

Abstract
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related death worldwide. Immune checkpoint blockade with anti-CTLA-4 and anti-PD-1 antibodies has shown promising results in the treatment of patients with advanced HCC. The anti-PD-1 antibody, nivolumab, is now approved for patients who have had progressive disease on the current standard of care. However, a subset of patients with advanced HCC treated with immune checkpoint inhibitors failed to respond to therapy. Here, we provide evidence of adaptive resistance to immune checkpoint inhibitors through upregulation of indoleamine 2,3-dioxygenase (IDO) in HCC. Anti-CTLA-4 treatment promoted an induction of IDO1 in resistant HCC tumors but not in tumors sensitive to immune checkpoint blockade. Using both subcutaneous and hepatic orthotopic models, we found that the addition of an IDO inhibitor increases the efficacy of treatment in HCC resistant tumors with high IDO induction. Furthermore, in vivo neutralizing studies demonstrated that the IDO induction by immune checkpoint blockade was dependent on IFN-γ. Similar findings were observed with anti-PD-1 therapy. These results provide evidence that IDO may play a role in adaptive resistance to immune checkpoint inhibitors in patients with HCC. Therefore, inhibiting IDO in combination with immune checkpoint inhibitors may add therapeutic benefit in tumors which overexpress IDO and should be considered for clinical evaluation in HCC.
Electronic supplementary material
The online version of this article (10.1007/s00262-018-2190-4) contains supplementary material, which is available to authorized users.
Keywords: Hepatocellular carcinoma, CTLA-4, PD-1, IDO, Adaptive resistance
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer and the second leading cause of cancer-related death worldwide [1]. HCC is largely considered an inflammation-induced cancer with many patients affected by underlying liver pathology who progress to develop cirrhosis and subsequently HCC. Therefore, immunotherapy may provide an ideal approach [2]. Immune checkpoint inhibitors have been approved for a variety of advanced malignancies [3, 4]. Recently, nivolumab, a monoclonal antibody which blocks PD-1 (anti-PD-1), has been approved for patients with advanced HCC who progressed on sorafenib based on the results of the phase I/II trial, CheckMate-040 [5].
Although great advances have been made with immune checkpoint inhibitors, the response rates remain low, and a significant proportion of patients with ranging pathologies exhibit primary or adaptive resistance to immune checkpoint blockade [6]. Our group utilized tremelimumab, a monoclonal antibody which blocks CTLA-4 (anti-CTLA-4), in combination with ablative therapies for patients with advanced HCC. We observed a partial response rate of 26%, while 63% of patients were deemed to have stable disease [7]. Primary resistance occurs when the tumor does not respond to an immunotherapy likely through lack of recognition by T cells. Adaptive or acquired resistance may occur when a tumor is recognized by the immune system, but protects itself from immune attack [6]. Understanding the underlying resistance mechanisms is needed to develop proper combination strategies to improve efficacy of checkpoint blockade for HCC patients.
Indoleamine 2,3-dioxygenase (IDO) is an enzyme in the kynurenine pathway that is responsible for the degradation of tryptophan and has been implicated in bolstering immune-inhibitory effects [8–10]. IDO exists in two forms, IDO1 and IDO2, derived from two separate genes [11]. IDO2 has been implicated to have a relatively weaker function compared to IDO1 and does not contribute to systemic tryptophan metabolism but its interplay in the tumor microenvironment (TME) is largely unknown [12, 13]. Many tumor types have been shown to over-express IDO [14]. Furthermore, IDO overexpression has largely been associated with a poor prognosis [10]. Immunohistochemical analysis of 138 tissue samples from HCC patients showed overexpression of IDO in 35% of tumor resection samples, where IDO-high tumors were associated with a worse overall survival than the IDO-low tumors [15, 16]. Previous studies have also shown that tumors modified to produce IDO exhibit more aggressive tumor growth as well as resistance to immunotherapy [14, 17–19]. However, gaps in knowledge persist on where IDO is expressed and active in the TME [13].
In this study, we report that immune checkpoint inhibitors can lead to an IFN-γ dependent increase in IDO expression by HCC tumor cells. This increase in tumor-derived IDO1 promoted resistance to single-agent anti-CTLA-4 therapy which was able to be overcome with the IDO inhibitor 1-methyl-d-tryptophan (1-d-MT). In addition, as nivolumab has been approved for advanced HCC, we showed a similar effect when anti-PD-1 was utilized as well as an alternative IDO inhibitor, epacadostat. Our results demonstrated that as a result of immune checkpoint inhibition, tumor cells can upregulate IDO1 providing a means of adaptive immune escape. These results help fill in the knowledge gap of the importance of tumor-derived IDO in the TME as a result of immune checkpoint inhibition. Therefore, inhibiting IDO in combination with immune checkpoint inhibitors should be further considered for clinical evaluation in HCC.
Materials and methods
Cell lines
The murine HCC cell lines, RIL-175 and BNL, were utilized for mouse experiments. RIL-175 cells were cultured in complete RPMI medium supplemented with 10% FBS (Gibco). The RIL-175 cell line possesses luciferase properties as previously utilized by our lab [20]. BNL tumor cells were cultured in DMEM supplemented with 10% FBS. The human HCC cell lines, Hep3B and HepG2, were used for human in vitro experiments. Hep3B and HepG2 tumor cells were cultured in DMEM supplemented with 10% FBS. Cells were tested to be mycoplasma free and cells from early passages were used for all experiments described.
Drugs
Anti-mouse CD152 (anti-CTLA-4) (Clone 9H10, GoInVivo™, BioLegend, CA, USA) was administered i.p. at 5 µg/g mouse body weight on days 8, 11, and 14 in 100 µL PBS [21]. Anti-mouse CD279 (anti-PD-1) antibody (Clone RMP1-14, GoInVivo™, BioLegend, CA, USA) was administered i.p. at 5 µg/g mouse body weight on days 8, 11, and 14 in 100 µL PBS. Anti-IFN-γ antibody (Clone XMG1.2, BioXCell, NH, USA) was administered i.p. at 25 µg/g mouse in 200 µL PBS [22]. Corresponding isotype controls were used: mouse IgG1 Isotype Ctrl Antibody and rat IgG2a Isotype Ctrl Antibody (BioLegend, GoInVivo™). 1-methyl-d-tryptophan (Sigma-Aldrich, MO, USA) was administered ad lib in drinking water of mice as previously described [14]. Mice drank approximately 2.5–3.5 mL of 1-d-MT supplemented water per day. Epacadostat (Selleckchem) was administered 300 mg/kg [23].
RNA isolation and real-time PCR
RNA was extracted from cell pellets, frozen tissue, or tumor with RNeasyMiniKit (Qiagen). Complementary DNA was synthesized by iScriptcDNA synthesis kit (BioRad). The reactions were run using iQSYBR green supermix kit (BioRad). The results were normalized to endogenous GAPDH expression levels [24]. The sequence of primers used for quantitative RT-PCR can be found in Supplementary Table 1 and Supplementary Table 2. Quantitative RT-PCR was performed on the ViiA™ 7 Real-Time PCR System (Life Technologies).
Mouse studies
C57BL/6 and BALB/C mice were purchased from Charles River Laboratories (VA, USA) at 8–10 weeks of age. Subcutaneous tumors were established by injection of 106 RIL-175 or BNL tumor cells into the left inguinal pocket of C57BL/6 mice or BALC/C mice, respectively. Four to five mice were randomized into each treatment group after tumor injection, and experiments were repeated for validation. The subcutaneous tumors were measured using calipers every 2–3 days and tumor volume was calculated as: (length × width2)/2 mm3 as previously reported [25]. Blinded measurements were utilized whenever possible.
Orthotopic tumors were induced by injecting 5 × 105 RIL-175 tumor cells under the capsule of the left liver via laparotomy of B6(Cg)-Tyr < c-2J>/J mice (B6-albino stock #000058) purchased from The Jackson Laboratory (Bar Harbor, USA). Tumor cells were injected in 20 µL of a 50:50 solution of PBS and Matrigel Matrix (Corning, MA, USA). Mice were anesthetized with 2% inhaled isoflurane in oxygen at 2 L/min. Tumor growth was monitored by bioluminescent imaging (BLI) with the Xenogen in vivo imaging system (IVIS Spectrum, Caliper Live Sciences, Hopkinton, MA). BLI was performed on days 7, 14, and 21. The CCD camera was cooled to between − 105 and − 120 °C and the field of view set to 25 cm. Mice were anesthetized with 2% isoflurane in oxygen at 2 L/min. Ten minutes after the mice received an intraperitoneal injection of 150 mg/kg of d-luciferin in PBS, bioluminescence images were acquired with an exposure time of 30 s, medium binning, 1.2 f/stop, with an open filter. A region of interest (ROI) was drawn around the tumor, and the bioluminescence signal was quantified as photons/sec/cm2/steradian (p/sec/cm2/sr).
Lymphocyte isolation
Single cell suspensions of lymphocytes were prepared from the spleen of sacrificed mice. Red blood cell lysis was performed with ACK Lysis Buffer (Quality Biologicals, MD, USA) [26]. Human blood samples (buffy coats) were obtained from the National Institutes of Health Blood Research Services for peripheral blood mononuclear cell (PBMC) isolation and prepared as previously described [27]. Fresh PBMCs were isolated by Ficoll density gradient centrifugation (Biochrom, Berlin, Germany). Splenocyte or PBMC cell activation was performed with the Cell Activation Cocktail without Brefeldin A (BioLegend) according to the manufacturer’s protocol. In brief, splenocytes or PBMCs were cultured in 1 mL medium with 2 µL of the Cell Activation Cocktail for 24 h. Cells and supernatant were then transferred from the six-well plate into an Eppendorf tube which was centrifuged and supernatant collected for co-culture with tumor cells.
Tumor cell isolation
Tumor cells were isolated from subcutaneous tumors via the autoMACS (Miltenyi Biotec) with negative selection utilizing a tumor dissociation kit and tumor cell isolation kit (Miltenyi Biotec) as per manufacturer’s protocol.
Statistical analysis
Sample sizes for animal studies were guided by previous studies in our laboratory in which the same mouse strains were used. Significance of the difference between groups was calculated by Student’s unpaired t test, one-way or two-way ANOVA (Tukey’s and Bonferroni’s multiple comparison test). Welch’s corrections were used when variances between groups were unequal. P < 0.05 was considered as statistically significant [24].
Results
Anti-CTLA-4 treatment increases IDO1 in resistant HCC tumors
A recent clinical trial by our group has shown promising results with anti-CTLA-4 therapy in patients with advanced HCC. However, a subset of patients proved to be resistant to the therapy [7]. To understand the resistance mechanism, we performed a pre-clinical study to examine changes in the HCC tumor environment upon anti-CTLA-4 therapy. RIL-175, a murine HCC cell line, was subcutaneously injected into female syngeneic C57BL/6 mice and then treated with anti-CTLA-4 therapy (Fig. 1a). A marginal decrease of tumor size was found in the anti-CTLA-4 treatment group compared to control (Fig. 1a, b). The lack of treatment response to anti-CTLA-4 by RIL-175 gave us the condition to study resistance. Indoleamine 2,3-dioxygenase (IDO), an immunomodulatory enzyme, has been found to be overexpressed in HCC tumors and confers a poor prognosis [16]. Anti-CTLA-4 treatment caused a substantial increase of IDO1 in RIL-175 HCC tumor tissue (Fig. 1c). The increase of IDO1 was found to be gender-independent as it was also found in male C57BL/6 mice (Sup. Figure 1a).
Fig. 1.
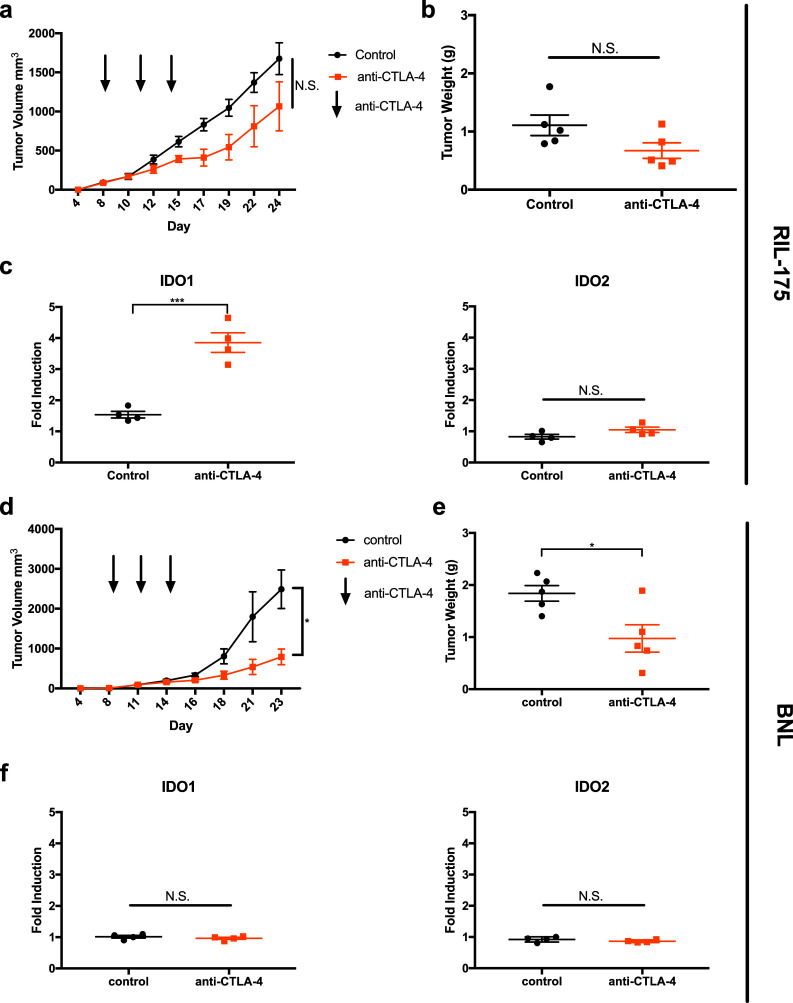
Mice were injected with 106 tumor cells in the left inguinal pocket. Mice received three i.p. injections of anti-CTLA-4 antibody administered on days 8, 11, and 14. Mice were sacrificed at which time tumor volume (a, d) and tumor weight (b, e) were recorded. RT-qPCR was performed measuring induction of IDO1 and IDO2 in RIL-175 (c) or BNL (f) tumors after anti-CTLA-4 treatment. *P < 0.05, ***P < 0.001
Next, we repeated the experiment using BNL, another murine HCC tumor line. Unlike RIL-175, anti-CTLA-4 effectively reduced subcutaneous BNL tumor growth (Fig. 1d, e). Interestingly, no IDO induction was found in BNL tumor tissue (Fig. 1f). In addition, baseline IDO expression was found to be higher in RIL-175 tumor cells than BNL tumor cells (Sup. Figure 1b). Our results indicate that IDO1 expression increases in anti-CTLA4 resistant but not sensitive HCC tumors.
IDO inhibition enhances the efficacy of anti-CTLA-4 treatment against resistant HCC
The IDO1 induction in anti-CTLA-4 resistant tumors prompted us to test the hypothesis that HCC tumors can develop an adaptive resistance to anti-CTLA-4 therapy by inducing IDO1 expression. RIL-175 tumor-bearing mice were treated with 1-methyl-d-tryptophan (1-d-MT), an IDO inhibitor, anti-CTLA-4, or the combination. 1-d-MT treatment alone had no effect on tumor growth. Again, anti-CTLA-4 monotherapy showed a small non-significant growth suppression. As expected, a potent reduction of tumor size was found in mice treated with combination therapy. The tumor inhibition was confirmed by the decrease of tumor weight (Fig. 2a, b and Sup. Figure 2a and 2b).
Fig. 2.

Tumor volume (a) was monitored in the high IDO inducible RIL-175 cell line for control mice, 1-d-MT alone, anti-CTLA-4 alone, or combination therapy. Tumors were established on day 0 and allowed to grow for 1 week. Anti-CTLA-4 was administered on days 8, 11, and 14. 1-d-MT was administered ad lib in drinking water of mice starting on day 9. Mean ± SEM of tumor sizes are shown from five mice per group. b Tumor weight was recorded for RIL-175 tumors as grams (g) of tumor tissue after mice were sacrificed. Orthotopic liver tumors were established and bioluminescent imaging was performed to monitor tumor growth reported in p/sec/cm2/sr (c, d). Mice with orthotopic tumors were sacrificed and tumors dissected away from normal liver and tumor weight recorded (e). *P < 0.05, ***P < 0.001
Subcutaneous HCC models lack the proper environment as HCC tumors arise in liver. To better mimic the HCC patient condition, an orthotopic HCC model was established. We took advantage that the RIL-175 tumors express luciferase which gave us the opportunity to monitor liver tumor growth kinetics in vivo by BLI [20]. Again, no significant tumor suppression was seen with 1-d-MT treatment alone (Sup. Figure 3a) and a mild effect was observed with anti-CTLA-4 monotherapy. In contrast, combination therapy caused the best impairment in tumor growth (Fig. 2c, d and Sup. Figure 3b). The observation of BLI was confirmed by the tumor weight (Fig. 2e). Together, the results from both subcutaneous and orthotopic HCC models demonstrate that IDO inhibition can sensitize the resistant HCC tumor to anti-CTLA-4 therapy and enhance treatment efficacy.
Anti-CTLA-4 treatment increases IDO in tumor cells via IFN-γ
Next, IDO expression in the tumor environment was studied. IDO has been shown not only to be inducible in tumor cells, but can also be expressed by tumor infiltrating leukocytes [8]. Therefore, the source of IDO induction was tested. Subcutaneous RIL-175 tumor cells were established and anti-CTLA-4 therapy was administered as in Fig. 1a. Tumor cells were then isolated to approximately 93% purity. The isolated RIL-175 tumor cells displayed a significant induction of IDO1 while the tumor associated leukocytes did not show a significant increase of IDO (Fig. 3a, b). This data demonstrates IDO1 induction may occur in the tumor cells after anti-CLTA-4 therapy.
Fig. 3.

Tumor cells were isolated from CD45+ cells. RT-qPCR was performed measuring induction of IDO1 and IDO2 in RIL-175 tumor cells (a) and CD45+ cells (b). ***P < 0.001
Anti-CTLA-4 therapy re-energizes T cells; therefore, we tested the effect of T cells on IDO expression in HCC tumor cells. RIL-175 or BNL tumors cells were cultured with splenocytes isolated from C57BL/6 or BALB/C mice, respectively. Indeed, an induction of IDO1 and IDO2 was found in RIL-175 but not BNL tumor cells (Fig. 4a, b). Furthermore, cultured RIL-175 tumors cells with a supernatant harvested from CD3/CD28 stimulated splenocytes caused a robust increase of IDO1 and IDO2 (Fig. 4c, d), indicating some soluble factor secreted from T cells mediates IDO induction. The supernatant also induced IDO expression in BNL tumor cells but at a much less extent. IFN-γ production after immune checkpoint blockade is well documented [28, 29]. Next, RIL-175 tumor cells were cultured with IFN-γ. A 27-fold induction of IDO1 was found in RIL-175 cells, while under the same condition, BNL tumor cells displayed much less IDO induction (~ fivefold) (Fig. 4e, f).
Fig. 4.
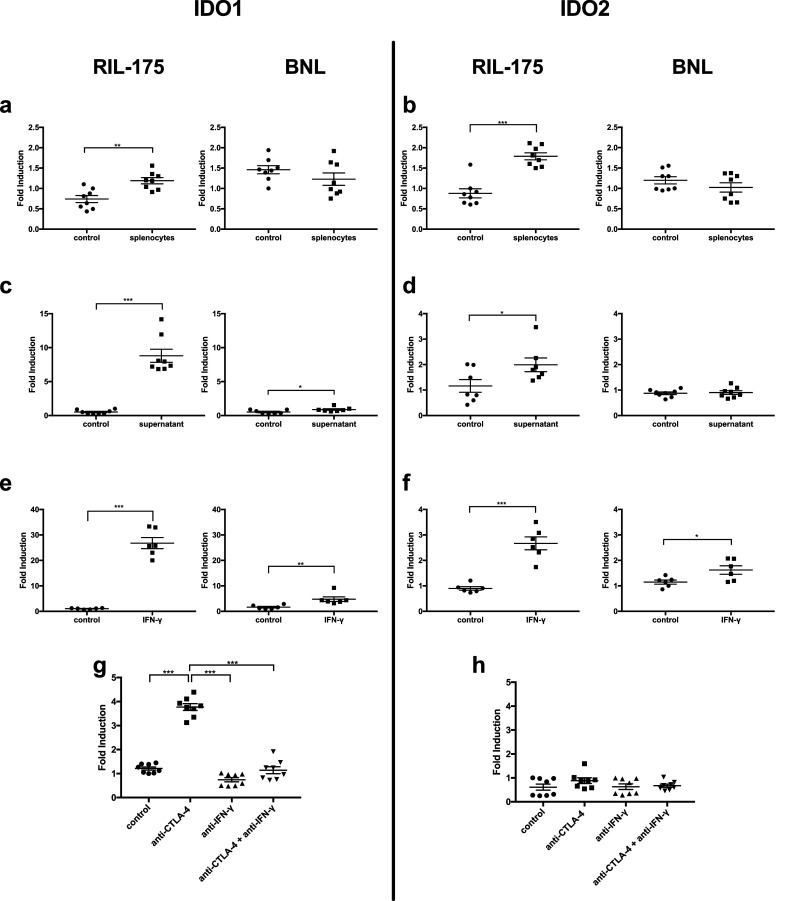
In vitro stimulation of murine tumor cells showed variable levels of IDO1 and IDO2 expression. 106 tumor cells were co-cultured with 107 splenocytes for 24 h and induction of IDO1 (a) and IDO2 (b) was measured by RT-qPCR. 107 splenocytes were cultured with PMA for 24 h after which supernatant was isolated and cultured with 106 tumor cells for 24 h and induction of IDO1 (c) and IDO2 (d) was measured by RT-qPCR. 106 tumor cells were treated with 750 unit IFN-γ for 24 h and induction of IDO1 (e) and IDO2 (f) was measured by RT-qPCR. Mice were injected with 106 tumor cells in left inguinal pocket. Subcutaneous RIL-175 tumors were established on day 0 and mice received i.p. injection of anti-IFN-γ on days 7, 10, and 13 and/or anti-CTLA-4 antibody on days 8, 11, and 14. Mice were sacrificed on day 15. Induction of IDO1 (g) and IDO2 (h) in RIL-175 with or without anti-IFN-γ was measured by RT-qPCR. *P < 0.05, **P < 0.01, ***P < 0.001
Next, in vivo IFN-γ blocking assay was performed to prove that anti-CTLA-induced IDO induction is mediated by IFN-γ. As expected, IFN-γ neutralizing antibody inhibited upregulation of IDO1 in RIL-175 tumors (Fig. 4g, h). This indicates that HCC tumor cells can utilize IFN-γ to induce IDO1 and develop an adaptive resistance against anti-CTLA-4 treatment.
IFN-γ-mediated IDO induction also occurs in human HCC tumor cells
IFN-γ induction is a general phenomenon upon immune checkpoint blockade treatment [28, 29]. Therefore, we tested whether the IFN-γ-mediated IDO induction also applies to the human setting. Human HCC cell lines, Hep3B and HepG2, were treated with IFN-γ. Similarly, IDO1 and IDO2 upregulation was observed (Fig. 5a, b). In addition, both cell lines displayed induction of IDO1 when cultured with cytokine rich supernatant harvested from stimulated health donor human PBMCs. These results suggest that human HCC tumor cells may also use the same strategy to acquire adaptive resistance against immune checkpoint blockade.
Fig. 5.

In vitro stimulation of human HCC tumor cells showed variable levels of IDO1 and IDO2 expression. Hep3B (a) or HepG2 (b) tumor cells (500,000 cells) cultured with 1000 unit IFN-γ or a cytokine rich supernatant isolated from 5 × 106 stimulated human PBMCs for 24 h. Induction of IDO1 and IDO2 was measured by RT-qPCR. **P < 0.01, ***P < 0.001
Anti-PD-1 treatment decreases tumor growth and induces IDO from tumors
Nivolumab, a monoclonal antibody which blocks PD-1, has recently been approved for patients with advanced HCC who progressed on sorafenib [5]. After administration of anti-PD-1 antibody, we observed similar results as with anti-CTLA-4, where anti-PD-1 therapy does indeed upregulate IDO1 and IDO2 in RIL-175 tumor cells (Fig. 6a, b). Furthermore, we observed a significant reduction in tumor volume with anti-PD-1 monotherapy and the best impairment in tumor growth with combination therapy in the RIL-175 model (Fig. 6c, d). In addition, we tested a different IDO inhibitor, epacadostat, and found similar result with the greatest tumor reduction seen with the combination therapy of anti-PD-1 and epacadostat (Fig. 6e, f). These data indicate that checkpoint inhibition with anti-CTLA-4 or anti-PD-1 may induce IDO and the addition of an IDO inhibitor could be beneficial.
Fig. 6.

Anti-PD-1 treatment effect on the induction of IDO1 (a) and IDO2 (b) in subcutaneous RIL-175 tumors as measured by RT-qPCR. c Tumor volume of RIl-175 tumors for control mice, anti-PD-1 alone, or combination therapy of anti-PD-1 and 1-d-MT. Means ± SEM of tumor sizes are shown from five mice per group. d Tumor weight was recorded for RIL-175 tumors as grams (g) of tumor tissue after mice were sacrificed. e Tumor growth kinetics of RIl-175 tumors for control mice, epacadostat, anti-PD-1, or combination therapy of anti-PD-1 and epacadostat. f Tumors weight was recorded as grams (g) of tumor tissue. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Discussion
Our current study has indicated that treatment of HCC tumors with immune checkpoint inhibitors may lead to IFN-γ induced IDO1 production from tumor cells which can provide a means of adaptive resistance to therapy. The RIL-175 tumor model, which overexpressed IDO1 both in vitro and in vivo when stimulated, did not have a significant reduction in tumor growth with anti-CTLA-4 alone. There was, however, a significant reduction in tumor growth with the addition of an IDO inhibitor. However, the BNL tumor model that did not upregulate IDO displayed a significant reduction in tumor growth with single-agent anti-CTLA-4 checkpoint blockade. We observed similar effects when the tumors were treated with anti-PD-1. In addition, a significant reduction in tumor growth was seen with anti-PD-1 monotherapy in the RIL-175 tumor model. This finding is not surprising as clinical trials have shown a greater response to anti-PD-1 therapy than anti-CTLA-4, allowing for the approval of nivolumab in patients with HCC who have progressed on sorafenib [5, 30]. Although other groups have also found IDO expression to render experimental mouse models resistant to checkpoint inhibition, our study provides evidence that immune checkpoint inhibitors have the capacity to induce IDO through IFN-γ, thereby providing a means of adaptive resistance to immune checkpoint blockade [18, 19, 31, 32]. In addition, our study highlights the importance of tumor-derived IDO1. Furthermore, we found IDO2 to be upregulated in vitro but not in vivo indicating the likely smaller impact of tumor-derived IDO2 in creating an immunosuppressive TME.
Tumors may not respond to immunotherapy via multiple mechanisms. For example, Restifo et al. found that tumor cells which lost beta2-microglobulin (ß2-m) can no longer be recognized by CD8+ T cells [33]. Gao et al. observed that patients with metastatic melanoma who were identified as non-responders to ipilimumab, an anti-CTLA-4 antibody, to have tumors with genomic defects in IFN-γ pathway genes [34]. Furthermore, Kulkarni et al. found upregulation of alternative immune checkpoints, notably TIM-3, in lung adenocarcinoma, suggesting the upregulation of alternative immune checkpoints may be associated with adaptive resistance to anti-PD-1 therapy and therefore may be a potential therapeutic target [35]. The difference in response to therapy between tumor types of different pathologies as well as within tumor types indicates both an intrinsic primary resistance and an acquired resistance to immunotherapy [36].
Treatment with immune checkpoint inhibitors have been shown to increase the production of IFN-γ [28, 29]. In this study, we demonstrated that by blocking IFN-γ in mice given anti-CTLA-4 therapy, we could suppress the induction of IDO1 in the TME. This finding supports a link between anti-CTLA-4 or anti-PD-1 blocking antibodies, IDO upregulation in the TME, and tumor resistance. The effector mechanism of the immune checkpoint blockade may in fact be mediated by IFN-γ [19]. However, as we have shown, IFN-γ may also contribute to immune escape by increasing IDO. It has been demonstrated that CD8+ T cells and IFN-γ can increase PD-L1 as well as IDO in the melanoma TME [32, 37]. This phenomenon could explain the added benefit of combination therapy of anti-CTLA-4 and anti-PD-1 in patients with advanced melanoma [38]. Blocking IFN-γ directly would not be a suitable strategy as it appears to be required for the initial function of the immune checkpoint inhibitor. Therefore, blockade of the downstream alternative checkpoints which are produced, such as IDO, appears to be necessary.
IDO is a complex molecule with production from multiple different cell types. Several groups have studied the effects of IDO deficiency with IDO deficient (Ido−/−) mice. Unlike CTLA-4−/− mice whose phenotype produces rampant inflammation and death, Ido−/− mice do not produce a severe pathologic phenotype [9, 39]. Holmgaard et al. used IDO deficient mice and found greater tumor reduction when anti-CTLA-4 was added, indicating the host-derived IDO was responsible for the suppressive activity of IDO in their B16 melanoma model [19]. In addition, they found that IDO deficient mice did not display delayed tumor growth without the addition of anti-CTLA-4. This corresponds to our findings, as well as others, that IDO inhibition with 1-d-MT alone does not significantly delay tumor growth [19, 32].
Conversely, Uyttenhove et al. studied the effect of IDO expression in tumor cells utilizing P815B cells transfected with Ido cDNA, using three clones with variable expression of IDO. Not only did they find that IDO-expressing tumor cells grow faster than non-IDO-expressing tumor cells but also immunized mice were able to reject non-IDO-expressing tumors. On the contrary, immunized mice were not able to reject IDO-expressing tumors, but this effect was able to be partially reversed with 1-MT [14]. Similarly, studies performed by Holmgaard et al. generated a B16 melanoma cell line to overexpress IDO where they found IDO overexpressing cells were able to recruit MDSCs and reduce tumor response to immunotherapy [17, 18]. Furthermore, Shibata et al. studied the role of IDO in HCC tumor carcinogenesis. They found HCC overexpressed IDO compared to surrounding normal liver tissue and HCC formation was greater in IDO wild-type than Ido−/− mice when challenged with diethylnitrosamine (DEN) [40]. Together, these results along with our own indicate IDO production in the tumor microenvironment is complex with host derived as well as tumor-derived IDO contributing to immune escape and potentially altering response to therapy.
We showed in a HCC model that tumor cells which produce IDO when treated with a checkpoint inhibitor may provide a means of adaptive resistance. Recent results found no difference in the progression-free survival of patients with advanced melanoma treated with the combination of the IDO inhibitor epacadostat and pembrolizumab versus pembrolizumab alone (ECHO-301/KEYNOTE-252) [41]. However, in patients with advanced melanoma, the 6-month progression-free survival rate to single-agent pembrolizumab was shown to be approximately 47% in the KEYNOTE-006 trial [42]. Therefore, as nearly 50% of patients with single-agent pembrolizumab demonstrated a 6-month progression-free survival, there may not have been much room for improvement over single-agent therapy. As response rates to immune checkpoint inhibitors tend to be lower for HCC compared to melanoma, the addition of an IDO inhibitor may prove to be beneficial [3]. Although patients with HCC generally have underlying liver disease and dysfunction, checkpoint inhibition appears safe and combination therapy should be considered for these patients [43]. In addition, a phase 3 study of nivolumab versus sorafenib as first line therapy is currently underway [44]. Based on our results, an IDO inhibitor may add additional benefit for patients with advanced HCC in combination with an immune checkpoint inhibitor.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank J. Berzofsky and his lab for their helpful discussion. We would also like to thank J.M. Hernandez for advising on performing intra-hepatic injections.
Abbreviations
- 1-d-MT
1-methyl-d-tryptophan
- BLI
Bioluminescent imaging
Author contributions
ZJB, SJY, and BH performed experiments. ZJB, CM, QF, FK, and TFG analyzed data. QF, MS, DA, and QZ assisted with experiments. ZJB and TFG conceived and designed the project. ZJB and TFG wrote the manuscript and all authors contributed to writing and providing feedback.
Funding
Tim F. Greten is supported by the NIH intra-mural program (ZIA BC 01134).
Compliance with ethical standards
Conflict of interest
The authors declare they have no conflicts of interest.
Animal experiments
All experiments were performed according to the institutional guidelines and approved by a NCI-Bethesda (Bethesda, MD, USA) Institutional Animal Care and Use protocol. Mice were purchased from Charles River Laboratories (VA, USA) or The Jackson Laboratory (Bar Harbor, USA). Human blood samples (buffy coat) were obtained from the National Institutes of Health Blood Research Services.
Cell lines
RIL-175 cell line was obtained from Dr Lars Zander (University Hospital of Tübingen, Germany), BNL cell line was provided by Dr. Jesus Prieto (University of Navarra, Spain), HepG2 (Cat no: HB-8065) and Hep3B (Cat no: HB-8064) was purchased from ATCC.
References
- 1.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi: 10.1038/nrdp.2016.18. [DOI] [PubMed] [Google Scholar]
- 2.Greten TF, Duffy AG, Korangy F. Hepatocellular carcinoma from an immunologic perspective. Clin Cancer Res. 2013;19:6678–6685. doi: 10.1158/1078-0432.CCR-13-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greten TF, Sangro B. Targets for immunotherapy of liver cancer. J Hepatol. 2017 doi: 10.1016/j.jhep.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark DP. Biomarkers for immune checkpoint inhibitors: the importance of tumor topography and the challenges to cytopathology. Cancer Cytopathol. 2018;126:11–19. doi: 10.1002/cncy.21951. [DOI] [PubMed] [Google Scholar]
- 5.El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017 doi: 10.1016/s0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duffy AG, Ulahannan SV, Makorova-Rusher O, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol. 2017;66:545–551. doi: 10.1016/j.jhep.2016.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Investig. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014;63:721–735. doi: 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jusof FF, Bakmiwewa SM, Weiser S, Too LK, Metz R, Prendergast GC, Fraser ST, Hunt NH, Ball HJ. Investigation of the tissue distribution and physiological roles of indoleamine 2,3-dioxygenase-2. Int J Tryptophan Res. 2017 doi: 10.1177/1178646917735098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metz R, Smith C, DuHadaway JB, et al. IDO2 is critical for IDO1-mediated T-cell regulation and exerts a non-redundant function in inflammation. Int Immunol. 2014;26:357–367. doi: 10.1093/intimm/dxt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prendergast GC, Mondal A, Dey S, Laury-Kleintop LD, Muller AJ. Inflammatory reprogramming with IDO1 inhibitors: turning immunologically unresponsive ‘Cold’ tumors ‘Hot’. Trends Cancer. 2018;4:38–58. doi: 10.1016/j.trecan.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 15.Korangy F, Hochst B, Manns MP, Greten TF. Immunotherapy of hepatocellular carcinoma. Expert Rev Gastroenterol Hepatol. 2010;4:345–353. doi: 10.1586/egh.10.18. [DOI] [PubMed] [Google Scholar]
- 16.Pan K, Wang H, Chen MS, et al. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134:1247–1253. doi: 10.1007/s00432-008-0395-1. [DOI] [PubMed] [Google Scholar]
- 17.Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine. 2016;6:5058. doi: 10.1016/j.ebiom.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, Merghoub T, Wolchok JD. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. 2015;13:412–424. doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–1402. doi: 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eggert T, Wolter K, Ji J, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30:533–547. doi: 10.1016/j.ccell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohlhapp FJ, Broucek JR, Hughes T, et al. NK cells and CD8+ T cells cooperate to improve therapeutic responses in melanoma treated with interleukin-2 (IL-2) and CTLA-4 blockade. J Immunother Cancer. 2015;3:18. doi: 10.1186/s40425-015-0063-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sell S, Dietz M, Schneider A, Holtappels R, Mach M, Winkler TH. Control of murine cytomegalovirus infection by gammadelta T cells. PLoS Pathog. 2015;11:e1004481. doi: 10.1371/journal.ppat.1004481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koblish HK, Hansbury MJ, Bowman KJ, et al. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol Cancer Ther. 2010;9:489–498. doi: 10.1158/1535-7163.MCT-09-0628. [DOI] [PubMed] [Google Scholar]
- 24.Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253–257. doi: 10.1038/nature16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu SJ, Yoon JH, Yang JI, et al. Enhancement of hexokinase II inhibitor-induced apoptosis in hepatocellular carcinoma cells via augmenting ER stress and anti-angiogenesis by protein disulfide isomerase inhibition. J Bioenerg Biomembr. 2012;44:101–115. doi: 10.1007/s10863-012-9416-5. [DOI] [PubMed] [Google Scholar]
- 26.Kapanadze T, Gamrekelashvili J, Ma C, et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol. 2013;59:1007–1013. doi: 10.1016/j.jhep.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, Korangy F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–243. doi: 10.1053/j.gastro.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 28.Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, Logothetis C, Sharma P. CTLA-4 blockade increases IFNgamma-producing CD4+ ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci USA. 2008;105:14987–14992. doi: 10.1073/pnas.0806075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 30.Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59:81–88. doi: 10.1016/j.jhep.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 31.O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. doi: 10.1016/j.ctrv.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. doi: 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst. 1996;88:100–108. doi: 10.1093/jnci/88.2.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J, Shi LZ, Zhao H, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397–404.e399. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16:121–126. doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller AJ, DuHadaway JB, Chang MY, et al. Non-hematopoietic expression of IDO is integrally required for inflammatory tumor promotion. Cancer Immunol Immunother. 2010;59:1655–1663. doi: 10.1007/s00262-010-0891-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibata Y, Hara T, Nagano J, et al. The role of indoleamine 2,3-dioxygenase in diethylnitrosamine-induced liver carcinogenesis. PLoS One. 2016;11:e0146279. doi: 10.1371/journal.pone.0146279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Broderick JM (2018) Pembrolizumab combo fails in melanoma. OncLive. https://www.onclive.com/web-exclusives/pembrolizumab-combo-fails-in-melanoma
- 42.Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 43.Brown ZJ, Heinrich B, Steinberg SM, Yu SJ, Greten TF. Safety in treatment of hepatocellular carcinoma with immune checkpoint inhibitors as compared to melanoma and non-small cell lung cancer. J Immunother Cancer. 2017;5:93. doi: 10.1186/s40425-017-0298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sangro B, Park J-W, Cruz CMD, Anderson J, Lang L, Neely J, Shaw JW, Cheng A-L (2016) A randomized, multicenter, phase 3 study of nivolumab vs sorafenib as first-line treatment in patients (pts) with advanced hepatocellular carcinoma (HCC): CheckMate-459. J Clin Oncol (suppl; abstr TPS4147)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.