity under basal conditions. These results reveal a significant role of MC1R signaling in maintaining nigrostriatal dopaminergic neuron survival. The trophic influence of MC1R may be due to its direct actions given its expression in dopaminergic neurons. Dopaminergic neuron–specific disruption of MC1R would differentiate whether local MC1R or rather systemic MC1R is responsible. Although MC1R appears to be expressed in other cells in the SN, numbers of nigral TH-negative neurons in MC1Re/e mice remain unchanged. Furthermore, the impaired dopaminergic neuronal integrity is accompanied by increased oxidative damage in the ventral midbrain but not in the striatum. These data suggest that the benefit of MC1R activity may be relatively specific to dopaminergic neurons. However, it is not clear whether MC1R disruption affects only the nigrostriatal system or whether it has broader influence on other dopaminergic cell populations or other central nervous system (CNS) regions. The greater oxidative damage in the midbrain is in agreement with greater oxidative damage in skin in redhead MC1Re/e mice,11 supporting a common antioxidant mechanism underlying MC1R-mediated protection in melanocytes and dopaminergic neurons. MC1Re/e mice do not develop dopaminergic deficits until adulthood, indicating age-dependent effects of endogenous MC1R signaling on the nigrostriatal dopaminergic pathway. Aging is a primary risk factor for PD.40 Further investigation is needed to elucidate potential interplay between MC1R and aging and age-related factors including oxidative stress. Nevertheless, the demonstrated nigrostriatal dopaminergic deficits in MC1Re/e mice suggest possible predisposition of these mice to parkinsonism as a result of MC1R disruption.
Redhead MC1Re/e mice display higher susceptibility to 6-OHDA and MPTP, two established neuronal toxins that are commonly used to model dopaminergic neurodegeneration of human PD.22 Their exacerbated dopaminergic deficits manifest at anatomical, neurochemical, and functional levels after intrastriatal infusion of 6- OHDA or systemic administration of MPTP. The greater susceptibility is likely related to preexisting dopaminergic neuron dysfunction and oxidative damage. In addition, MC1R has been reported to play a central role in inflammatory responses.23,34 Oxidative stress and inflammatory responses are known to be involved in 6-OHDA and MPTP toxicity as well as in the pathophysiology of human PD.23,34 Dopaminergic neurodegeneration in the context of MC1R disruption may also directly or indirectly involve pigmentation pathway itself. A recent study intriguingly reported increased SN echogenicity in fair skin human subjects.41 Further investigations are essential to answer important questions regarding downstream intracellular pathways and mediators.
The enhanced vulnerability of redhead MC1R mutant mice in models of PD is consistent in parallel with previously reported lower threshold for melanoma induction in these mice,11 supporting protective effects of MC1R in both melanocytes and dopaminergic neurons against tumorigenesis and neurodegeneration, respectively. Together with the epidemiological associations of redhead MC1R loss-of-function variants and high risk of melanoma,7,8 and possibly high risk of PD,15–21 the predisposition of MC1R redhead mutation to melanoma and PD in animal models supports a potential role of MC1R as the biological basis of the melanoma and PD link. Future studies should focus on epidemiological, biological, and clinical interactions between MC1R and PD genetic factors,2 particularly α-synuclein, 42 and leucine-rich repeat kinase 2 (LRRK2).43 α-Synuclein has been implicated in melanoma pathophysiology, 44,45 and LRRK2 is the most commonly mutated among PD-associated genes in melanomas.46
Identifying the role of endogenous MC1R in adult dopaminergic system maintenance and defense presents a unique opportunity to explore the potential of MC1R as a novel therapeutic target for PD. BMS-470539, a selective MC1R agonist23 with modest blood–brain barrier penetrance, is found to protect dopaminergic neurons against MPTP-induced cell loss in the SN and DA depletion in the striatum. The neuroprotective action of pharmacological MC1R activation provides further evidence that compromised dopaminergic neuronal integrity in redhead MC1R mutant mice and their exacerbated dopaminergic neurotoxicity in models of PD are likely to be MC1R specific. Studies are underway to investigate specificity and efficacy of MC1R neuroprotection by complementary genetic MC1R overexpression. MC1R agonists are currently undergoing clinical testing for depigmentation disorders and an acute phototoxicity related condition.47,48 Whether systemic MC1R activation is required for its CNS effects, more CNS-penetrant versions may be needed to target neuronal MC1R for the treatment of PD.
Our findings that genetic inactivation of MC1R signaling impairs nigrostriatal dopaminergic neuron survival and that pharmacological activation of MC1R is neuroprotective uncover a protective role of MC1R in the nigrostriatal dopaminergic system. They provide evidence for targeting MC1R as a novel therapeutic strategy for PD. Together with epidemiological associations between MC1R, melanoma,7,8 and PD,15–21 and the known roles of MC1R in melanocytes and melanoma pathogenesis,1,5,60,11 our data support a shared mechanistic link between melanoma and PD.
Acknowledgments
This work is supported by The National Institute of Neurological Disorders and Stroke (1R21NS090246- 01A1 to X. C.), National Natural Science Foundation of China (81471293 to X.C.), the RJG Foundation (2011D004473 to X. C.), the Michael J. Fox Foundation (9908 to X. C.), the Milstein Medical Asian American Partnership Foundation (2015 to fellow W. C. and mentor X. C.), The National Institute of Neurological Disorders and Stroke (K24NS060991 to M.A.S.), and the US Department of Defense (W81XWH-11-1-0150, M.A.S.). We thank Y. Xu for animal breeding and genotyping.
Footnotes
Author Contributions
X.C., H.C., D.E.F., and M.A.S. contributed to conception and design of the study; X.C., H.C., W.C., M.M., B.Y., F.Z., R.L., H.L., K. R., C.R.V., Y.Y., and Y.W. contributed to acquisition and analysis of data; X.C. wrote the article.
Potential Conflicts of Interest
Nothing to report.
References
- 1.Liu R, Gao X, Lu Y, Chen H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology. 2011;76:2002–2009. doi: 10.1212/WNL.0b013e31821e554e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Devine MJ, Plun-Favreau H, Wood NW. Parkinson’s disease and cancer: two wars, one front. Nat Rev Cancer. 2011;11:812–823. doi: 10.1038/nrc3150. [DOI] [PubMed] [Google Scholar]
- 3.Gao X, Simon KC, Han J, et al. Family history of melanoma and Parkinson disease risk. Neurology. 2009;73:1286–1291. doi: 10.1212/WNL.0b013e3181bd13a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kareus SA, Figueroa KP, Cannon-Albright LA, Pulst SM. Shared predispositions of parkinsonism and cancer: a population-based pedigree-linked study. Arch Neurol. 2012;69:1572–1577. doi: 10.1001/archneurol.2012.2261. [DOI] [PubMed] [Google Scholar]
- 5.Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer. 2012;12:349–361. doi: 10.1038/nrc3218. [DOI] [PubMed] [Google Scholar]
- 6.Wolf Horrell EM, Boulanger MC, D’Orazio JA. Melanocortin 1 receptor: structure, function, and regulation. Front Genet. 2016;7:95. doi: 10.3389/fgene.2016.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raimondi S, Sera F, Gandini S, et al. MC1R variants, melanoma and red hair color phenotype: a meta-analysis. Int J Cancer. 2008;122:2753–2760. doi: 10.1002/ijc.23396. [DOI] [PubMed] [Google Scholar]
- 8.Williams PF, Olsen CM, Hayward NK, Whiteman DC. Melanocortin 1 receptor and risk of cutaneous melanoma: a meta-analysis and estimates of population burden. Int J Cancer. 2011;129:1730–1740. doi: 10.1002/ijc.25804. [DOI] [PubMed] [Google Scholar]
- 9.Robbins LS, Nadeau JH, Johnson KR, et al. Pigmentation phenotypes of variant extension locus alleles result from point mutations that alter MSH receptor function. Cell. 1993;72:827–834. doi: 10.1016/0092-8674(93)90572-8. [DOI] [PubMed] [Google Scholar]
- 10.D’Orazio JA, Nobuhisa T, Cui R, et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature. 2006;443:340–344. doi: 10.1038/nature05098. [DOI] [PubMed] [Google Scholar]
- 11.Mitra D, Luo X, Morgan A, et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature. 2012;491:449–453. doi: 10.1038/nature11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salazar-Onfray F, López M, Lundqvist A, et al. Tissue distribution and differential expression of melanocortin 1 receptor, a malignant melanoma marker. Br J Cancer. 2002;87:414–422. doi: 10.1038/sj.bjc.6600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong KY, Rajora N, Boccoli G, et al. A potential mechanism of local anti-inflammatory action of alpha-melanocyte-stimulating hormone within the brain: modulation of tumor necrosis factor-alpha production by human astrocytic cells. Neuroimmunomodulation. 1997;4:37–41. doi: 10.1159/000097313. [DOI] [PubMed] [Google Scholar]
- 14.Xia Y, Wikberg JE, Chhajlani V. Expression of melanocortin 1 receptor in periaqueductal gray matter. Neuroreport. 1995;6:2193–2196. doi: 10.1097/00001756-199511000-00022. [DOI] [PubMed] [Google Scholar]
- 15.Gao X, Simon KC, Han J, et al. Genetic determinants of hair color and parkinson’s disease risk. Ann Neurol. 2009;65:76–82. doi: 10.1002/ana.21535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tell-Marti G, Puig-Butille JA, Potrony M, et al. The MC1R melanoma risk variant p. R160W is associated with Parkinson disease. Ann Neurol. 2015;77:889–894. doi: 10.1002/ana.24373. [DOI] [PubMed] [Google Scholar]
- 17.Dong J, Gao J, Nalls M, et al. Susceptibility loci for pigmentation and melanoma in relation to Parkinson’s disease. Neurobiol Aging. 2014;35:1512.e5–1512.e10. doi: 10.1016/j.neurobiolaging.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lubbe SJ, Escott-Price V, Brice A, et al. Is the MC1R variant p.R160W associated with Parkinson’s? Ann Neurol. 2016;79:159–161. doi: 10.1002/ana.24527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gan-Or Z, Mohsin N, Girard SL, et al. The role of the melanoma gene MC1R in Parkinson disease and REM sleep behavior disorder. Neurobiol Aging. 2016;43:180.e7–180.e13. doi: 10.1016/j.neurobiolaging.2016.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorenzo-Betancor O, Wszolek ZK, Ross OA. Rare variants in MC1R/TUBB3 exon 1 are not associated with Parkinson’s disease. Ann Neurol. 2016;79:331. doi: 10.1002/ana.24581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tell-Martí G, Puig-Butille JA, Potrony M, et al. Reply: To PMID 25631192. Ann Neurol. 2015;78:153–154. doi: 10.1002/ana.24418. [DOI] [PubMed] [Google Scholar]
- 22.Blesa J, Przedborski S. Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front Neuroanat. 2014;8:155. doi: 10.3389/fnana.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herpin TF, Yu G, Carlson KE, et al. Discovery of tyrosine-based potent and selective melanocortin-1 receptor small-molecule agonists with anti-inflammatory properties. J Med Chem. 2003;46:1123–1126. doi: 10.1021/jm025600i. [DOI] [PubMed] [Google Scholar]
- 24.Chen JF, Beilstein M, Xu YH, et al. Selective attenuation of psychostimulant-induced behavioral responses in mice lacking A(2A) adenosine receptors. Neuroscience. 2000;97:195–204. doi: 10.1016/s0306-4522(99)00604-1. [DOI] [PubMed] [Google Scholar]
- 25.Jiao Y, Sun Z, Lee T, et al. A simple and sensitive antigen retrieval method for free-floating and slide-mounted tissue sections. J Neurosci Methods. 1999;93:149–162. doi: 10.1016/s0165-0270(99)00142-9. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Burdett TC, Desjardins C, et al. Disrupted and transgenic urate oxidase alter urate and dopaminergic neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:300–305. doi: 10.1073/pnas.1217296110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kachroo A, Schwarzschild MA. Adenosine A2A receptor gene disruption protects in an α-synuclein model of Parkinson’s disease. Ann Neurol. 2012;71:278–282. doi: 10.1002/ana.22630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao J, Wan L, Hacker E, et al. MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Mol Cell. 2013;51:409–422. doi: 10.1016/j.molcel.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong HK, Veremeyko T, Patel N, et al. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum Mol Genet. 2013;22:3077–3092. doi: 10.1093/hmg/ddt164. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Yuan B, Guerrero C, et al. Quantification of oxidative DNA lesions in tissues of Long-Evans Cinnamon rats by capillary high-performance liquid chromatography-tandem mass spectrometry coupled with stable isotope-dilution method. Anal Chem. 2011;83:2201–2209. doi: 10.1021/ac103099s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, Wales P, Quinti L, et al. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS One. 2015;10:e0116919. doi: 10.1371/journal.pone.0116919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc. 2007;2:141–151. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- 33.Chen JF, Steyn S, Staal R, et al. 8-(3-Chlorostyryl) caffeine may attenuate MPTP neurotoxicity through dual actions of monoamine oxidase inhibition and A2A receptor antagonism. J Biol Chem. 2002;277:36040–36044. doi: 10.1074/jbc.M206830200. [DOI] [PubMed] [Google Scholar]
- 34.Schapira AH, Tolosa E. Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat Rev Neurol. 2010;6:309–317. doi: 10.1038/nrneurol.2010.52. [DOI] [PubMed] [Google Scholar]
- 35.Doyle JR, Fortin JP, Beinborn M, Kopin AS. Selected melanocortin 1 receptor single-nucleotide polymorphisms differentially alter multiple signaling pathways. J Pharmacol Exp Ther. 2012;342:318–326. doi: 10.1124/jpet.112.194548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritter SL, Hall RA. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Biol. 2009;10:819–830. doi: 10.1038/nrm2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hazell GG, Hindmarch CC, Pope GR, et al. G protein-coupled receptors in the hypothalamic paraventricular and supraoptic nuclei—serpentine gateways to neuroendocrine homeostasis. Front Neuroendocrinol. 2012;33:45–66. doi: 10.1016/j.yfrne.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sánchez-Laorden BL, Jiménez-Cervantes C, García-Borrón JC. Regulation of human melanocortin 1 receptor signaling and trafficking by Thr-308 and Ser-316 and its alteration in variant alleles associated with red hair and skin cancer. J Biol Chem. 2007;282:3241–3251. doi: 10.1074/jbc.M606865200. [DOI] [PubMed] [Google Scholar]
- 39.Beaumont KA, Shekar SN, Newton RA, et al. Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles. Hum Mol Genet. 2007;16:2249–2260. doi: 10.1093/hmg/ddm177. [DOI] [PubMed] [Google Scholar]
- 40.Reeve A, Simcox E, Turnbull D. Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res Rev. 2014;14:19–30. doi: 10.1016/j.arr.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rumpf JJ, Schirmer M, Fricke C, et al. Light pigmentation phenotype is correlated with increased substantia nigra echogenicity. Mov Disord. 2015;30:1848–1852. doi: 10.1002/mds.26427. [DOI] [PubMed] [Google Scholar]
- 42.Dehay B, Bourdenx M, Gorry P, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14:855–866. doi: 10.1016/S1474-4422(15)00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallings R, Manzoni C, Bandopadhyay R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015;282:2806–2826. doi: 10.1111/febs.13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuo Y, Kamitani T. Parkinson’s disease-related protein, α-synuclein, in malignant melanoma. PLoS One. 2010;5:e10481. doi: 10.1371/journal.pone.0010481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan T, Zhu J, Hwu WJ, Jankovic J. The role of alpha-synuclein in melanin synthesis in melanoma and dopaminergic neuronal cells. PLoS One. 2012;7:e45183. doi: 10.1371/journal.pone.0045183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inzelberg R, Samuels Y, Azizi E, et al. Parkinson disease (PARK) genes are somatically mutated in cutaneous melanoma. Neurol Genet. 2016;2:e70. doi: 10.1212/NXG.0000000000000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haddadeen C, Lai C, Cho SY, Healy E. Variants of the melanocortin-1 receptor: do they matter clinically? Exp Dermatol. 2015;24:5–9. doi: 10.1111/exd.12540. [DOI] [PubMed] [Google Scholar]
- 48.Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for erythropoietic protoporphyria. N Engl J Med. 2015;373:48–59. doi: 10.1056/NEJMoa1411481. [DOI] [PMC free article] [PubMed] [Google Scholar]