

Abstract
Development of therapeutic strategies against RAS-driven cancers has been challenging due in part to a lack of understanding of the biology of the system and the ability to design appropriate assays and reagents for targeted drug discovery efforts. Recent developments in the field have opened up new avenues for exploration both through advances in the number and quality of reagents as well as the introduction of novel biochemical and cell-based assay technologies which can be used for high-throughput screening of compound libraries. The reagents and assays developed at the NCI RAS Initiative offer a suite of new weapons that could potentially be used to enable the next generation of RAS drug discovery efforts with the hope of finding novel therapeutics for a target once deemed undruggable.
Keywords: assay development, drug discovery, RAS, cell-based assays, biochemical assays
1. Introduction
It has now been five decades since RAS oncogenes were first identified, and still, RAS-driven cancers have managed to elude effective therapeutic attack. RAS genes are frequently mutated in human cancers [1], and these RAS-driven cancers have proven to be extremely challenging to treat and are often are excluded from conventional therapies [2]. For decades, RAS proteins were considered “undruggable” based on a long line of failures of drugs which targeted mutant RAS proteins or their downstream effectors. In general, gaps in our understanding of the details of RAS biology are probably responsible for the lack of development of novel therapeutic strategies. In the past decade, better cellular and biochemical understanding of some elements of the RAS signaling pathway has begun to offer new opportunities for targeting RAS [2-4], but many hurdles remain. In particular, most of the biochemical and biophysical data surrounding RAS interactions with downstream effector proteins has been derived from work on soluble domains of RAS proteins which do not interact with membranes. As RAS is not believed to signal within the cell in the absence of membrane localization, this presents a highly biased picture of how RAS carries out its role in activation of signal transduction. In addition, much of the biochemical drug screening work which has been carried out has also been done outside of this membrane context, potentially missing out on a wide range of therapeutic reagents which may be specific to the structure or interactions of RAS and its partners in their native membrane environment. Even structural information about RAS proteins have mostly ignored components which were involved in membrane interaction, making it extremely difficult to understand the structural rationale of cell-based assays using membrane-localized RAS. Taken together, these issues argue strongly for the need for new reagents and assays which are designed to examine the role of RAS in its native environment, bound to the plasma membrane and interacting with effectors which signal from that membrane environment. One of the main goals of the NCI RAS Initiative at the Frederick National Laboratory for Cancer Research has been to develop such new reagents and tools to enable a new attack on the well-defended RAS proteins, with the hope of finding ways to penetrate their defenses and permit development of therapeutics to deal with this unmet clinical need.
2. Reagents
A review of the RAS-related literature quickly demonstrates that there is a very limited amount of consistency in DNA, protein, and cell line reagents, which may account for some of the confusing and contradictory data over the first several decades of RAS research. Notably, RAS proteins are often generated with different amino and carboxy termini, or with various tags for solubility, purification, and detection. Purity levels are often hard to assess in the literature, and supporting biochemical and biophysical data is often difficult to interpret in the absence of quality control measurements. DNA reagents also suffer from inconsistency, not only from errors in cDNA sequences used as templates, but a failure to consider isoform differences or alternative splicing, or lack of clarity in which forms of genes are utilized. Finally, cell based reagents perhaps suffer the most in terms of quality control issues—often comparisons are made between two completely different cell lines which are simply defined by their RAS mutational status as G12D or Q61R, with total ignorance of the fact that these cell lines likely differ not only in overall mutational status, but even in cell type and tissue of origin. Taken together, these issues strongly argue for a more rational and quality-focused approach to designing RAS reagents that are essential for future drug discovery efforts.
2.1 Nucleic Acid Reagents
Early in the development of the NCI RAS Initiative, a crowdsourced effort was started by Dr. Frank McCormick and Dr. Bob Stephens to identify the genes which likely represented the whole of the RAS pathway, beyond the relatively simple set of genes already well established to be involved with RAS signaling through the main MAP kinase and PI3 kinase pathways. The initial efforts identified a set of 211 genes determined to be involved in RAS signaling, with 178 of these upstream of the transcription factors that were often the endpoint of the signaling cascade (www.cancer.gov/research/key-initiatives/ras/ras-central/blog/2015/ras-pathway-v2). While all 178 of these genes were present in cDNA libraries, and more than 95% of them were represented in “sequence-validated” ORFeome collections, we were surprised to find that in more than 24% of these genes, the validated commercially available clones did not match the reference sequence of the most commonly expressed isoform of the gene of interest in cancer cell lines. In 24 cases, the ORFeome or commercially available cDNA was for a splice variant which represented a very small proportion of the observed transcripts, while 4 genes had only ORFeome or cDNA clones which were no longer even supported in Genbank as likely transcripts in the cell. In another 15 cases without validated ORFeome clones, the only commercially available cDNA clones contained mutations varying from the reference sequence—while some of these may well be SNPs, in most cases they appear to be PCR-derived errors which are not supported by any other bioinformatics as being acceptable mutations. It is likely that many of these incorrect cDNA and ORFeome clones have been used in experiments, and while a single point mutation may not be a significant issue in all cases, the potential for lack of correlation of results due to DNA sequence alterations should not be considered acceptable. The NCI RAS Initiative RAS Reagents Core generated fully sequence validated ORFeome clones of all 178 most common transcript forms, and has distributed these reagents to the Addgene repository (www.addgene.org/cancer/ras-pathway) to ensure that all RAS researchers can access validated clones for the pathway components.
2.2 Protein Reagents
RAS proteins were first crystallized in 1990 by the Wittinghofer group, who generated crystal structures of human HRAS bound to both GDP and GTP [5]. Subsequently, a number of structures of mutant RAS proteins were generated, including structures with RAS family members bound to small domains of effectors including RAF1 [6], RASA1 [7] and SOS1 [8]. However, all of these early structures of RAS proteins either used truncated proteins lacking the C-terminal hypervariable region, or failed to show any structure in this region likely due to a lack of proper protein processing. Thus, for decades, nearly all work on RAS proteins was carried out using soluble proteins lacking the membrane interacting regions essential for most of the signaling functions of the protein. Likewise, much of the work done on the development of compounds targeting RAS utilized these non-natural substrates. In recent years, several groups began to look more carefully at the interaction of RAS on membrane surfaces utilizing chemical ligation or tethering methods to construct surrogate RAS molecules which could bind artificially to membranes. The Ikura group developed a technique for using maleimide linkages to tether the truncated RAS proteins directly in lipid nanodiscs [9], while other groups developed protein ligation strategies to mimic the true in vivo form of RAS [10]. Historically, several groups had attempted to produce more naturally processed KRAS, either by partial post-translational modification using farnesyltransferase enzymes, or by developing eukaryotic expression systems to make the proteins directly in their native form [11, 12]. While these were successful, the protein yields and quality were poor and beyond some basic biophysical work, it was not possible to generate enough good quality protein for structural biology or drug screening efforts. Recent engineering of insect cells finally permitted the high-yield production of fully processed KRAS, which led to the generation of the first crystal structure which showed the entirety of native KRAS, including the hypervariable region, the farnesyl group, and the carboxyterminal methyl group [13]. While this structure does not show significant differences within the G-domain region of KRAS, the extended alpha helical structure of the hypervariable region now allows more accurate modeling of the potential binding of KRAS to membranes. This protein-membrane interface could serve as a novel target for drug screening efforts to identify inhibitors of RAS binding to the membrane.
Key to these efforts was the development of a suite of quality control measurements to ensure that the protein was fit for purpose (Fig. 1). These QC steps began with mass spectrometry to verify the molecular mass of the protein, and to confirm the exact processing of the C-terminus. Secondary structure evaluation of the KRAS proteins by circular dichroism spectroscopy was consistent with published reports [14]. Determination of the melting temperature of KRAS proteins using intrinsic fluorescence or environmentally sensitive dyes (such as Sypro Orange) provided additional validation of the integrity of the protein, while dynamic light scattering was used to measure the polydispersity of the protein and provide information on its aggregation state. Finally, analytical ultracentrifugation was used to confirm the monomeric status of the protein. Ensuring that this full suite of QC tools is used for all protein reagents within the program has greatly enhanced our confidence in the protein reagents which are generated, especially controlling for batch-to-batch variability which is often an issue in any biological expression system. While many of these methods have been used in the literature in isolation, it is clear that consistently applying the entire suite to each protein generated has greatly benefited our program, and has identified issues with protein batches which might not have been seen if a limited subset of testing was carried out.
Fig. 1.
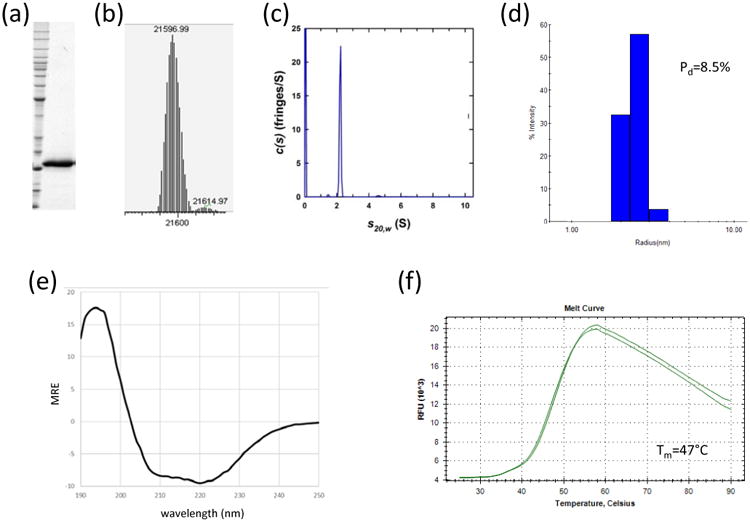
Representative quality control measures for protein reagents produced in the NCI RAS Initiative. All proteins undergo extensive quality control similar to the data shown here for an oncogenic mutant of KRAS4b. These data include (a) SDS-PAGE analysis, (b) intact mass determination by electrospray ionization mass spectrometry (ESI-MS), (c) analytical ultracentrifugation (AUC), (d) dynamic light scattering (DLS), (e) circular dichroism spectroscopy (CD), and (f) thermal shift assay.
Another improvement in protein production efforts around RAS drug discovery has come from dramatic advances in technologies surrounding protein expression in alternate host systems. While early work was almost entirely dependent on expression in bacterial systems (primarily E. coli), these systems were frequently unable to deal with the complexity of post-translationally modified human proteins, or multiprotein complexes. In addition, designing proteins with tags for many biochemical assays often led to issues with expression, solubility, or stability of the expressed proteins. In the past decade, major improvements have been made to common eukaryotic expression systems which have opened the door for production of proteins which could not be made in E. coli. Improvements to baculovirus-based insect cell expression systems [15], as well as heavily engineered mammalian systems based on HEK293 and CHO cells [16], increase the size of the protein production toolbox considerably. This opens up new avenues to approach multiprotein complexes and heavily modified proteins (such as phosphorylated MAP kinase pathway proteins).
2.3 Cell Line Reagents
One of the challenges in designing reagents for cell-based assays is identifying cell lines which accurately recapitulate the biology of the system under study. In the case of RAS, the literature is full of confusion regarding the actual dependency of certain cell lines on RAS mutations [17], and in many cases, cell lines designated as mutant for a particular RAS allele also carry wild type copies of the gene or have other mutations present which could affect interpretation of the results. To properly carry out cell-based assays comparing cell lines, having a cell line which is isogenic or as close to isogenic as possible would be highly beneficial. One option for this includes CRISPR modification of cell lines to remove endogenous KRAS genes or to introduce specific oncogenic mutations. Some commercial vendors such as Horizon have developed cell lines modified in this way, although in many cases they utilize cell lines which have defects in DNA repair pathways which enhance the gene editing efficiency. These may not be the best models for cell-based assays, and in many cases, these cell lines are not RAS-dependent. For this reason, we chose to further develop a mouse embryonic fibroblast (MEF) cell line panel containing single RAS transgene alleles by utilizing the RAS-dependent MEF system developed by Matthias Drosten and Mariano Barbacid [18]. These MEFs are endogenous Hras, Nras and Kras null, and their growth is completely dependent on exogenous RAS or activated MAPK pathway genes. By adding back specific isoforms of RAS genes or other pathway activators, cell lines can be developed which are completely dependent on these genes, but maintain as close to an isogenic background as possible. This makes the use of pairs of these cells in cell-based assays highly valuable for examining the effects of drugs targeting RAS or the pathway. The panel of cells we have generated currently includes all of the major oncogenic mutants of KRAS4b as well as wild type alleles of the other RAS isoforms, and a control line driven by mutant BRAF (V600E).
The workflow to generate these MEF lines requires removal of endogenous floxed KRAS with Cre recombinase, leading to the cells arresting in the G1 phase of the cell cycle (Fig. 2, panel a). Proliferation is resumed only through the delivery of the transgene to the cells using lentiviral transduction. Clonal cell lines are derived from initial pools and are thoroughly characterized by a wide variety of QC assays. These include confirmation of endogenous KRAS gene removal, identification of the transgene insertion site(s), calculation of proliferation rates and doubling times, determination of RAS protein expression level, analysis of signaling pathways, and response to tool compounds. Finally, the cell lines are exome sequenced to exclude lines with mutations in cancer relevant genes. This panel is a comprehensive resource that can be used to test allele and isoform specific hypotheses, and in addition, screen for RAS inhibitors and determine novel compound specificity (Fig. 2, panel b). It is important to note that RAS-dependent MEF cells do not capture all aspects of KRAS driven tumor biology. In fact, MEF cells are neither tumor nor human cells, but murine derived fibroblasts that express a single RAS isoform in isolation. There are likely multiple genetic, epigenetic and lineage related variables that alter tumor biology in human patients that are not captured by any engineered cell line. However, the RAS-dependent MEF system is remarkably simple and useful as a model to assess on-target activity of putative RAS and MAPK pathway inhibitors. Additionally, these cell lines are currently being used to evaluate gene-expression profiles, to screen antibodies, to generate effector binding profiles, and to characterize GTP-loading of RAS alleles. These RAS-dependent MEFs can also aid our understanding of RAS biology by elucidating mutations in RAS or pathway genes which can or cannot permit rescue of the cells.
Fig. 2.
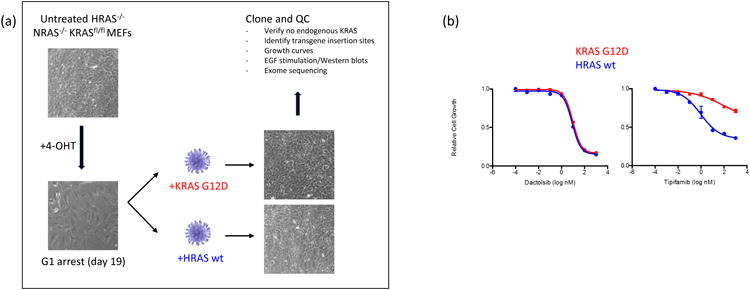
The RAS-dependent mouse embryonic fibroblast (MEF) system. Panel a shows a schematic for the production of RAS-dependent MEF lines, starting with untreated MEFs which lack HRAS and NRAS alleles and have floxed KRAS. When treated with 4-hydroxytamoxifen (4-OHT), the cells lose KRAS and enter G1 arrest. Transduction with lentiviruses carrying single alleles of RAS isoforms (KRAS G12D mutant and HRAS wild type are shown here as an example) allows a return to growth, now dependent entirely on the added RAS transgene. Clones can be selected from these pools and subject to rigorous QC as described. Panel b shows a set of dose-response curves using the two single-isoform RAS-dependent MEF lines. The Y-axis readout is cell viability based on Cell-Titer Glo assay. Dactolsib (left) is an PI3K inhibitor which should have no differential effect on different RAS isoforms. Tipifarnib (right) is a farnesyltransferase inhibitor which is expected to be highly selective for inhibition of HRAS over KRAS.
3. Biochemical Assays
It has long been recognized that the primary mechanism of KRAS oncogenesis occurs through mutations at codons 12, 13 or 61, which disrupt GAP-dependent GTP hydrolysis and thereby maintain KRAS proteins in a GTP-bound state. KRAS-GTP complexes readily associate with and activate effector proteins such as RAF, PI3K and RALGDS to promote cell cycle progression. Historical efforts to target oncogenic RAS proteins have not proven clinically tractable due to: 1) the high affinity for and high concentration of small molecule cofactors; 2) relatively shallow binding pockets and low affinity of small molecules that bind directly to RAS proteins; or 3) redundant signaling pathways and feedback mechanisms that render oncogenic KRAS cells resistant to inhibitors of prenylation or downstream effectors [4]. However, recent preclinical success developing catalytic inhibitors that covalently modify the oncogenic cysteine present in KRAS G12C [19], and advances in screening technologies, chemical library composition, and a more thorough understanding of KRAS biology have renewed interest in direct targeting of KRAS with small molecules. A major focus of any RAS drug discovery effort should be to develop biochemical assays and reagents that can be used in high-throughput screening efforts to identify small molecule inhibitors of oncogenic KRAS or oncogenic KRAS dependent cellular growth. At the NCI RAS Initiative, these assays are used in-house and in collaboration with pharmaceutical companies interested in targeting KRAS, to identify hit and lead molecules that disrupt oncogenic KRAS dependent cellular growth.
3.1 GTP binding and hydrolysis assays
Historically, the GTPase activity of RAS proteins was measured by monitoring the release of 32P from the single turnover hydrolysis of 32P labeled GTP [20]. This assay provides a sensitive and direct assay of KRAS GTPase activity. Additional coupled assays using fluorescence and absorbance read-outs have been developed that circumvent the need to use radioactivity. Use of a coumarin modified phosphate binding protein (PiBP) that increases fluorescence on binding inorganic phosphate released from GTP hydrolysis provides a simple homogeneous assay to measure KRAS activity [21]. Fusion of PiBP to a fluorescent protein enables FRET to be used to detect the release of free inorganic phosphate in KRAS GTPase reactions [22]. In addition absorbance measurement of 2-amino-6-mercapto-7-methylpurine that is converted by purine nucleoside phosphorylase from 2-amino-6-mercapto-7-methylpurine riboside in the presence of phosphate released from GTP hydrolysis has been used to measure KRAS GTPase activity [23]. Alternatively, KRAS GTPase activity has been measured using luminescence to detect the ATP levels, that are enzymatically converted from the GTP remaining and detected using luciferin/luciferase. Finally, identification of a GTP-specific antigen-binding fragment (Fab) coupled with GTP-Eu chelate and a fluorescence quencher has been used to detect soluble inorganic GTP and measure conversation of GTP to GDP [24]. Enzyme-linked immunosorbent assays (ELISA) have also been used to measure KRAS-GTP levels with purified proteins or in cellular lysates. Immobilization of RAF1 RBD domain will bind specifically to RAS-GTP, and not RAS-GDP or apo-RAS. The amount of RAS-GTP is then measured by addition of HRP or fluorescent conjugated antibody that binds total RAS. In all of these cases, a key initial step is to exchange KRAS into a non-hydrolysable or modified GTP analog for biochemical assays. GppNHp (5′-guanylyl imidodiphosphate) can be used as a suitable GTP mimetic that is resistant to hydrolysis for hours at 37C. Exchange of bound GDP for GppNHp is facilitated by removal of the magnesium at the catalytic site of KRAS using EDTA coupled with hydrolysis of the GDP using alkaline phosphatase [25]. Following GppNHp exchange it is important to validate the efficiency of exchange using a quantitative HPLC assay [25].
3.2 Proximity assays to interrogate protein-protein interactions
One of the challenges of designing RAS therapeutics relates to the difficulty in targeting protein-protein interactions which make up the bulk of the important signaling events driven by oncogenic RAS. To probe these interactions and potentially screen for drugs which can inhibit them, a series of assay technologies have been utilized, mostly focused on proximity-based methods that detect the physical interaction of two protein partners within a certain distance. The most popular assay technologies for proximity measurement are fluorescence assays including fluorescence resonance energy transfer (FRET), time resolved fluorescence (TRF) or singlet oxygen diffusion (Alpha, AlphaLISA), and these assays have all been used to detect KRAS-RBD interactions in solution. In addition, all of these assays have been widely adopted for drug screening and are amenable to high-throughput approaches in formats as small as 1536 well plates. All of the proximity assay formats involve incorporation of an affinity or fluorescence tag on a donor and acceptor protein (typically KRAS and RAF1-RBD or other RAS-binding protein) and require direct contact or close proximity (10-100 nm). However, singlet oxygen can diffuse up to 100-200 nm and react with acceptor lanthanide chelate beads, allowing for detection of larger molecular complexes (Fig. 3). This format has been used to detect KRAS-membrane and membrane-KRAS-RAF1 RBD complexes [26]. The variety of possible assay formats is only limited to the number of purified tagged proteins that can be generated, and multiple tags are available which can be detected using the Alpha chemistry, including FLAG, His6, GST, and Strep2. In many cases, these proteins need to be analyzed with tags on both N and C termini, since without structural data, it is hard to predetermine which terminus will be most readily available for a proximity based assay. In some cases, a particular end of the protein may not be available for tagging due to either inability to express or purify the tagged protein or interference with proper protein processing (as is the case for C-terminal fusions to RAS proteins). These assays are also completely dependent on production of high quality recombinant proteins, which can make them challenging for membrane-associated proteins or proteins like RAF1 which require chaperones or higher order complexes that are recalcitrant to in vitro production or purification.
Fig. 3.
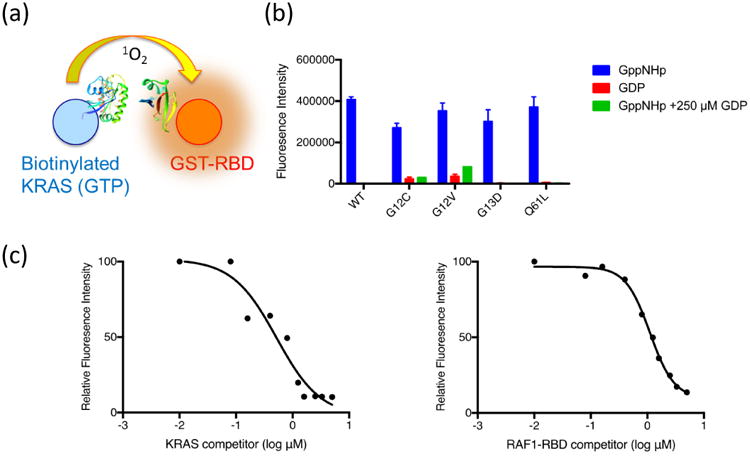
Schematic of the KRAS:RAF1 Alpha assay incorporating the RAS-binding domain of RAF1 (fused to a GST tag), KRAS-GTP (biotinylated on an Avi tag), and Alpha beads (panel a). The assay was validated with 4 oncogenic KRAS mutations (G12C, G12V, G13D, and Q61L). Addition of 250 uM GDP is used as a negative control (panel b). Full-length KRAS and RAF-RBD lacking the detection tags were used to disrupt the KRAS:RBD interaction and demonstrate specificity of binding and Alpha signal detection (panel C).
3.3 Surface plasmon resonance and other biosensor assays
Application of SPR based biosensors has long been used as an assay to measure direct interaction of small molecules with target proteins [27]. Using this assay, the equilibrium binding constant (KD) along with the association and dissociation rate constants for the binding of small molecules with KRAS is measured directly. Specifically, capture of Avi-tagged KRAS onto a neutravidin sensor chip surface provides an orientated KRAS protein surface that binds to the Ras-binding domain (RBD) of RAF1 in a nucleotide dependent manner (Fig. 4, panels a-c), consistent with published work. In the same type of experiment, no binding is observed with Avi-RHOA providing additional specificity on the interaction of RBD with surface captured small GTPases. Validation of this experimental design using RAF1-RBD ensures that the KRAS-GppNHp is available to bind small molecule ligands and inclusion of RHOA-GppNHp can be used as a control to demonstrate KRAS specificity. Small molecule KRAS binders can also be assayed for their activity in inhibiting KRAS-RBD interactions. KRAS-GppNHp binding to Avi-RAF1-RBD that is captured on a neutravidin sensor chip surface can be measured. Incubation of a fixed KRAS-GppNHp concentration with increasing concentrations with RBD completely inhibits binding to Avi-RBD captured on the chip surface (Fig. 4, panels d and e). In this same assay design, KRAS small molecule binders can be evaluated for their activity in inhibiting binding to RBD. This assay is a complementary tool to the proximity assays mentioned above. In addition to SPR, there are other new biosensor-based methods for identifying allosteric binders which can modulate RAS activity. One such platform takes advantage of an optical phenomenon called Second Harmonic Generation (SHG) to measure sub-Å conformational changes in a protein upon compound binding [28, 29]. In such an assay, KRAS can be labeled with an SHG sensitive dye and tethered to a lipid bilayer. The intensity of the SHG signal is highly sensitive to the orientation of the dye relative to the surface. A screen of a fragment collection identified a series of fragments that caused dose dependent shifts in the KRAS SHG signal, and subsequently hits were validated using SPR to measure direct binding to captured KRAS (Vo, unpublished data).
Fig. 4.
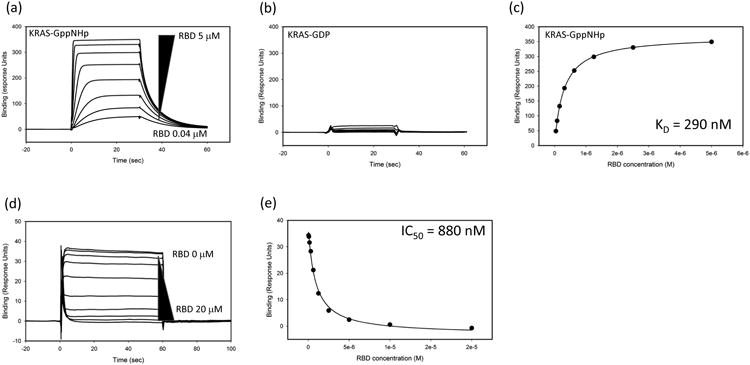
Surface Plasmon Resonance (SPR) analysis of KRAS:RBD interactions. Increasing concentrations of RAF1-RBD (5 – 0.04 uM; 2-fold dilutions) were flowed over KRAS-GppNHp (panel a) or KRAS-GDP (panel b). The binding response to KRAS-GppNHp can be fit to determine the equilibrium dissociation constant (KD) of the interaction (panel c). Inhibition of the interactions can be measured using variable amounts (20 – 0 uM; 2-fold dilutions) of Avi-RBD incubated with 300 nM of KRAS-GppNHp. In this case, unbound KRAS-GppNHp binds to captured Avi-RBD on the chip surface (panel d). Panel e shows the inhibition profile and calculation of an IC50 value.
4. Cell-based Assays
The purified and homogenous biochemical assays described in Section 3 are missing many components from both the normal cell, and importantly, from the cancer cell harboring mutant RAS. These could include unknown components that are themselves important and tractable targets, or that modulate the sensitivity of RAS to compounds. By building cell-based assays centered around some of the key insights of the RAS field, one can produce assays that are optimized to identify molecules that target the disease relevant functions of RAS. These assays could, in addition to identifying direct binders, identify compounds that for example, inhibit RAS-processing or lead to post-translational modifications of RAS, that inhibit the interaction of RAS with key effectors, that inhibit the interactions of RAS with scaffolding or chaperone molecules, or disrupt RAS function through some novel mechanism. Some of these assays which have been used to screen for potential RAS drugs are described below.
4.1 RAS localization assay
All the RAS isoforms, including the oncogenic variants, must localize to the plasma membrane to initiate downstream signaling; consequently, identifying small molecules that selectively target and disrupt the localization of mutant KRAS4b is one strategy for targeting the activity of this important oncogene. For correct trafficking and plasma membrane localization to occur, RAS proteins undergo several post-translational modifications [30]. Newly synthesized RAS molecules in the cytosol are farnesylated at the C-terminal CAAX motif. On the cytosolic face of the ER, RAS molecules are further processed through proteolytic cleavage of the three terminal amino acids, and methylation on the terminal carboxyl group. Additional and isoform specific modifications on residues in the C-terminal hyper variable region (HVR) occur in different compartments of the cell, and require different trafficking and processing machinery. KRAS4a, NRAS and HRAS all can be reversibly palmitoylated at one or two additional cysteines within the C-terminus. In contrast, KRAS4b contains a series of charged lysine residues. These positively charged lysines interact with negatively charged phospholipids in cell membranes, and evidence suggests that the trafficking of KRAS4b to the plasma membrane and its exchange to and from different membrane compartments in the cell and the cytosol is mediated by chaperones, including PDE-delta [13, 31] and calmodulin [32]. However, the details of KRAS4b transit to and from the plasma membrane have not been fully clarified.
To identify inhibitors of this potentially rich KRAS4b-specific target space, a high content assay (HCA) and image analysis pipeline have been developed to screen for molecules that disrupt the PM localization of KRAS4bG12D (Fig. 5). The assay is based on a clonally derived, doxycycline-inducible GFP-KRASG12D expressing HeLa cell line, and uses confocal microcopy to automatically image cells at sub-micron resolution in multi-well glass bottom plates. We chose HeLa cells for two reasons. First, HeLa cells are wild type for RAS isoforms. When we overexpress the mutant GFP-KRAS4b G12D protein, they show activation of the relevant MAPK pathway; consequently, this demonstrates that the RAS molecules that we express in these cells are functional in a background of wild type RAS. Second, HeLa cells are epithelial in origin; and are, therefore, suitable for imaging fluorescently tagged RAS molecules in the plasma membrane. The plasma membrane in HeLa cells is an unambiguous structure, and is visible, when stained appropriately, as a clear margin around the edge of the cell in confocal images. To specifically counterstain the PM compartment and nucleus, we use fluorescently labelled Concanavalin A (ConA) and Hoechst (a DNA-specific dye) respectively. An image analysis pipeline was developed to segment and quantitate the levels of GFP-KRASG12D in the PM compartment. Imaging for the localization assay consists of acquiring an image of the nucleus, the GFP-RAS and the plasma membrane from 12 random locations within each well of the assay plate. To identify the relevant signal above background, a supervised machine learning algorithm is run on 3-4 random images for both the nuclear or plasma membrane channels. This training is then used to create a binary mask for every nuclear and plasma membrane image, while the raw GFP-RAS images, as wells as the binary masks, are imported into CellProfiler (http://cellprofiler.org/) for data processing. Calculated Z′ factors using the mean values per well of membrane localized GFP-KRASG12D in doxycycline treated and untreated cells show acceptable values for screening. A counter-screen approach was developed using a doxycycline-inducible GFP-HRASWT expressing HeLa cell line. This allows us to identify molecules that are selectively active in disrupting KRAS localization, since HRAS and NRAS trafficking is distinct from KRAS, as described above.
Fig. 5.

Localization Assay for RAS at the plasma membrane. A high content imaging assay (panel a) was developed to detect molecules that differentially inhibit KRAS rather than wild-type HRAS plasma membrane localization using doxycycline inducible GFP-KRAS G12D and GFP-HRAS WT cell lines. Nuclei and membranes are counterstained with Hoechst (blue) and Concanavalin A (red) respectively. Image analysis is used to segment the plasma membrane compartment (Con A) and to quantify GFP from the RAS fusion proteins which are colocalized to the plasma membrane. The GFP signal can be normalized to on-plate controls (cells treated with only dox and untreated cells) to minimize signal variation. L778123 is a dual farnesyl and geranylgeranyl transferase inhibitor. As shown in panel b, this compound inhibits both HRAS and KRAS localization to the plasma membrane in the localization assay.
4.2 Resonance transfer assays
Once at the plasma membrane, mutant and GTP-loaded KRAS recruits effectors such as RAF, and activates downstream signaling. Disrupting RAS-dependent activation of effector signaling is another key strategy for targeting oncogenic RAS. However, how GTP-loaded RAS proteins activate effectors is not entirely understood. Conformational changes to RAS result in binding of RAF, but whether RAF dimerization is the result of RAS clustering or even RAS dimerization, or conversely, whether RAF dimerization leads to assemblage of a higher order RAS oligomer or cluster is not known. Furthermore, it may be that RAS solely functions to bring RAF to the membrane, or it may be that binding of RAS results in some conformational changes in RAF which are required for signaling.
The length scales of these interactions require assays that read out nanometer scale interactions, well below the limits of detection in conventional light microscopes. Förster resonance energy transfer (FRET) is a non-radiative transfer of energy between two chromophores: a donor and an acceptor. The exchange of the energy between the photo excited donor and the acceptor only occurs when the molecules are close enough for efficient transfer of the energy (inversely proportional to the sixth power of distance), and therefore can be exploited as a sensitive ruler of molecular interaction at the nanometer scale [33]. Bioluminescence resonance energy transfer (BRET) eliminates the requirement for external illumination, which reduces background, photo bleaching and bleed through of signals, and results in assays with significantly better signal to background ratios [34].
This technology has been exploited to develop assays for reporting protein-protein interactions of RAS molecules with itself and effector molecules in living cells. These assays are suitable for high throughput screening, and can be used to identify molecules that disrupt these interactions, and to probe the surfaces in the proteins responsible for the observed protein-protein interactions. NanoLuc is a 19 kDa engineered, luciferase protein that uses a chemical substrate (furimazine, a coelenterazine derivative) to generate sustained, high-intensity luminescence [35]. The intensity and narrow emission bandwidth of NanoLuc make it an ideal donor for resonance energy transfer (RET). In NanoBRET, Halo-tagged proteins are stoichiometrically and irreversibly labeled with an organic fluorophore, NCT, that acts as the RET acceptor [34]. The emission profiles of the donor and acceptor in NanoBRET are well separated increasing the signal to background performance of the assay.
Using a variety of fusion constructs to express tagged, full-length KRAS4b proteins in transiently transfected HEK293 cells, donor saturation assays demonstrate that increasing concentrations of the acceptor-tagged KRAS molecules will saturate the donor-tagged KRAS4b molecules, and that the interaction can be competed with expression of untagged KRAS4b (Fig. 6). The KRAS4b C185S mutant, which cannot be farnesylated, shows poor localization to the membrane, will not associate and accept energy from the WT and membrane localized donor, and serves as a negative control for the assay. Using this assay format, extensive analysis of RAS/RAS and HVR/HVR interactions can be performed to show evidence of close association of these molecules in the membrane of cells. In addition to identifying RAS/RAS interactions, these same assays can be applied to RAS interactions with effectors, such as RAF and RALGDS, where much more is known about the surfaces and residues in the proteins that are responsible for binding and association than in the case of the putative RAS/RAS interactions. We have exploited this prior knowledge to test the feasibly of using BRET to both evaluate the biochemistry of these interactions in the context of a living cell, and to develop assays suitable for high-throughput screening. Using both oncogenic and wild type NanoLuc-KRAS as donors, this assay can identify increased association of oncogenic KRAS molecules with RAF1. Conversely, these assays can be used to show that mutations in either Switch I of KRAS, or in the RBD domain of RAF disrupt the interaction.
Fig. 6.
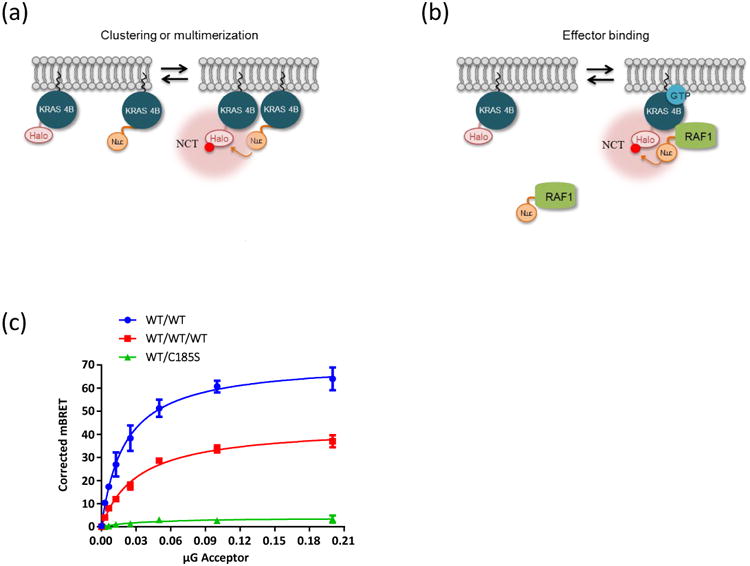
A Bioluminescence Resonance Energy Transfer (BRET) assay for measuring RAS:RAS and RAS:effector interactions in vivo. Panel a shows a scheme for examining the in vivo clustering of RAS molecules. The donor KRAS4b is tagged with NanoLuc, which upon interaction with the furimazine substrate produces bioluminescence as a byproduct of catalysis. If close enough, the energy from the donor can be transferred to the acceptor fluorophore, NCT, which is covalently linked to the Halotag on the second molecule of KRAS4b. Both NCT and fumarizine are fully membrane permeable and can be used with live cells. Panel b shows a similar scheme to examine RAS:effector interactions, now using a NanoLuc donor on the RAF1 protein to read out proximity to Halo tagged KRAS4b in the membrane. Panel c shows a donor saturation curve of HEK293 cells transfected with a constant amount of the donor NanoLuc-KRAS4b, and increasing amounts of the acceptor, either Halo-KRAS4b or Halo-KRAS4b C185S mutant which cannot bind to the membrane. One set of reactions (red) was competed with a constant concentration of untagged KRAS4b. The Y-axis reads out the level of BRET energy transfer.
The BRET assay has been thoroughly evaluated for high-throughput screening and found to have excellent performance characteristics. A library of ∼1500 chemically diverse molecules from the NCI (Diversity Set V from the Developmental Therapeutics Program) was tested on two days in a RAS/RAS BRET pair to check the reproducibility of the assay, and a very good correlation was found between runs, Z′ factors and signal to background ratios. Cell-based and phenotypic assays that report on the modulation or inhibition of mutant RAS within the physiological milieu of the cell are important tools for RAS drug discovery. They can serve as primary assays to identify tool compounds and potential lead molecules for drug development, and can be used to probe the biology of RAS in the whole cell context with quantitative readouts.
4.3 Proliferation assays
Loss of function RNAi and CRISPR “drop-out” screens have identified genetic targets that are synthetic lethal with KRAS mutation status, and identified several cell models that are highly dependent on mutated KRAS for growth and proliferation [36, 37]. Evaluating a small molecule inhibitor across a large panel of KRAS-dependent and KRAS-independent cancer cell lines has been one strategy to demonstrate on-target activity for putative KRAS inhibitors [38] or for inhibition of synthetic lethal targets [39-41]. However, this requires a relatively large number of cell lines to achieve statistical significance. Furthermore, KRAS dependence can vary in 2-D vs 3D culture environments, and toxicity due to off-target activity can easily confound results. Generation of the “RAS-less MEF” cells has been one useful way to address these problems using just 2 isogenic cell lines. Barbacid et al demonstrated on-target and off-target toxicity by treating the quiescent “RAS-less” cells with different MEK inhibitors [42]. Inhibitors that are highly selective for MEK should have no effect on MEF cells in G1 arrest due to loss of KRAS and MAPK activity. Conversely, compounds that induce toxicity in RAS-less MEF cells, which do not activate MEK or the MAPK pathway, are likely to be acting independent of activity for MEK1/MEK2. Similarly, even highly selective MEK inhibitors merely induce G1 arrest in the MEF cells that express KRAS, and do not cause toxicity. Any cell death observed after inhibitor treatment is also likely due to off-target compound activities. This system has also been used to demonstrate RAS-selective activity for putative KRAS inhibitors.
Generating the panel of KRAS-dependent MEF cells has also allowed not just the opportunity to evaluate existing RAS or MAPK selective inhibitors (Fig. 2, panel b), but also to conduct high-throughput phenotypic screens to identify small molecules that inhibit growth of cells harboring oncogenic KRAS, but not alter proliferation of MEF cells harboring wild-type KRAS, HRAS, BRAF V600E or any other isogenic MEF cell. Proliferation can be measured using any cellular viability measurement, including SRB, Cell-titer GLO, or any other viability measurement assay. Alternatively, MAPK pathway activity can be measured directly by ELISA, HTRF, Alpha, or other comparable assay format to measure MEK or ERK phosphorylation. With an approximate doubling time of 24 hours, the RAS-dependent MEF cells perform well in 2 or 3-day growth assays and can be optimized for 96, 384 or 1536 well formats.
5. Conclusions
Clearly, the difficulty involved in identifying therapeutics to directly target RAS-driven cancers has been borne out by the lack of success in a field full of highly competent researchers from all corners of the scientific world. However, whereas a few years ago RAS was often deemed “undruggable”, new research has begun to identify novel ways that we might be able to attack this formerly intractable problem. Coupling some of the technologies described here, particularly the use of high quality, well controlled reagents to design and screen biochemical and cell-based assays, there seems to be the proverbial light at the end of the tunnel. Continued development of new assay approaches which can each probe a different portion of RAS biology should ultimately permit a broader understanding of how RAS operates, and hopefully identify where it's weaknesses lie. Once those weapons begin to chip away at the armor surrounding RAS, it should be possible to bring the full power of the arsenal of 21st century structure-based drug design to bear on the problem with the hope of finally striking the fatal blow against RAS-driven cancers.
Acknowledgments
We thank the many members of the NCI RAS Initiative at the Frederick National Laboratory for Cancer Research for providing reagents and carrying out assay development work described in this manuscript. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129(7):1287–92. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25(3):272–81. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 3.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2(3):359–72. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCormick F. KRAS as a Therapeutic Target. Clin Cancer Res. 2015;21(8):1797–801. doi: 10.1158/1078-0432.CCR-14-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pai EF, Kabsch W, Krengel U, Holmes KC, John J, Wittinghofer A. Structure of the guanine nucleotide binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature. 1989;341:209–214. doi: 10.1038/341209a0. [DOI] [PubMed] [Google Scholar]
- 6.Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature. 1995;375(6532):554–60. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- 7.Scheffzek K, Lautwein A, Kabsch W, Ahmadian MR, Wittinghofer A. Crystal structure of the GTPase-activating domain of the human p120GAP and implications for the interaction with Ras. Nature. 1996;384:591–596. doi: 10.1038/384591a0. [DOI] [PubMed] [Google Scholar]
- 8.Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature. 1998;394(6691):337–43. doi: 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- 9.Mazhab-Jafari MT, Marshall CB, Smith MJ, Gasmi-Seabrook GM, Stathopulos PB, Inagaki F, Kay LE, Neel BG, Ikura M. Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1419895112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen YX, Koch S, Uhlenbrock K, Weise K, Das D, Gremer L, Brunsveld L, Wittinghofer A, Winter R, Triola G, Waldmann H. Synthesis of the Rheb and K-Ras4B GTPases. Angew Chem Int Ed Engl. 2010;49(35):6090–5. doi: 10.1002/anie.201001884. [DOI] [PubMed] [Google Scholar]
- 11.Page MJ, Hall A, Rhodes S, Skinner RH, Murphy V, Sydenham M, Lowe PN. Expression and characterization of the Ha-ras p21 protein produced at high levels in the insect/baculovirus system. JBiol Chem. 1989;264(32):19147–54. [PubMed] [Google Scholar]
- 12.Lowe PN, Page MJ, Bradley S, Rhodes S, Sydenham M, Paterson H, Skinner RH. Characterization of recombinant human Kirsten-ras (4B) p21 produced at high levels in Escherichia coli and insect baculovirus expression systems. J Biol Chem. 1991;266(3):1672–8. [PubMed] [Google Scholar]
- 13.Dharmaiah S, Bindu L, Tran Timothy H, William PHF, Gillette K, Ghirlando Rodolfo, Nissley Dwight V, Esposito Dominic, McCormick Frank, Stephen Andrew G, Simanshu Dhirendra K. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc Natl Acad Sci U S A Early on line publication. 2016 doi: 10.1073/pnas.1615316113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Matthews CR. Ligand binding is the principal determinant of stability for the p21(H)-ras protein. Biochemistry. 1998;37(42):14881–90. doi: 10.1021/bi9811157. [DOI] [PubMed] [Google Scholar]
- 15.Mehalko JL, Esposito D. Engineering the transposition-based baculovirus expression vector system for higher efficiency protein production from insect cells. J Biotechnol. 2016;238:1–8. doi: 10.1016/j.jbiotec.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain NK, Barkowski-Clark S, Altman R, Johnson K, Sun F, Zmuda J, Liu CY, Kita A, Schulz R, Neill A, Ballinger R, Patel R, Liu J, Mpanda A, Huta B, Chiou H, Voegtli W, Panavas T. A high density CHO-S transient transfection system: Comparison of ExpiCHO and Expi293. Protein Expr Purif. 2017;134:38–46. doi: 10.1016/j.pep.2017.03.018. [DOI] [PubMed] [Google Scholar]
- 17.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6):489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, Barbacid M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29(6):1091–104. doi: 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–51. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.John J, Frech M, Wittinghofer A. Biochemical Properties of Ha-ras Encoded p21 Mutants and Mechanism of the Autophosphorylation Reaction. Journal of Biological Chemistry. 1988;263:11792–11799. [PubMed] [Google Scholar]
- 21.Shutes A, Der CJ. Real-time in vitro measurement of GTP hydrolysis. Methods. 2005;37(2):183–89. doi: 10.1016/j.ymeth.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 22.Biancucci M, Dolores JS, Wong J, Grimshaw S, Anderson WF, Satchell KJ, Kwon K. New ligation independent cloning vectors for expression of recombinant proteins with a self-cleaving CPD/6xHis-tag. BMC Biotechnol. 2017;17(1):1. doi: 10.1186/s12896-016-0323-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chavan TS, Meyer JO, Chisholm L, Dobosz-Bartoszek M, Gaponenko V. A novel method for the production of fully modified K-Ras 4B. Methods Mol Biol. 2014;1120:19–32. doi: 10.1007/978-1-62703-791-4_2. [DOI] [PubMed] [Google Scholar]
- 24.Kopra K, Ligabue A, Wang Q, Syrjanpaa M, Blazevits O, Veltel S, van Adrichem AJ, Hanninen P, Abankwa D, Harma H. A homogeneous quenching resonance energy transfer assay for the kinetic analysis of the GTPase nucleotide exchange reaction. Anal Bioanal Chem. 2014;406(17):4147–56. doi: 10.1007/s00216-014-7795-7. [DOI] [PubMed] [Google Scholar]
- 25.Smith SJM, Rittinger K. Preparation of GTPases for Structural and Biophysical Analysis. In: Manser EJ, Leung T, editors. GTPase Protocols: The Ras Superfamily. 2002. pp. 13–24. [DOI] [PubMed] [Google Scholar]
- 26.Gillette WK, Esposito D, Abreu Blanco M, Alexander P, Bindu L, Bittner C, Chertov O, Frank PH, Grose C, Jones JE, Meng Z, Perkins S, Van Q, Ghirlando R, Fivash M, Nissley DV, Mc Cormick F, Holderfield M, Stephen AG. Farnesylated and methylated KRAS4b: high yield production of protein suitable for biophysical studies of prenylated protein-lipid interactions. Sci Rep. 2015;5:15916. doi: 10.1038/srep15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myszka DG. Analysis of small-molecule interactions using Biacore S51 technology. Anal Biochem. 2004;329(2):316–23. doi: 10.1016/j.ab.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 28.Moree B, Connell K, Mortensen RB, Liu CT, Benkovic SJ, Salafsky J. Protein Conformational Changes Are Detected and Resolved Site Specifically by Second-Harmonic Generation. Biophys J. 2015;109(4):806–15. doi: 10.1016/j.bpj.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moree B, Yin G, Lazaro DF, Munari F, Strohaker T, Giller K, Becker S, Outeiro TF, Zweckstetter M, Salafsky J. Small Molecules Detected by Second-Harmonic Generation Modulate the Conformation of Monomeric alpha-Synuclein and Reduce Its Aggregation in Cells. J Biol Chem. 2015;290(46):27582–93. doi: 10.1074/jbc.M114.636027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13(1):39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, Ismail SA, Hedberg C, Hanzal-Bayer M, Venkitaraman AR, Wittinghofer A, Bastiaens PI. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2012;14(2):148–58. doi: 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
- 32.Villalonga P, Lopez-Alcala C, Bosch M, Chiloeches A, Rocamora N, Gil J, Marais R, Marshall CJ, Bachs O, Agell N. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol Cell Biol. 2001;21(21):7345–54. doi: 10.1128/MCB.21.21.7345-7354.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel SS, Thaler C, Koushik SV. Fanciful FRET. Sci STKE. 2006;2006(331):re2. doi: 10.1126/stke.3312006re2. [DOI] [PubMed] [Google Scholar]
- 34.Machleidt T, Woodroofe CC, Schwinn MK, Mendez J, Robers MB, Zimmerman K, Otto P, Daniels DL, Kirkland TA, Wood KV. NanoBRET--A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem Biol. 2015;10(8):1797–804. doi: 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- 35.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7(11):1848–57. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168(5):890–903 e15. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Downward J. RAS Synthetic Lethal Screens Revisited: Still Seeking the Elusive Prize? Clin Cancer Res. 2015;21(8):1802–9. doi: 10.1158/1078-0432.CCR-14-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6(3):316–29. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 39.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Frohling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, Weir BA, Reiling JH, Sheng Q, Gupta PB, Wadlow RC, Le H, Hoersch S, Wittner BS, Ramaswamy S, Livingston DM, Sabatini DM, Meyerson M, Thomas RK, Lander ES, Mesirov JP, Root DE, Gilliland DG, Jacks T, Hahn WC. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, Dohner K, Bullinger L, Sandy P, Boehm JS, Root DE, Jacks T, Hahn WC, Gilliland DG. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 42.Urosevic J, Sum EY, Moneo V, Drosten M, Dhawahir A, Becerra M, Carnero A, Barbacid M. Using cells devoid of RAS proteins as tools for drug discovery. Mol Carcinog. 2009;48(11):1038–47. doi: 10.1002/mc.20555. [DOI] [PubMed] [Google Scholar]