

Abstract
Purpose of review
The present review aims to describe the epigenetic alterations observed in oral cancer linked to the exposure to alcohol and/or tobacco.
Recent Findings
Recent findings emphasize the importance of epigenetics in oral cancer progression and in how risk factors (as tobacco and alcohol) affect the basal epigenetic profiles. Deeper techniques and detailed approaches allowed the perception that individual CG changes and even subtle changes may represent important epigenetic alterations resulting in expression changes and other carcinogenic consequences. New classes of epigenetic alterations including non-coding RNAs have been gaining attention.
Summary
Many epigenetic alterations have been described in oral carcinoma progression induced by tobacco and/or alcohol, including: promoter hypermethylation in genes with tumor suppressive activity, global (genome-wide) hypomethylation, change in methylation patterns throughout the genes, alteration in non-coding RNAs and histones modifications. These changes represent progress in the knowledge of how these risk factors act in a molecular level. There is an urgent need for large independent studies to move these potential makers further and validate them in order to identify risk assessment, early diagnostic markers, and therapeutic targets, as well as to be the base for prevention and intervention techniques.
Keywords: Epigenetics, Oral Carcinoma, Methylation, Tobacco, Alcohol
Introduction
Oral carcinoma is the most common neoplastic disease in the head and neck region and there are over 50,000 new cases in USA expected for 2018 (considering oral cavity and pharynx together) and over 10,000 estimated deaths due to this tumor type [1]. Oral squamous cell carcinoma (OSCC) is commonly preceded by oral premalignant lesions (OPML) such as oral leukoplakia, eritroplakia and actinic cheilitis [2]. OPML have a variable histopathological presence of epithelial dysplasia, and clinical and histological approaches are unable to predict lesion progression and malignant transformation. No screening tool suggested so far by the literature has succeed to improve OPML diagnosis and prognosis, or helped to reduce oral cancer [3]. OPML clinical features can be heterogeneous and difficult to evaluate risk. Despite various classifications of dysplasia grading available, they fail to identify the one at high risk of malignant transformation [2]. The biological understanding/characterization of oral carcinoma and its different stages of progression offers opportunities to identify key players that could be used as molecular markers.
In this review article, we focus on epigenetic changes described in oral carcinoma, related to two very important established risk factors: tobacco and alcohol.
Epigenetics refers to mechanisms that alter the phenotype without changing the DNA sequence. These phenomena comprise DNA methylation (methylation on the cytosine bases of the DNA, on CG dinucleotides, commonly referred to as CpG), noncoding RNAs (miRNA, lnRNA, etc), histone posttranslation modifications (e.g. acetylation, methylation). These are important mechanisms for gene regulation in normal cells, they are key in the normal development process; they are physiologically dynamic and may present a tissue specific patter. Alteration in epigenetic machineries and/or patterns is observed in multiple conditions, including diseases such as cancer. As an example, changes in DNA methylation in regulatory regions (promoters) of genes may result in altered behavior in oncogenes and tumor suppression genes.
Therefore, the study of epigenetic alterations on cancer progression and dysplastic lesions helps to understand oral carcinogenesis. Promoter hypermethylation on genes with tumor suppressive characteristics are observed in healthy tissue adjacent to tumors and OPML, suggesting that these epigenetic modifications are early events in oral carcinogenesis [4, 5]. Hypermethylation of CpG islands in promoter regions is often associated with gene silencing and contributes to the typical hallmarks of a cancer cell that result from tumor suppressor gene inactivation [6]. The fact that epigenetic alterations are reversible offers an attractive opportunity of candidates for therapeutic targets (not discussed in the current review).
Epigenetic changes may occur in response to exposure to risk factors. It can be caused by external factors including tobacco, alcohol, diet, pharmacological treatments. The relationship between tobacco consumption and genetic and epigenetic changes has been explored in several studies. Alexandrov et al. observed a mutational signature for each cancer type depending on smoking status, the duration of the exposure (pack years), and pattern of exposure [7]. Smoking is usually associated with altered methylated CpG loci, which is best reported in lung tissue of smokers [8], or transversion cytosine to adenine (C>A) [9].
Chronic alcohol consumption can also induce a sequence of epigenetic alterations. It causes the inhibition of the ubiquitin-proteasome pathway in the nucleus, which leads to changes in the turnover of transcriptional factors, histone-modifying enzymes, and, therefore, altered epigenetic mechanisms, this has been shown in humans and animal models [10–12].
The most recent common epigenetic alterations induced by tobacco and/or alcohol in the scenario of oral squamous cell carcinoma progression (OSCC) and its progression are summarized in Table 1 and Figure 1.
Table 1.
Summary of recently described epigenetic alterations in oral carcinoma induced by tobacco and/or alcohol. lnc-RNA, Long non-coding RNA.
| Epigenetic Mechanism |
ID | Proposed Biological function |
Associations | Tobacco association |
Alcohol association |
References |
|---|---|---|---|---|---|---|
| Promoter/Regulatory region DNA hypermethylation | CDKN2a/P16 | Cell cycle | Cancer progression, increased lymph node invasion and local recurrence | yes | yes | Hasegawa et al. 2002; Kulkarni and Saranath 2004; |
|
| ||||||
| CDH1 | E-cadherin Cell adhesion | Decreased disease-free survival, invasion and metastasis | yes | yes | Hasegawa et al. 2002; Supic et al. 2009 | |
|
| ||||||
| P15 | Cell growth | Cancer progression | yes | yes | Chang et al. 2004 | |
|
| ||||||
| DAPK | Apoptosis regulation | Metastasis, lymph node involvement | yes | yes | Liu et al. 2012 | |
|
|
|
|||||
| Altered methylation pattern in gene body and introns | FGF3 | Cell survival | Cancer progression | yes | Sun et al. 2017 | |
|
| ||||||
| VAMP3 | Vesicle associated membrane protein | Cancer Progression | yes | Sun et al. 2017 | ||
|
| ||||||
| LINE1 | Repetitive element | Cancer Progression | yes | yes | Wangsri et al. 2012; Smith et al. 2007 | |
|
|
||||||
| microRNA | miR-30a, miR-934, miR-3164, miR-3178 | Non-coding RNA | Cellular proliferation | yes | Saad et al. 2015 | |
|
|
||||||
| lnc-RNA | lnc-PSD4-1 lnc-NETO1-1 | Non-coding RNA | Cancer progression | yes | Yu et al. 2016 | |
|
|
||||||
| Histone Modification | H3K9me3 H3K27me3 H3K9/14ac H3K27ac | Histone methylation | Cancer Initiation | yes | Urvalek et al. 2015 | |
| Histone Acetilation | ||||||
|
|
||||||
CDH1, cadherin 1; CDKN2a/P16, cyclin dependent kinase inhibitor 2A; DAPK, death associated protein kinase 1; FGF3, fibroblast growth factor 3; LINE-1, retrotransposable element 1; lnc-RNA, long non-coding RNA; VAMP3, vesicle associated membrane protein 3.
Figure 1.
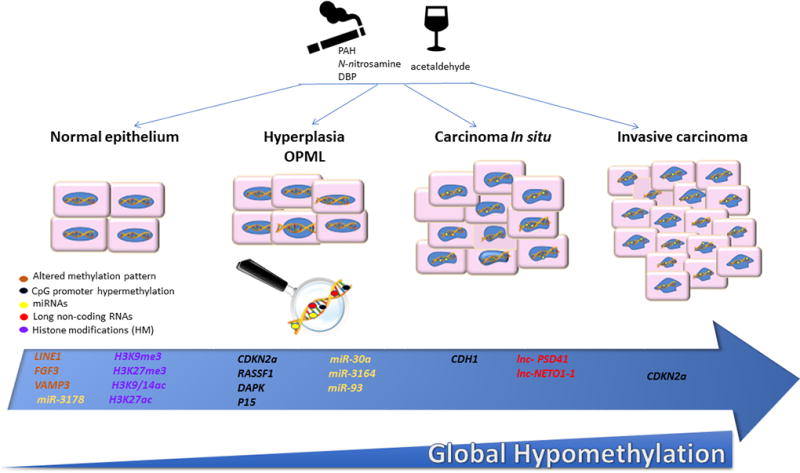
Recently described epigenetic changes in oral carcinoma induced by Alcohol and/or Tobacco. The representative progression model of oral carcinoma shows some of the epigenetic alterations described in different stages of oral carcinoma progression. Genes that have altered methylation patterns throughout the gene are in orange, genes described to be subject to promoter hypermethylation are in black, microRNAs (miRNAs) in yellow and long non-coding RNAs (lncRNAs) in red, modified histones are in purple (me, methylation; ac, acetylation). OPML, Oral pre-malignant lesions. The lower blue panel represents the loss of global (genome wide) methylation (hypomethylation) throughout the progression. These alterations/changes have been described influenced by the consumption of ethanol (represented by the wine glass cartoon) and/or tobacco (represented by the cigarette cartoon). This figure is original and was created by the authors Y. Ghantous, Juliana L Schussel and Mariana Brait.
Epigenetic carcinogenic changes in OSCC induced by Tobacco
Tobacco consumption is the main risk factor for developing oral squamous cell carcinoma, at least one third of oral cancers are attributed to tobacco usage [13]. The relative risk of oral cancer for smokers compared to non-smokers is between 2 and 3 [14]. Tobacco, both in its chewable and smoking forms, contains more than 60 carcinogens; a quarter of those carcinogens are proven to be carcinogenic in humans, with polycyclic aromatic hydrocarbons (PAHs) and N-nitrosamines being the most critical ones [15]. Tobacco participates in early carcinogenic stages inducing allelic losses at 3p11, 5g11, 9p21, 17p13, 18q12, gains at 11q13, amplification of the CCND1 gene, loss of p16, p15 and TP53 mutations.
The main carcinogenic mechanism by tobacco is the formation of covalent bonds between the different carcinogens and DNA, which lead to the development of tobacco-related DNA adducts, and consequently the accumulation of several mutations in critical genes such as oncogenes and tumor-suppressor genes [16]. Covalent DNA adducts are considered a major DNA damage, which has been shown to alter DNA methylation by various mechanisms associated with the formation of DNA lesions or by inhibition of DNA methyltransferases (DNMTs). Cigarette smoke causes hypomethylation by inducing chronic methylation via the reactive oxygen species [17]. (The relationship between cigarette smoking and DNA methylation are revised in [18]).
Dibenzo[def,p]chrysene (DBP) is the most potent carcinogenic polycyclic aromatic hydrocarbons (PAH) found in tobacco that induces oral cancer in animal models [19]. Mice treated with DBP developed maximum DNA damage, which included 30 and 48 differentially methylated CpG sites [20]. These methylated sites were mapped to genes known to be involved in cancer-related pathways. The most significant finding was intronic hypomethylation of FGF3 (correlating with gain of expression), observed in an early stage of the OSCC development but not evident in control oral mucosa not treated with DBP, and also intronic hypermethylation of VAMP3 that was inversely correlated with its expression. These results indicate that tobacco carcinogen DBP plays an essential role in oral carcinogenesis induced by tobacco and that it affects methylation with the gene body, changing expression patterns and this epigenetic changes may serve as a potential biomarker for early detection of OSCC [20].
An inverse correlation was found between cigarette smoking and global or genome-wide DNA methylation (shown by Guerrero-Preston et al.[21]). These could be accounted for gene body mehtylation as well as gene-specific hypomethylation that has been associated with activation of oncogenes and loss of genomic integrity. Hypomethylation can also lead to inappropriate recombinations resulting in defects in cell cycle monitoring, checkpoint genes, and genes involved in chromosome condensation and stability [21].
A recurrent finding of a conexion between promoter methylation and cigarette consumptions is on the CDKN2a (p16) gene. A significant correlation was found between promoter methylation of CDKN2a and tobacco carcinogens, with a significantly higher proportion of promoter methylation in patients who started smoking younger [22]. CDKN2a regulates the RB pathway, leading to inhibition of cell cycle progression in the G1-S transition, and therefore acts as a tumor suppressor that is implicated in the prevention of carcinogenesis. Its inactivation is widely prevalent in head and neck squamous cell carcinoma, and it plays a critical role in the carcinogenesis process [23]. Chewing tobacco was also found to cause concurrent aberrant methylation in the promoter regions of CDKN2a detected by methylation-specific PCR assay. This methylation pattern was observed in both cancerous cells and adjacent normal mucosa of smokers. The overall methylation in the primary tumors was 86.7%, compared to 76% in adjacent healthy tissues. On the other hand, no aberrant methylation was detected in buccal scrapings from healthy individuals with no tobacco habits [24].
E-cadherin is encoded by the CDH1 gene, and it is a transmembrane glycoprotein, with five extracellular and one cytoplasmic domain. This molecule plays a vital role in creating adhesive junctions, cell polarity, and intracellular signaling. It acts as a tumor suppressor gene, and its absence is related to cell invasion and metastasis [25]. E-cadherin was also found hypermethylated in OSCC cells related to tobacco consumption. The promoter methylation was further associated with increased pack-years smoked [22]. Methylation of CpG sites in E-Cadherin expands through time and the longest time of exposure will lead to a more considerable amount of de-novo methylations, which explains the higher portion of promoter methylation in E-Cadherin in patients with higher pack-years of smoking.
P15 is an effector of transforming growth factor (TGF-β)-induced cell cycle arrest. Inactivation of P15 by promoter hypermethylation has been observed in many human malignancies, suggesting that its downregulation may be essential in neoplastic transformation. Aberrant methylated P15 gene was found in higher proportion in the oral mucosa of healthy smokers (68%), compared with non-smokers mucosa (8%). It was also found methylated in 64% of OSCC patients, who were heavy smokers [26]. Smoking was also shown to affect methylation patterns throughout the repetive element LINE-1 [27]. LINE-1 is usually assessed to measure global methylation, because it is found thousands of time along the genome. The interesting observation by Wanhsri and the study group was that certain locus gained while other lost methylation in smokers when compared to never smokers. These alterations seem to be proportional to the amount of cigarette esmoked and seem to persist even after one year of stopping smking. When the patients also consumed alcohol, the pattern did not particularly change, although gain of overall methylation was observed. These observations highlight that the influences of these risk factors in the epigenetic patterns need to be assessed in details in order to undersand their real efects.
Tobacco carcinogens play an essential role in the development of oral carcinoma in its early stages and causes different epigenetic alterations. Recognizing these alterations can potentially lead to earlier diagnosis and targeted treatment.
Tobacco cessation
Smoking induced epigenetic changes have been recently found to be dynamic and partially reversible by genome-wide methylation profiling. A study conducted by Guida and colleagues showed that there are two distinct classes of CpG sites which were affected by smoking cessation [28]. Sites which methylation reverts to levels typical of never smokers within decades after smoking cessation, and sites remaining differentially methylated, even more than 35 years after smoking cessation [28, 29].
These findings indicate the presence of historical tobacco signatures, which could be used as biomarkers to refine individual risk profiling of smoking-induced chronic disease such as OSCC.
These epigenetic data highlight the importance of smoking cessation education for the benefit of the patient and even further emphasize that the most important strategy is preventing the start of smoking, once some of these studied epigenetic marks are not reversible even after years of smoking cessation.
Epigenetic carcinogenic changes in OSCC induced by Alcohol
Alcohol consumption is a known etiological factor for many diseases, and is responsible for a large number of deaths worldwide [30]. It has been classified as a carcinogenic factor by IARC (International Agency for Research on Cancer), and is associated with cancer of the oral cavity, pharynx, larynx, esophagus, liver, pancreas, colorectum and others [30]. Patient attributable risk for head and neck cancer for patients smoking and drinking was reported as high as 74% (80% for men and 61% for women), confirming a multiplicative joint effect between tobacco and alcohol [31]. Alcohol consumption alone has a comparatively lower risk for head and neck cancer, usually associated with high doses and with worse impact on pharyngeal and laryngeal cancers [32]. Shingler and colleagues [33] reported that three alcoholic drinks per day (considered as a high dose) in non-smoker patients could increase 2.04 the risk of head and neck cancer. In addition, patients who continue to drink after treatment have a higher risk of recurrence and second primary tumors [34].
The mechanisms associated with alcohol carcinogenic effect might include DNA damage induced by ethanol and its metabolite, acetaldehyde. DNA methylation is affected by altering folate metabolism and transmethylation reactions [35]. It also can alter DNMT activity; therefore, chronic low dose of alcohol intake is associated with global hypomethylation, and higher doses induce gene specific DNA hypermethylation, both showing carcinogenic consequences.
Acetaldehyde is produced in the oral cavity, as well in the kidney and the liver, and has a mutagenic effect. Studies on long non-coding RNAs (lncRNA) have indicated its association with cancer progression on different types of epithelial cancer, including HSNCC [36]. Yu et al. [37] reported the role of lncRNAs on the pathogenesis of alcohol-associated HNSCC (head and neck squamous cell carcinoma) and demonstrated the dysregulation of lnc-PSD4-1 and lnc-NETO1-1 in oral keratinocytes exposed to alcohol and acetaldehyde. Alcohol has also been shown to influence another type of non-coding RNAs, micro RNAs (miRNAs). Saad et al. [38] used a cohort to identify epigenetic alterations associated with alcohol consumption in non-smoker patients. Four miRNA (miR-30a, miR-934, miR-3164, and miR-3178) were upregulated in oral keratinocytes exposed to ethanol and acetaldehyde, suggesting participation on early events of oral carcinogenesis. They showed that the expression of miR-30a and miR-934 promotes the induction of BCL-2, an anti-apoptotic gene, as well as increased cellular proliferation. These findings point to more relevant roles of alcohol as an independent risk factor on HNSCC carcinogenesis.
Alcohol has already been associated with DNA methylation, histone acetylation and histone methylation during carcinogenesis. Urvalek and collaborators [39] found global carcinogenic epigenetic alterations associated to 4-NQO (4-nitroquinoline-1-oxide, a carcinogen) and ethanol exposure. They observed that the exposure to these substances increases H3K9/14 and H3K27 acetylation, and methylation on H3K27 and H3K9. They report global increase in histone modifications during oral carcinogenesis, associated to poor prognosis, increased metastasis, and increased recurrence rates.
Smith et al. [40] demonstrated a global hypomethylation associated with smoking and/or alcohol, and they point the difficulty to separate tobacco and alcohol effect as they can be confounding in characteristic in HNSCC patients. Another study investigated methylation of 5 tumor suppressor genes’ regulatory/promoter region (P16, DAPK, APC, CDH1 and MGMT) in OSCC and found a higher proportion of multiple gene hypermethylation in male patients and a trend of association between concurrent methylation and alcohol consumption [41]. They did not show single gene association but demonstrated as within this group of genes, they could observe higher methylation proportional to the alcohol consumption data.
Alcohol cessation decreases HNSCC risk annually and the risk becomes similar to never drinkers after 20 years of quitting. More accurate data can be difficult to be analyzed due tobacco frequent concomitant use [42].
Conclusion and Vision
The study of epigenetic alterations on oral cancer progression and dysplastic lesions helps to understand oral carcinogenesis. Oral cancer and oral cancer progression are intimately associated with tobacco and alcohol consumption and epigenetic aberrations induced by these factors are being described. Oral cancer is the most common head and neck cancer and was chosen as the specific focus of this review.
There are several data showing that epigenetic changes occur in early stages of oral carcinogenesis, and even in adjacent normal tissue. Coupled with the malleability of these changes, that supports the idea of the possibility of reversing those alterations. Understanding the molecular consequences of known risk factors such as alcohol and tobacco are the foundation for choosing and implementing strategies to prevent the exposure to such risk factors, to identify and put in practice intervention actions, to create and implement effective risk assessment approaches, to support awareness campaigns. Building the molecular knowledge of the consequences on a specific tissue type will aid the translational scientific community to identify markers that could open doors for risk assessment, early diagnosis, follow-up and development of therapy targets and prevention tactics. There is a lot of accumulated knowledge on the epigenetics consequences of tobacco and/or alcohol consumption in the scenario of oral carcinoma that could benefit of large validation studies on prospective cohorts with follow-up after intervention and exposure/behavioral changes to allow these findings to progress to translational opportunities and advance the field of personalized medicine.
Key Points.
Alcohol and Tobacco induce epigenetic changes in oral carcinoma progression.
Smoking cessation reverts certain epigenetic marks acquired throughout the years of exposure and these should reinforce the importance of cessation.
Not all epigenetic alterations induced by alcohol and tobacco are reversible after many years of cessation; therefore prevention should be considered the best strategy.
The amount of exposure to risk factors has been associated with levels of observed changes.
Understanding and validating specific epigenetic alterations in oral cancer may lead to the identification of markers for risk assessment, early diagnosis, and therapy targets.
Acknowledgments
Funding
This work was supported by a career enhancement program awarded to M.B. from the Specialized Program of Research Excellence (SPORE) in Head and Neck P50DE019032 from the United States National Institutes of Health (NIH)/National Institute of Dental and Craniofacial Research (NIDCR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
None.
Footnotes
Conflict of Interest: None. The authors declare no conflict of interest.
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. Ca-Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2*.Morandi L, Gissi D, Tarsitano A, et al. CpG location and methylation level are crucial factors for the early detection of oral squamous cell carcinoma in brushing samples using bisulfite sequencing of a 13-gene panel. Clin Epigenetics. 2017;9:85. doi: 10.1186/s13148-017-0386-7. The authors used next-generation sequencing to evaluate 18 target genes for DNA methylation, and highlight the importance of CpG location and DNA methyation level to improve oral squamous cell carcinoma diagnosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brocklehurst P, Kujan O, O'Malley LA, et al. Screening programmes for the early detection and prevention of oral cancer. Cochrane Database Syst Rev. 2013;(11):CD004150. doi: 10.1002/14651858.CD004150.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishra R. Biomarkers of oral premalignant epithelial lesions for clinical application. Oral Oncol. 2012;48(7):578–584. doi: 10.1016/j.oraloncology.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 5.Shridhar K, Walia GK, Aggarwal A, et al. DNA methylation markers for oral pre-cancer progression: A critical review. Oral Oncol. 2016;53:1–9. doi: 10.1016/j.oraloncology.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 7.Alexandrov LB, Ju YS, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354(6312):618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8):e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13(2):97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 10.Bardag-Gorce F, French BA, Joyce M, et al. Histone acetyltransferase p300 modulates gene expression in an epigenetic manner at high blood alcohol levels. Exp Mol Pathol. 2007;82(2):197–202. doi: 10.1016/j.yexmp.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliva J, Dedes J, Li J, et al. Epigenetics of proteasome inhibition in the liver of rats fed ethanol chronically. World J Gastroenterol. 2009;15(6):705–712. doi: 10.3748/wjg.15.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aguilera O, Fernandez AF, Munoz A, Fraga MF. Epigenetics and environment: a complex relationship. J Appl Physiol (1985) 2010;109(1):243–251. doi: 10.1152/japplphysiol.00068.2010. [DOI] [PubMed] [Google Scholar]
- 13.Petti S. Lifestyle risk factors for oral cancer. Oral Oncol. 2009;45(4–5):340–350. doi: 10.1016/j.oraloncology.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 14.Anantharaman D, Marron M, Lagiou P, et al. Population attributable risk of tobacco and alcohol for upper aerodigestive tract cancer. Oral Oncol. 2011;47(8):725–731. doi: 10.1016/j.oraloncology.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Pfeifer GP, Denissenko MF, Olivier M, et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21(48):7435–7451. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 16.Grigoryeva ES, Kokova DA, Gratchev AN, et al. Smoking-related DNA adducts as potential diagnostic markers of lung cancer: new perspectives. Exp Oncol. 2015;37(1):5–12. [PubMed] [Google Scholar]
- 17.Iyama T, Wilson DM., 3rd DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 2013;12(8):620–636. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:132. doi: 10.3389/fgene.2013.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Bayoumy K, Chen KM, Zhang SM, et al. Carcinogenesis of the Oral Cavity: Environmental Causes and Potential Prevention by Black Raspberry. Chem Res Toxicol. 2017;30(1):126–144. doi: 10.1021/acs.chemrestox.6b00306. [DOI] [PubMed] [Google Scholar]
- 20*.Sun YW, Chen KM, Imamura Kawasawa Y, et al. Hypomethylated Fgf3 is a potential biomarker for early detection of oral cancer in mice treated with the tobacco carcinogen dibenzo[def,p]chrysene. PLoS One. 2017;12(10):e0186873. doi: 10.1371/journal.pone.0186873. This paper demonstrates the cumulative effect of tobacco carcinogens exposure, which could lead to the development of OSCC. They describe Fgf3 intronic hypomethylation induces its expression and may serve as a potential biomarker for early detection of OSCC. They also show that intronic hypermethylation of Vamp3 diminishes its expression and is a consequence of tobacco exposure in oral tissue. This data not only gives importance to these genes but also emphasizes the importance of intronic methylation assessment and its importance in expression control. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerrero-Preston R, Baez A, Blanco A, et al. Global DNA methylation: a common early event in oral cancer cases with exposure to environmental carcinogens or viral agents. P R Health Sci J. 2009;28(1):24–29. [PubMed] [Google Scholar]
- 22.Hasegawa M, Nelson HH, Peters E, et al. Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene. 2002;21(27):4231–4236. doi: 10.1038/sj.onc.1205528. [DOI] [PubMed] [Google Scholar]
- 23.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61(8):3225–3229. [PubMed] [Google Scholar]
- 24.Kulkarni V, Saranath D. Concurrent hypermethylation of multiple regulatory genes in chewing tobacco associated oral squamous cell carcinomas and adjacent normal tissues. Oral Oncol. 2004;40(2):145–153. doi: 10.1016/s1368-8375(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 25.Vleminckx K, Vakaet L, Jr, Mareel M, et al. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66(1):107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- 26.Chang HW, Ling GS, Wei WI, Yuen AP. Smoking and drinking can induce p15 methylation in the upper aerodigestive tract of healthy individuals and patients with head and neck squamous cell carcinoma. Cancer. 2004;101(1):125–132. doi: 10.1002/cncr.20323. [DOI] [PubMed] [Google Scholar]
- 27.Wangsri S, Subbalekha K, Kitkumthorn N, Mutirangura A. Patterns and possible roles of LINE-1 methylation changes in smoke-exposed epithelia. PLoS One. 2012;7(9):e45292. doi: 10.1371/journal.pone.0045292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guida F, Sandanger TM, Castagne R, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet. 2015;24(8):2349–2359. doi: 10.1093/hmg/ddu751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ambatipudi S, Cuenin C, Hernandez-Vargas H, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8(5):599–618. doi: 10.2217/epi-2016-0001. [DOI] [PubMed] [Google Scholar]
- 30.Cao Y, Willett WC, Rimm EB, et al. Light to moderate intake of alcohol, drinking patterns, and risk of cancer: results from two prospective US cohort studies. BMJ. 2015;351:h4238. doi: 10.1136/bmj.h4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashibe M, Brennan P, Chuang SC, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. 2009;18(2):541–550. doi: 10.1158/1055-9965.EPI-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashibe M, Brennan P, Benhamou S, et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007;99(10):777–789. doi: 10.1093/jnci/djk179. [DOI] [PubMed] [Google Scholar]
- 33.Shingler E, Robles LA, Perry R, et al. Tobacco and alcohol cessation or reduction interventions in people with oral dysplasia and head and neck cancer: systematic review protocol. Syst Rev. 2017;6(1):161. doi: 10.1186/s13643-017-0555-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayne ST, Cartmel B, Kirsh V, Goodwin WJ., Jr Alcohol and tobacco use prediagnosis and postdiagnosis, and survival in a cohort of patients with early stage cancers of the oral cavity, pharynx, and larynx. Cancer Epidemiol Biomarkers Prev. 2009;18(12):3368–3374. doi: 10.1158/1055-9965.EPI-09-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang TH, Hsia SM, Shih YH, Shieh TM. Association of Smoking, Alcohol Use, Betel Quid Chewing with Epigenetic Aberrations in Cancers. Int J Mol Sci. 2017;18(6) doi: 10.3390/ijms18061210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochim Biophys Acta. 2014;1839(11):1097–1109. doi: 10.1016/j.bbagrm.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 37.Yu V, Singh P, Rahimy E, et al. RNA-seq analysis identifies key long non-coding RNAs connected to the pathogenesis of alcohol-associated head and neck squamous cell carcinoma. Oncol Lett. 2016;12(4):2846–2853. doi: 10.3892/ol.2016.4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saad MA, Kuo SZ, Rahimy E, et al. Alcohol-dysregulated miR-30a and miR-934 in head and neck squamous cell carcinoma. Mol Cancer. 2015;14:181. doi: 10.1186/s12943-015-0452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urvalek AM, Osei-Sarfo K, Tang XH, et al. Identification of Ethanol and 4-Nitroquinoline-1-Oxide Induced Epigenetic and Oxidative Stress Markers During Oral Cavity Carcinogenesis. Alcohol Clin Exp Res. 2015;39(8):1360–1372. doi: 10.1111/acer.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith IM, Mydlarz WK, Mithani SK, Califano JA. DNA global hypomethylation in squamous cell head and neck cancer associated with smoking, alcohol consumption and stage. Int J Cancer. 2007;121(8):1724–1728. doi: 10.1002/ijc.22889. [DOI] [PubMed] [Google Scholar]
- 41.Supic G, Kozomara R, Brankovic-Magic M, et al. Gene hypermethylation in tumor tissue of advanced oral squamous cell carcinoma patients. Oral Oncol. 2009;45(12):1051–1057. doi: 10.1016/j.oraloncology.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 42**.Kawakita D, Matsuo K. Alcohol and head and neck cancer. Cancer Metastasis Rev. 2017;36(3):425–434. doi: 10.1007/s10555-017-9690-0. This article reviews the association between alcohol drinking and head and neck cancer and reports the participation of competing causes on mortality of this cancer type. [DOI] [PubMed] [Google Scholar]