

Abstract
Mucus secretion accumulation in the airways may act as a contributing factor for the development of airflow limitation in severe fetal asthma patients. Accumulated evidences showed that transforming growth factor beta (TGF-β) plays a regulatory role in airway remodeling including mucus hyper-secretion in asthma. However, the detailed molecular mechanisms of TGF-β3 induced MUC5AC hyper-expression in airway epithelium remains unclear. Here, we demonstrated the pivotal roles of autophagy in regulation of MUC5AC hyper-production induced by TGF-β3 in airway epithelium. Our experimental data showed that inhibiting autophagy pathway in repeated ovalbumin (OVA) exposed mice exhibited decreased airway hyper-response and airway inflammation, diminishing the expression of Muc5ac and TGF-β3. Furthermore, our studies demonstrated that autophagy was induced upon exposure to TGF-β3 and then mediated MUC5AC hyper-expression by activating the activator protein-1 (AP-1) in human bronchial epithelial cells. Finally, Smad2/3 pathway was involved in TGF-β3-induced MUC5AC hyper-expressions by promoting autophagy. These data indicated that autophagy was required for TGF-β3 induced airway mucous hyper-production, and that inhibition of autophagy exerted therapeutic benefits for TGF-β3 induced airway mucus secretion.
Keywords: TGF-β3, MUC5AC, Autophagy, Smad2/3
Highlights
-
•
TGF-β3 may affect mucus hyper-production in bronchial asthma via autophagy pathway.
-
•
Smad2/3 serves as a regulator of TGF-β3-induced autophagy activity and MUC5AC hyper-expressions in bronchial epithelium.
Research in context.
Mucous secretion accumulation in the airways may be a contribution for the development of airflow limitation of severe fetal asthma. Accumulating evidence suggests that autophagy plays important roles in asthma. Here, we confirmed the importance of TGF-β3-induced autophagy in mucus hypersecretion in asthma. Smad2/3 is confirmed as a regulator for TGF-β3-induced autophagy accompanied by mucus hyper-secretion, which is likely involved in activator protein-1 (AP-1) pathway. Our study indicates that autophagy is required for TGF-β3 induced airway mucous hyper-production, and that inhibition of autophagy exerts therapeutic benefits for TGF-β3 induced airway inflammation and airway mucous secretion.
1. Introduction
Asthma has become a global health issue affecting approximately 1–18% of the world population [1]. By the year 2025, it is estimated that >100 million individuals suffer from asthma [2]. Airway epithelial barrier fragility is central to the pathogenesis of asthma [3]. Repeated epithelial injury and repair contributed to histological changes and functional abnormalities in the airway mucosal epithelium [4]. Airway remodeling involves airflow obstruction which results from mucus hyper-secretion and impaired mucus transport [5, 6], subsequently becoming important pathological features of severe asthma and even fetal asthma [7]. Goblet cells are the major secretory cells involved in airway epithelium [8]. Abnormalities in the goblet cell number are accompanied by changes in stored and secreted mucus [6]. The mucin glycoproteins are the major macromolecular components of mucus. Among these 14 mucins, MUC5AC and MUC5B are the major gel-forming mucins [8]. MUC5AC is produced in the goblet cells from the surface epithelium, while MUC5B is largely produced in the sub-mucosal glands [6]. MUC5AC is upregulated by a wide variety of environmental and host factors relevant to chronic inflammatory airway diseases, including proteases, microbes, reactive oxygen species, air pollutants, cigarette smoke and cytokines [8]. Interleukin 13 (IL-13), a central mediator of asthma, upregulates MUC5AC via mitogen-activated protein kinase 13 (MAPK13), MEK/EKR1/2 and STAT6/SPDEF signaling pathways [9, 10].
Transforming growth factor beta (TGF-β) is produced in the airway by inflammatory cells infiltrated in the bronchial mucosal, as well as by structural cells of the airway wall including epithelial and endothelial cells [11]. Increased levels of TGF-β were observed in the bronchoalveolar lavage fluid of asthmatic patients [11]. Several studies have demonstrated that TGF-β isoforms participated in the regulation of airway inflammation and remodeling process [12, 13], especially mucus hyper-secretion in the airway epithelial cells [[14], [15], [16], [17]]. TGF-β1 was identified as an important factor in the regulation of MUC5AC through Smad4 and Sp1 in a co-operative manner to transactivate Muc5ac promoter activity [14]. However, there is a distinct observation that TGF-β1-Smad3/4 signaling act as a negative regulator for nontypeable haemophilus influenzae-induced MUC5AC transcription via a MAPK phosphatase-1-dependent inhibition of MAPK14 [15]. In addition, the topic of the effect of TGF-β2 isoform on MUC5AC expression is also controversial. A previous study has showed that TGF-β2 caused a decrease in both MUC5AC and MUC5B mRNA and protein in human bronchial epithelial (HBE) cells, which also could partially reduce IL-13-induced MUC5AC mucin production through Smad4 binding to MUC5AC promoter [17]. Another study suggested that IL-13 could induce TGF-β2 expression in vitro and the argument TGF-β2 could promote mucin expression in airway epithelial cells [16]. TGF-β3 isoform in house dust mite (HDM) model is implicated in the development of a severe phenotype of chronic airway remodeling [13]. However, there is a little direct and clear evidence that focused on whether TGF-β3 could regulate MUC5AC expressions.
Autophagy pathway induced by several environment factors such as respiratory infection, smoking, pollutants etc. contribute to the severity of asthma, affecting the pathological process of asthma [18, 19]. Excessive autophagy plays an essential role in airway mucus hyper-secretion and airway remodeling in chronic lung diseases [20, 21]. Depletion autophagy elements could block MUC5AC hyper-expression induced by cytokines or environment factors [19, 20, 22]. Autophagy pathway is required for both IL-13-mediated MUC5AC secretion and reactive oxygen species (ROS) activity in the airway bronchial epithelial cells [20]. And the underlying mechanism of IL-13-mediated increase in superoxide levels is that autophagy pathway could direct Dual oxidase 1 (DUOX1) to the apical surface of the airway epithelium [23]. Interleukin 4 (IL-4), a key effector Th2 cytokine in allergic asthma, was critical for autophagy induction in B cells and hence sustained B cell survival that enhanced IgE production and antigen presentation [24]. TGF-β1 can promote autophagy in different cell types, including cancer cell lines and lung fibroblasts, which has shown a response to promoting cells invasion, epithelial-mesenchymal transition (EMT) and the synthesis of extracellular matrix proteins [[25], [26], [27], [28]]. TGF-β1 may regulate autophagy through pRb/E2F1-dependent transcriptional activation of multiple autophagy-related genes in cancer cell lines of various origins [29]. TGF-β1 also activates protective autophagy by AEG-1 association with promoting EMT [27]. Moreover, upregulation of autophagy was accompanied by TGF-β1 hyper-production [21]. TGF-β2 could initiate autophagy to promote glioma cells' invasion via Smad and JNK pathway [30]. However, whether TGF-β3 induces autophagy and this process involved in regulation of mucus hyper-secretion remains unclear.
We hypothesized that TGF-β3 could induce autophagy and regulate mucus secretion. Hence, pharmacological and genetic experiments are conducted to address the molecular mechanisms mediating the effects of autophagy in TGF-β3 induced mucus secretion. Our results demonstrated that TGF-β3 isoforms trigger autophagy activity and autophagy flow via Smad2/3 activity, which is essential for TGF-β3 in modulating MUC5AC expression. During this process, the activation of activator protein-1 (AP-1) was also involved in as a downstream of TGF-β3-induced autophagy in regulation of MUC5AC expression. These study suggesting that autophagy pathway play a role in TGF-β3-modulation MUC5AC expression in airway bronchial epithelial cells.
2. Materials & Methods
2.1. Reagents
mCherry-EGFP-LC3 lentivirus vectors, ATG5-siRNA lentivirus vectors and BECN1-siRNA lentivirus vectors were purchased from GeneChem Technology (Shanghai, China). The control siRNA, Smad2/3 siRNA and c-Jun siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). 3-methyladenine (3-MA, M9281) was purchased from Sigma-Aldrich. Bafilomycin A1 (Baf A1; S1413) was purchased from Selleck. TGF-β1 ELISA reagents (MB100B) and TGF-β2 ELISA reagents (MB200) were purchased from R&D systems. TGF-β3 ELISA reagents (E-EL-M1192) was purchased from Elabscience Biotechnology. Recombinant human TGF-β1 (100-21) and recombinant human TGF-β3 (100-36E) were purchased from PeproTech.
2.2. Animal Models
Female C57BL/6 J mice at 6 to 8 weeks of age under specific pathogen-free conditions were purchased from Tengxin Biotechnology Co., Ltd. (Chongqing, China). All mice were maintained in a specific pathogen-free facility in the Animal Experimental Center of Southwest Medical University. All experimental protocols were approved by and performed in accordance with the guidelines of the Committee of Animal Experiments Center of Southwest Medical University and the National Institute of Health guidelines on the care and use of animals.
To establish a mouse model of allergic airway inflammation, mice were intraperitoneally sensitized on days 1 and 8 with 20 μg ovalbumin (OVA) and 1 mg aluminum hydroxide. From day 15 to 21, the mice were challenged by intratracheal instillation 10 μg OVA (in 50 μl PBS) per day for 7 d [31]. Control groups were given PBS alone. 3-MA (1 g/kg body weight) was given to each mice by intragastric administration 2 h before the challenge with OVA in OVA-3-MA mice. Mice were sacrificed 48 h after the last challenge. For bronchoalveolar lavage fluid (BALF), the lungs were slowly lavaged five times with 0.8 ml PBS. Then the total number of BALF cells was counted. Smears of bronchoalveolar lavage cells were fixed and stained with Wright-Giemsa stain (Leagene, DM0007), and differential counts were determined by counting 200 total cells.
2.3. Measurement of Airway Hyperresponsiveness
Evaluations of airway hyperresponsiveness (AHR) were performed as previously described [32]. Twenty-four hours after the final challenge, whole body plethysmography (Buxco Europe Ltd., Winchester, UK) was used to assess total respiratory system resistance after administration of increasing doses of methacholine (0, 6.25, 12.5, 25, and 50 mg/ml). Data are reported as peak Penh values.
2.4. Histological Analyses
The lungs of mice were collected and fixed in formalin. These Specimens were then embedded in paraffin and stained with hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS) following standard protocols. Inflammation was assessed using previously described methods [33].
2.5. Immunohistochemistry (IHC)
IHC assay was performed using antibodies to TGF-β1 (1:200; Santa Crus, SC-146), TGF-β2 (1:200; Santa Crus, SC-90) or TGF-β3 (1:200; Santa Crus, SC-82) according to the manufacturer's instructions by Specialized Histopathology Laboratory of Affiliated Hospital of Southwest Medical University using the Envision detection kit (Dako, K5007). The stained slides were analyzed with microscope (Leica, Germany).
2.6. Cell Culture, Transfection and Lentiviral Transduction
Human bronchial epithelial cells (16HBE cells) were used in our study [33]. These cells were cultured in Dulbecco's modified Eagle's medium (HyClone, SH30022.01) supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified 5% CO2 atmosphere. Cells were plated at 1.5 × 105–2 × 105 per ml in 6-well plates in antibiotic-free medium and transfected the following day with siRNA using Lipofectamine 2000 according to the manufacturer's standard protocol. The infection media was changed 6–8 h after transfection. 16HBE cells (2 × 105/ml) were transduced with lentivirus at a multiplicity of infection (MOI) of 5–10 in 8 μg/ml polybrene (Sigma-Aldrich, H9268). Viral supernatant was exchanged for fresh media 12 h after inoculation. To generate stable cDNA expressing cell lines, the infected cells were selected with 1 μg/ml puromycin. To explore the role of TGF-β3 in autophagy and MUC5AC hyper-expressions, the cells were treated with TGF-β3 at concentrations of 0, 2.5, 5, and 10 ng/ml for a certain time (see Figure Legends). To explore the role of autophagy in MUC5AC hyper-expressions, the cells were co-treated with 3-MA (or Baf A1), and the final concentrations in the culture medium were 5 μM (10 nM for Baf A1).
2.7. Real-Time PCR
Total RNA was isolated with TRIzol Reagent (Invitrogen), and cDNA was synthesized using PrimeScript™ RT Master Mix (Takara, China). The real-time PCR (RT-PCR) analysis was performed with SYBR Advantage qPCR Premix (Clontech, USA). Primer sequences are listed in the Supplemental Table S1.
2.8. Western Blotting
Western Blotting was performed using standard methods. After treatment with the indicated drugs, cells were washed with cold PBS and lysed in RIPA buffer (Thermo Fisher Scientific, 87787) supplemented with protease inhibitors cocktail and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 78420). Lysates were centrifuged at 12,000 ×g for 15 min at 4 °C, and protein concentrations were determined by BCA assay (Beyotime, P0010). Proteins (30 μg) were electrophoresed on 10–12% SDS-PAGE and transferred onto a polyvinylidene difluoride membranes (Merck Millipore, ISEQ00010). After blocking with 5% skim milk in Tris-buffered saline for 2 h, the membrane was incubated with the primary antibodies for overnight at 4 °C. The following primary antibodies were used: LC3B antibody (1:1000; Sigma-Aldrich, L7543), BECN1 antibody (1:1000; MBL, PD017), ATG5 antibody (1:1000; Sigma-Aldrich, A0856), SQSTM1 antibody (1:1000; MBL, PM045), Smad2 antibody (1:800; CST, 5339S), phospho-Smad2 antibody (1:800; CST, 3108S), Smad3 antibody (1:1000; Abcam, ab28379), phospho-Smad3 antibody (1:800; Abcam, ab52903), c-Jun antibody (1:800; CST, 9165S), phospho-c-Jun antibody (1:800; CST, 3270S), GAPDH antibody (1:1000; CST, 97166S). Secondary antibodies were labeled with HRP and the signals were detected using ECL Kit (Biorad, 170-5061).
2.9. Immunofluorescence Microscopy
For immunofluorescence (IF) analysis, cells were plated in the chamber slides and sections of lung tissues were fixed with 4% formaldehyde, and permeabilized with 0.3% Triton X-100 in PBS for 10 min at room temperature. After blocking with 1% BSA, the specimens were incubated with primary antibodies to MUC5AC (1:100; Abam, ab24070) or LC3B (1:100; Sigma-Aldrich, L7543) overnight at 4 °C. Subsequently, Alexa Fluor 555 or Alexa Fluor 488 conjugated secondary antibody (1:500; Invitrogen, A32727, A32732, A28175) was used to probe the primary Ab. DAPI was used for nuclear staining. IF samples were then imaged and quantitated using SP5 Leica confocal microscope with Leica Application Suite Software (Version number: 14.0.0.162, Leica, German).
2.10. Statistical Analysis
Statistical analyses were carried out using SPSS 16.0 software. All data are presented as mean ± s.d. Statistically significant differences were determined by one-way ANOVA test, Student's t-test and nonparametric Mann-Whitney test. P values <0.05 were considered statistically significant.
3. Results
3.1. Autophagic Inhibition Ameliorated Airway Hyper-Responsive and Mucus Hyper-Secretion in Allergic Mice
Autophagy plays an essential role in lung diseases and in the pathogenesis of asthma [34]. Airway inflammation, airway hyperresponse and airway mucus hyperproduction have played a central role in the development of asthma [13]. Severe asthma patients manifest higher levels of autophagic activation than healthy persons [35]. 3-methyladenine (3-MA), a class III group of phosphoinositide 3-kinase (PI3K) inhibitors, that has been widely used as autophagy inhibitor in the studies of autophagy related pathways [36]. In the present study, we used 3-MA to prove that the autophagy pathway possibly affected the pathological progression of allergic airway inflammation in the mice models (Fig. 1a). The bronchial airway hyper-response has been markedly elevated in the mice that are repeatedly exposed to OVA. The mice were treated with methacholine with the concentrations of 12.5 mg/ml, 25 mg/ml and 50 mg/ml, especially with 50 mg/ml. However, the airway response were declined in the mice exposed to OVA in addition to 3-MA compared with the mice exposed to OVA, when the mice were atomized with methacholine (Fig. 1b). Also the cell counts were detected in the alveolar lavage fluid of the mice. Interestingly, the total cells were declined in the allergic mice models together treated with 3-MA. The neutrophil as well as eosinophil (EOS) cell counts of BALF are obviously increased in the allergic mice models when exposed to OVA than the cell counts of BALF in the mice exposed to PBS. Moreover, 3-MA could decrease OVA induced neutrophil cell counts and EOS cell counts in the BALF (Fig. 1c). Thus, these results suggested that 3-MA could relieve OVA-induced airway inflammation. This conclusion was proved by HE staining of the airway in the mice. In the present study, we found less inflammatory cell infiltration around the airway in the mice exposed to OVA in addition to 3-MA treatment than in the mice exposed to OVA. Quantification of airway inflammation index also proved that 3-MA suppressed OVA-induced inflammation (Fig. 1d–e).
Fig. 1.

Autophagic inhibition ameliorated airway hyper-responsive and mucous hyper-secretion in allergic mice model. (a) Schematic diagram of the experimental protocol for sensitization and challenge with OVA. (b) Airway hyperresponsiveness was measured using plethysmographs at 24 h after the final challenge. (c) Quantification of inflammatory cell infiltration in BALF was performed by Wright-Giemsa stain. (d-e) Representative images of H&E-stained lung tissue sections of PBS-mice, OVA-mice, 3-MA-mice and 3-MA-OVA-mice. The inflammatory index demonstrated inflammatory cell infiltration, which were statistically analyzed by non-parametric Kruskal-Wallis test. (f-g) Representative images of PAS-stained lung tissue sections of PBS-mice, OVA-mice, 3-MA-mice and 3-MA-OVA-mice. The goblet cell percentage was quantified in 10 random fields. (h-i) Representative immunofluorescence images of Muc5ac protein in lung tissues of PBS-mice, OVA-mice, 3-MA-mice and 3-MA-OVA-mice. Quantitation of fluorescence intensity of Muc5ac in 10 random fields. (j) Real-time PCR was performed to detect the expression of Muc5ac gene. (k) The expression of LC3B was detected using western blot assays. (l) Relative changes in the density of LC3B II. (m-n) Representative immunofluorescence images of LC3B protein in lung tissues of PBS-mice, OVA-mice, 3-MA-mice and 3-MA-OVA-mice. Quantitation of fluorescence intensity of LC3B in 10 random fields. Data are representative of three independent experiments and are presented as means ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001, determined by one-way ANOVA with Tukey-Kramer post-test.
In addition, the goblet cell and mucus secretion were also reduced by 3-MA. The total area of periodic acid-Schiff (PAS) staining measured as a percent of total airway epithelium in the mice exposed to OVA and revealed greater goblet cell and mucus secretion than control mice exposed to PBS. However, the total area of PAS staining measured as a percent of total airway epithelium in the mice exposed to OVA in addition with 3-MA was smaller than the mice exposed to OVA (Fig. 1f–g). Consistent with the airway PAS staining results, the mice exposed to OVA in the presence of 3-MA reduced Muc5ac expression. In mice exposed to OVA, immunofluorescence staining of the airway Muc5ac showed significant increase than control mice exposed to PBS, suggesting that 3-MA reversed OVA-induced Muc5ac hyper-expression (Fig. 1h–i). Moreover, 3-MA could significantly reduce OVA-indcued Muc5ac gene expression (Fig. 1j).
As anticipated, western blotting results showed that the levels of microtubule-associated protein 1 light chain 3β II (LC3B-II) were markedly enhanced in the lung tissues of the mice exposed to OVA than those exposed to OVA in addition to 3-MA (Fig. 1k–l). Additionally, the levels of SQSTM1 protein were significantly reduced in the lung tissues of the mice exposed to OVA than those of control mice treatment with PBS. Moreover we observed that the reduction of LC3B-II protein expression treatment with 3-MA correlated with the increased protein levels of SQSTM1 (Fig. 1k–l). Simultaneously, immunofluorescence staining of LC3B was greater in the airway of the mice exposed to OVA than control mice exposed to PBS. But 3-MA treatment relieved LC3B expression of OVA-induced airway by immunofluorescence staining and quantified immunofluorescence intensity of LC3B showed consistent results (Fig. 1m–n). These results suggest that autophagic pathway plays a role in allergic airway inflammation mice. Autophagy activation may contribute to the hyper-responsiveness, inflammation and mucus secretion of the airway.
3.2. TGF-β3 Activated Autophagy of Airway Epithelial Cells
Elevated levels of TGF-β was correlated with epithelial layer damage, goblet cell hyperplasia and induced apoptosis in airway epithelial cells [37]. In the present study, an increased expression of TGF-β3 was observed in the lung tissues of the mice exposed to OVA than the mice exposed to PBS (Fig. 2a). Despite increased expression of TGF-β3 in the lung tissues of mice treated with OVA, BALF from these mice actually contained greater TGF-β3 production as compared to mice in the presence of PBS (Fig. 2b). We also have found a markedly increasing in the expression of TGF-β1 and TGF-β2 in the lung tissue of the mice exposed to OVA (Fig. S1a). Consistent with the results, the expression of TGF-β1 and TGF-β2 were up-regulated in the BALF of the mice exposed to OVA (Fig. S1b-S1c). But 3-MA have no effect on the expression of TGF-β isoforms respectively both in the lung tissues and in the BALF of the mice exposed to OVA (Fig. 2a–b, Fig. S1a-S1c). A previous study have demonstrated that TGF-β1 could induced epithelial-to-mesenchymal transition (EMT) via activating autophagy [27]. These findings suggested that OVA treatment induced autophagy and TGF-β3 hyper-expression. However, there is little study involvement in the effect of TGF-β3 on autophagy in asthma. So, we also determined whether TGF-β3 was dependent on autophagy pathway for pathological development of asthma. The effect of TGF-β3 on LC3B by immunoblotting was monitored firstly. Results revealed that TGF-β3 could increase the levels of LC3B II when treated at concentrations of 2.5 ng/ml, 5 ng/ml and 10 ng/ml but not at 0 ng/ml in 16HBE cells (Fig. 2c–d). Moreover, TGF-β3-mediated LC3B II demonstrated a concentration dependent increase at 10 ng/ml (Fig. 2d). In addition, Western blotting analysis demonstrated a time-dependent induction of LC3B II by TGF-β3 in 16HBE cells. We observed that prolonged treatment with TGF-β3 for 24 h significantly increased LC3B II to GAPDH ratio in 16HBE cells (Fig. 2e–f). Consistent with these consequences, we found that BECN1 and ATG5 are also increased in the presence of TGF-β3 at 10 ng/ml for 24 h. Also SQSTM1 have manifested a decreasing trend induced by TGF-β3 (Fig. 2c–f). We further confirmed these findings by transfecting 16HBE cells with mCherry-EGFP-LC3 lentivirus. Results showed an increased autophagy flux when treated with TGF-β3 (Fig. 2g–h), suggesting that TGF-β3 was capable of stimulating autophagy.
Fig. 2.

TGF-β3 activated autophagy of airway epithelial cells. (a) Representative immunohistochemistry of TGF-β3 proteins in lung tissues from PBS-mice, OVA-mice, 3-MA-mice and 3-MA-OVA-mice. (b) TGF-β3 in BALF was detected by ELISA. (c-d) 16HBE cells were treated with different concentrations of TGF-β3 for 24 h. Autophagy and target proteins were detected by western blotting in 16HBE cells. (e-f) 16HBE cells were treated with 10 ng/ml TGF-β3 at indicated time points. Autophagy and target proteins were detected by western blotting in 16HBE cells. (g) 16HBE cells that stably expressed mCherry-EGFP-LC3 fusion protein were treated with TGF-β3. In green and red-merged images, autophagosomes are shown as yellow puncta (i.e.,mCherry+EGFP+), while autolysosomes are shown as red puncta (i.e.,mCherry+EGFP−). Autophagic flux was increased when both yellow and red puncta are increased in the cells. Confocal microscopic analysis was shown (2000× magnification). Bar scale, 5 mm. (h) Quantification of the number of LC3 puncta (each group n = 10 images for quantification). Data are representative of three independent experiments and are presented as means ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001, determined by Student's t-test.
3.3. Autophagy is Required for TGF-β3-Induced Elevation of Mucin MUC5AC Levels in 16HBE Cells
As known to all, mucus hyper-secretion contributes to multiple clinical abnormalities in patients with asthma. MUC5AC was predominantly produced in the secretory cells present on the airway surface [38]. TGF-β1 was identified as an important factor in the regulation of MUC5AC [14, 15]. We have found that TGF-β1 could induce MUC5AC expression in 16HBE cells (Fig. S2a-S2b). There were little reports demonstrating that TGF-β3 could induce mucus secretion in 16HBE cells. In the present study, we found that TGF-β3 not only induced autophagy, but also up-regulated the expression of MUC5AC (Fig. S2c-S2d). These results implied that autophagy has a connection with elevated MUC5AC expressions in TGF-β3-mediated 16HBE cells. So, we studied the function of autophagy in the regulation of TGF-β3-induced mucus production by knock down of autophagy proteins ATG5 or BECN1, respectively (Fig. 3a–d). Results revealed that ATG5 and BECN1 siRNAs not only blocked the induction of LC3B II but also dramatically inhibited the upregulation of MUC5AC induced by TGF-β3 (Fig. 3a–f). Simultaneously, blockade of ATG5 or BECN1 significantly reduced the mRNA levels of MUC5AC gene (Fig. 3g). Then, 16HBE cells treated with TGF-β3 combined with 3-MA, an autophagy inhibitor (Fig. 3h). Addition of 3-MA blocked TGF-β3-induced upregulation of LC3B II and simultaneously increased SQSTM1 protein expression in 16HBE cells (Fig. 3h–i). Consistent with these findings, the protein levels and the mRNA expression of MUC5AC were also reduced in 16HBE cells treated with TGF-β3 and 3-MA (Fig. 3l–n). Furthermore, we treated 16HBE cells with Baf A1, an autophagy lysosomal inhibitor (Fig. 3j). Baf A1 treatment with TGF-β3 led to a markedly elevated expression of LC3B II by western blot analysis compared with TGF-β3 treatment or Baf A1 treatment (Fig. 3j–k). In addition, the decrease in SQSTM1 levels caused by TGF-β3 was effectively reversed by BafA1 cotreatment (Fig. 3j). It was indicated that the TGF-β3-induced autophagosomes that fuse with lysosomes to form autolysosomes were inhibited by BafA1. Therefore, we revealed that TGF-β3 indeed stimulated autophagy because of an impaired autophagosome-lysosome fusion. To determine the effects of autophagy on TGF-β3 induced MUC5AC expression, we detected MUC5AC expression via immunofluorescence staining and tested the mRNA levels of MUC5AC in 16HBE cells stimulated with Baf A1 and TGF-β3. Surprisingly, 16HBE cells treated with Baf A1 significantly inhibited TGF-β3 induced MUC5AC expressions (Fig. 3l–n), confirming that autophagy flux was required for TGF-β3 induced mucus secretion.
Fig. 3.

Autophagy is required for TGF-β3-induced elevated mucin MUC5AC levels in 16HBE cells. (a-b) 16HBE cells were transfected with ATG5-siRNA lentivirus. After treating the cells with TGF-β3 (10 ng/ml) for 24 h, ATG5, LC3B and SQSTM1 were detected by western blot. Relative changes in the density of LC3B II were detected. (c-d) 16HBE cells were transfected with BECN1-siRNA lentivirus. After treating the cells with TGF-β3 (10 ng/ml) for 24 h, BECN1, LC3B and SQSTM1 were detected by western blot. Relative changes in the density of LC3B II were detected. (e) Representative immunofluorescence images of TGF-β3-induced MUC5AC in 16HBE cells transduced with siRNA lentivirus were observed. (f) Quantitation of the fluorescence intensity of MUC5AC (each group n = 10 images for quantification). (g) 16HBE cells were transfected with ATG5-siRNA lentivirus or BECN1-siRNA, respectively. Real-time PCR was performed to detect the expression of MUC5AC gene after treated with TGF-β3 (10 ng/ml). (h-k) 16HBE cells were treated with TGF-β3 in the presence of 3-MA (h) or Baf A1 (j). Then, the expression of LC3B and SQSTM1 was detected using western blot assays. And relative changes in the density of LC3B II were detected. (l) Representative immunofluorescence images of TGF-β3-induced MUC5AC in 16HBE cells treated with 3-MA or Baf A1. (m) Quantitation of the fluorescence intensity of MUC5AC (each group n = 10 images for quantification). (n) Real-time PCR was performed in 16HBE cells to detect the expression of MUC5AC gene after treated with TGF-β3 (10 ng/ml) in the presence of 3-MA or Baf A1. Data are representative of three independent experiments and are presented as means ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001, determined by one-way ANOVA with Tukey-Kramer post-test.
3.4. Smad2/3 Pathway is Involved in TGF-β3 Induced Autophagy and MUC5AC Expression
Our current study reported that TGF-β3 could activate autophagy in the airway epithelial cells accompanied by mucus hypersecretion. Next, we studied the exact mechanism of the process involved. Usually, TGF-β signals go downstream of Smad pathways, but this pathway was involved in the autophagy induction of TGF-β2 influencing glioma invasion and metabolism [30]. In the present study, we found that the levels of Smad2 and Smad3 phosphorylation were enhanced in response to TGF-β3 treatment in 16HBE cells (Fig. 4a). Hence, we used western blotting to investigate whether Smad2/3 pathway was involved in TGF-β3 induced autophagy activity. We analyzed LC3B II levels in Smad2/3-siRNA-transfected 16HBE cells in the presence of TGF-β3. Results showed that TGF-β3 increased LC3B II expression in wild-type Smad2 and Smad3 expressing 16HBE cells (Fig. 4a), while knock down of Smad2 and Smad3 blocked TGF-β3-induced LC3B II expression (Fig. 4a). To examine whether the autophagy elements such as ATG5 and BECN1 were affected by Smad2/3 pathway in 16HBE cells induced by TGF-β3, we analyzed ATG5 and BECN1 expression in Smad2/3-siRNA-transfected 16HBE cells. Consistent with the LC3B II levels, the levels of ATG5 and BECN1 also attenuated by blocking Smad2/3 siRNAs (Fig. 4a). To investigate whether Smad2 and Smad3 affect the autophagy, we used mCherry-EGFP-LC3 lentivirus to transfect 16HBE cells. Immunofluorescence staining of LC3 puncta showed that accumulation of autophagosomes was reduced in Smad2/3 siRNA transfected 16HBE cells induced by TGF-β3 compared to 16HBE cells induced by TGF-β3 only (Fig. 4c–d). These data strongly suggest that Smad2 and Smad3 participated in TGF-β3 induced autophagy pathway and affected autophagic pathway associated proteins such as ATG5 and BECN1. So, further discussion of the actual significance of Smad2 and Smad3 with MUC5AC expressions in 16HBE cells is warranted. According to our results, the mRNA levels of MUC5AC were decreased in the Smad2/3 siRNA transfected 16HBE cells stimulated with TGF-β3 (Fig. 4e). Furthermore, immunofluorescence staining revealed that MUC5AC protein was also significantly reduced in Smad2/3 siRNA transfected 16HBE cells (Fig. 4f-g). Collectively, these studies indicated that Smad2/3 pathway was involved in TGF-β3 induced MUC5AC hyper-expressions by promoting autophagy.
Fig. 4.

Smad2/3 pathway is involved in TGF-β3 induced autophagy and MUC5AC. (a) 16HBE cells were transfected with Smad2/3-siRNA. After treating the cells with TGF-β3 (10 ng/ml) for 24 h, LC3B, BECN1, ATG5, phospho-Smad2, Smad2, phospho-Smad3 and Smad3 were detected by western blot. (b) Relative changes in the density of LC3B II were detected. (c) 16HBE cells that stably expressed mCherry-EGFP-LC3 fusion protein were transfected with Smad2/3-siRNA. After treating with TGF-β3 (10 ng/ml) for 24 h, autophagosomes were observed under confocal microscope (2000× magnification) in 16HBE cells. Bar scale, 5 mm. (d) Quantification of the number of LC3 puncta (each group n = 10 images for quantification). (e) 16HBE cells were transfected with Smad2/3-siRNA. Real-time PCR was performed to detect the expression of MUC5AC gene after treated with TGF-β3 (10 ng/ml). (f) Representative immunofluorescence images of TGF-β3-induced MUC5AC in 16HBE cells transfected with Smad2/3-siRNA. (g) Quantitation of the fluorescence intensity of MUC5AC (each group n = 10 images for quantification). Data are representative of three independent experiments and are presented as means ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001, determined by one-way ANOVA with Tukey-Kramer post-test.
3.5. TGF-β3-Induced Autophagy Contributed to Increased MUC5AC Production by Activating AP1
Activating protein-1 (AP-1) is a transcription factor complex that consists of heterodimers or homodimers of Fos, Jun, ATF, and MAF protein families [39]. AP-1 is also known to mediate mucus production in Cigarette Smoke-Induced airway epithelium [19]. In the present study, we proved that the induction of p-c-jun by TGF-β3 is autophagy dependent (Fig. 5a–f). Since blocking autophagy elements of ATG5 or BECN1 could reduce the levels of c-jun phosphorylation induced by TGF-β3, indicating an important role of autophagy regulation of AP-1 activity in 16HBE cells induced by TGF-β3 (Fig. 5a–b). Then, we used autophagy inhibitor or lysosome inhibitor to further prove the effect of autophagy flux on the modulation of c-jun activity by TGF-β3. The induction of c-jun phosphorylation was inhibited by in addition of Baf A1 or 3-MA (Fig. 5c–d), demonstrating the blockage of autophagy activity and inhibition of autophalysosome suppressed TGF-β3-induced c-jun phosphorylation. According to the requirement for Smad2/3 in autophagy activation, we further investigated the effect of Smad2/3 on the phosphorylation levels of c-jun induced by TGF-β3. The results showed that genetic inhibition of Smad2/3 could down-regulate the phosphorylation levels of c-jun induced by TGF-β3 (Fig. 5e). In line with this, we further discussed whether the transcription factor AP-1 participated in autophagy-mediated MUC5AC expression in 16HBE cells induced by TGF-β3. In the study, we found that both the increased protein levels and mRNA levels of MUC5AC by TGF-β3 was reduced in c-jun siRNA transfected 16HBE cells (Fig. 5f–i). These results suggested that the activity of AP-1 is as a downstream regulator of TGF-β3/autophagy signaling pathway in modulating MUC5AC hyperexpression in 16HBE cells.
Fig. 5.

TGF-β3-induced autophagy contributed to increased MUC5AC production by activating the AP1. (a-b) 16HBE cells were transduced with ATG5-siRNA lentivirus (a) and BECN1-siRNA lentivirus (b), respectively. After treating the cells with TGF-β3 (10 ng/ml) for 24 h, phospho-c-Jun and c-Jun were detected by western blot. (c-d) 16HBE cells were treated with TGF-β3 in the presence of 3-MA (c) or Baf A1 (d). Then, the expression of phospho-c-Jun and c-Jun were detected using western blot assay. (e) 16HBE cells were transfected with Smad2/3-siRNA. After treating the cells with TGF-β3 (10 ng/ml) for 24 h, phospho-c-Jun and c-Jun were detected by western blot. (f) 16HBE cells were transfected with c-Jun-siRNA, after treating with TGF-β3 (10 ng/ml) for 24 h, and then phospho-c-Jun and c-Jun were detected by western blot. (g) Representative immunofluorescence images of TGF-β3-induced MUC5AC in 16HBE cells were transfected with c-Jun-siRNA. (h) Quantitation of fluorescence intensity of MUC5AC (each group n = 10 images for quantification). (i) 16HBE cells were transfected with c-Jun-siRNA. Real-time PCR was performed to detect the expression of MUC5AC gene after treated with TGF-β3 (10 ng/ml). Data are representative of the three independent experiments and are presented as means ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001, determined by one-way ANOVA with Tukey-Kramer post-test.
4. Discussion
Autophagy plays an essential role in numerous lung diseases including chronic obstructive pulmonary disease (COPD), acute lung injury (ALI) and pulmonary infectious diseases [40]. At present, more attention should be paid on the pathological process of asthma. In our present study, we demonstrated that autophagy was important for bronchial epithelial mucus production in OVA-induced allergic mice and TGF-β3-induced mucus hyper-production in 16HBE cells. TGF-β3 exposure initially triggered Smad2/3 phosphorylation and subsequently elicited autophagy, regulating MUC5AC expression through the activation of transcription factor AP-1. Our results revealed that autophagy plays a vital role in TGF-β3-induced MUC5AC expression in the airway epithelium (Fig. 6).
Fig. 6.
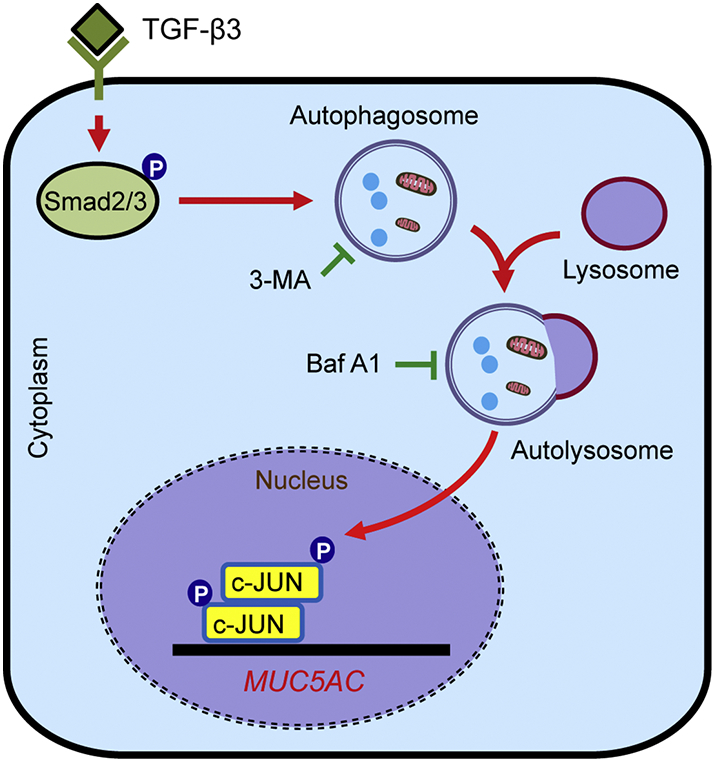
Schematic diagram of the mechanisms of Smad2/3-dependent autophagy in the regulation of TGF-β3-induced MUC5AC in airway epithelial cells. TGF-β3 exposure initially triggered Smad2/3 phosphorylation and subsequently elicited autophagy, promoting MUC5AC expression through activation of AP-1 transcription factor.
Autophagy of airway cells plays an important role in the pathogenesis of COPD including exuberant mucus production, inflammatory responses, oxidative injury and endoplasmic reticulum (ER) stress responses [41]. A couple of studies have demonstrated autophagy mediated regulation of mucus secretion [20, 22]. Autophagy-related genes deficiency displayed a markedly reduced number of goblet cells and attenuated the levels of MUC5AC in response to cigarette smoke exposure [19]. In this study, we inactivated autophagy using 3-MA to examine the role of autophagy in airway hyper-response, airway inflammation and airway mucus secretion in allergen-induced mice. Compared with OVA treated mice, the presence of OVA and 3-MA treated mice exhibited decreased airway hyper-response and airway inflammation, diminishing airway epithelial expression of mucus and Muc5ac. According to the latest research, Follistatin-like 1 (FSTL1) may induce EMT and airway remodeling by activating autophagy [42]. And IL-13 activated autophagy in airway epithelial cells to regulate MUC5AC secretion [20]. However, the mechanism affecting the autophagy of epithelial cells has not been elucidated in asthma.
Cytokine regulation of autophagy occurs widely. IL-13, a Th2-type cytokines, was elevated in asthma, inducing autophagy regulation of airway epithelial mucus production [20]. TGF-β1 is one of the known isoforms that promote EMT resulting in airway remodeling [43]. Epithelial TGF-β2 expression levels were increased and were significantly correlated with mucin expression [16], and promoted glioma cell invasion though regulation of autophagy [30]. Whereas, TGF-β3 expression levels were increased in the epithelial biopsy tissues of repeated OVA-induced mice in the present study, which was consistent with increased autophagy-related genes and mucus secretion. Thus, we used 16HBE cells exposed to TGF-β3 to uncover the phenomenon whether TGF-β3 induced autophagy activity and regulated MUC5AC expression in epithelial cells. Chemical inhibitor of autophagy significantly reduced TGF-β3-induced expression of MUC5AC in 16HBE cells. Whereas interference of the autophagic flux with Baf A1 accumulated autophagosomes and intriguingly attenuated MUC5AC hyper-expression. Autophagy-related (Atg) proteins are involved in autophagosome formation and these are dependent on each other for the recruitment of autophagosomes [44, 45]. The most studied Atg proteins were BECN1, ATG5 and ATG7 and are involved in the activation of autophagosome formation [46]. Beclin1 and ATG5 affected airway inflammation in asthma [35]. In the study, TGF-β3 exposure induced a significant elevation of Atg protein expression including BECN1 and ATG5, except for SQSTM1 which was reported to bind to LC3 and preferentially degraded by autophagy [47, 48]. Genetic blockage of Atg proteins exhibited a decrease of MUC5AC expression and diminished LC3B II puncta. Thus, these results support that TGF-β3 could induce autophagy and autophagic flux, which are required for TGF-β3-induced MUC5AC hyper-expression.
Previous study indicated that TGF-β-induced autophagy was regulated by Disabled-2 (Dab2) [49]. Mir-34a inhibited macroautophagy activation by directly targeting Smad4 through TGF-β/Smad4 pathway [50]. TGF-β could activate p38 MAP kinase to mediate Smad-independent TGF-β responses [51]. TGF-β1 induces autophagy in mouse mesangial cells via TAK1-MKK3-p38 signaling pathway [52]. The expression levels of phosphorylated Smad2 in bronchial biopsy were elevated and were clearly correlated with airway remodeling in asthmatic individuals [53]. Collectively, previous studies urged us to investigate the mechanisms of TGF-β3 mediating autophagy activity and mucus hyper-secretion. In our study, knock down of Smad2/3 blocked TGF-β3 induced autophagy activity and MUC5AC expression in 16HBE cells. TGF-β3 exposure to 16HBE cells significantly aggregated Smad2/3 phosphorylation and Atg proteins expression, which simultaneously exhibited elevated LC3 puncta and MUC5AC expression. Therefore, these results indicated that TGF-β3-induced autophagy activity was Smad2/3 signaling dependent.
Accumulated studies showed that autophagy could induce mucus production through activation of AP-1 [19, 22]. AP-1 activation by oxidative stress in alveolar epithelial cells may be considered as a potential mechanism in the gene transcription of lung inflammation [54]. Airway mucus hypersecretion of severe pneumonia induced by respiratory virus (RSV) might be corrected by the activation of JNK/AP-1 signaling pathway [55]. We further showed that autophagy activity and autolysosomes regulated MUC5AC expression via triggering AP-1 in human bronchial epithelial cells. Down-regulation of ATG protein and inhibition of autophagy decreased c-jun phosphorylation, whereas knock down of c-jun ameliorated TGF-β3 induced MUC5AC expression in 16HBE cells. Collectively, all the above results suggest that autophagy activity and autophagy flux plays a regulatory role of TGF-β3-induced MUC5AC hyper-expression in 16HBE cells.
Our observations have suggested that autophagy is considered as an important aspect of biological effects of TGF-β3 in modulating airway mucus hyper-secretion. Moreover, in this study we find evidence supporting in the contribution of ameliorating airway mucous accumulation by targeting inhibition of autophagy.
Disclosure of Potential Conflicts of Interest
There were no potential conflicts of interest to be disclosed.
Author Contributions
Conceived and designed the study: Xing Wang. Performed the experiments: Yun Zhang, Hongmei Tang, Xiefang Yuan, Qin Ran, Xiaoyun Wang, Qi Song, Lei Zhang and Yuhuan Qiu. Wrote the paper: Xing Wang and Yun Zhang. Analyzed the data: Yun Zhang, Hongmei Tang and Xing Wang. All authors read and approved the final manuscript.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NO. 81600024) and the Health and Family Planning Commission of Sichuan Province (NO. 16PJ543). Funders had no role in study design, data collection, data analysis, interpretation, writing of the report.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2018.06.032.
Appendix A. Supplementary data
Supplementary material
References
- 1.Bruton A., Lee A., Yardley L., Raftery J., Arden-Close E., Kirby S. Physiotherapy breathing retraining for asthma: a randomised controlled trial. Lancet Respir Med. 2018;6(1):19–28. doi: 10.1016/S2213-2600(17)30474-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nunes C., Pereira A.M., Morais-Almeida M. Asthma costs and social impact. Asthma Res Pract. 2017;3(1) doi: 10.1186/s40733-016-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holgate S.T. The airway epithelium is central to the pathogenesis of asthma. Allergol Int: Off J Jpn Soc Allergol. 2008;57(1):1–10. doi: 10.2332/allergolint.R-07-154. [DOI] [PubMed] [Google Scholar]
- 4.Gon Y., Hashimoto S. Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol Int: Off J Jpn Soc Allergol. 2018;67(1):12–17. doi: 10.1016/j.alit.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Ferreira D.S., Carvalho-Pinto R.M., Gregorio M.G., Annoni R., Teles A.M., Buttignol M. Airway pathology in severe asthma is related to airflow obstruction but not symptom control. Allergy. 2018 Mar;73(3):635–643. doi: 10.1111/all.13323. [DOI] [PubMed] [Google Scholar]
- 6.Bonser L.R., Erle D.J. Airway mucus and asthma: the role of MUC5AC and MUC5B. J Clin Forensic Med. 2017;6(12) doi: 10.3390/jcm6120112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grainge C.L., Lau L.C., Ward J.A., Dulay V., Lahiff G., Wilson S. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. 2011;364(21):2006–2015. doi: 10.1056/NEJMoa1014350. [DOI] [PubMed] [Google Scholar]
- 8.Ma J., Rubin B.K., Voynow J.A. Mucins, mucus, and goblet cells. Chest. 2017 Nov 21 doi: 10.1016/j.chest.2017.11.008. (pii: S0012-3692(17)33080-5) [DOI] [PubMed] [Google Scholar]
- 9.Alevy Y.G., Patel A.C., Romero A.G., Patel D.A., Tucker J., Roswit W.T. IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J Clin Invest. 2012;122(12):4555–4568. doi: 10.1172/JCI64896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu H., Li Q., Kolosov V.P., Perelman J.M., Zhou X. Interleukin-13 induces mucin 5AC production involving STAT6/SPDEF in human airway epithelial cells. Cell Commun Adhes. 2010;17(4–6):83–92. doi: 10.3109/15419061.2010.551682. [DOI] [PubMed] [Google Scholar]
- 11.Duvernelle C., Freund V., Frossard N. Transforming growth factor-beta and its role in asthma. Pulm Pharmacol Ther. 2003;16(4):181–196. doi: 10.1016/S1094-5539(03)00051-8. [DOI] [PubMed] [Google Scholar]
- 12.Bottoms S.E., Howell J.E., Reinhardt A.K., Evans I.C., McAnulty R.J. Tgf-Beta isoform specific regulation of airway inflammation and remodelling in a murine model of asthma. PLoS One. 2010;5(3) doi: 10.1371/journal.pone.0009674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li G., Fox J., 3rd, Liu Z., Liu J., Gao G.F., Jin Y. Lyn mitigates mouse airway remodeling by downregulating the TGF-beta3 isoform in house dust mite models. J Immunol. 2013;191(11):5359–5370. doi: 10.4049/jimmunol.1301596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jonckheere N., Van Der Sluis M., Velghe A., Buisine M.P., Sutmuller M., Ducourouble M.P. Transcriptional activation of the murine Muc5ac mucin gene in epithelial cancer cells by TGF-beta/Smad4 signalling pathway is potentiated by Sp1. Biochem J. 2004;377(Pt 3):797–808. doi: 10.1042/BJ20030948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jono H., Xu H., Kai H., Lim D.J., Kim Y.S., Feng X.H. Transforming growth factor-beta-Smad signaling pathway negatively regulates nontypeable Haemophilus influenzae-induced MUC5AC mucin transcription via mitogen-activated protein kinase (MAPK) phosphatase-1-dependent inhibition of p38 MAPK. J Biol Chem. 2003;278(30):27811–27819. doi: 10.1074/jbc.M301773200. [DOI] [PubMed] [Google Scholar]
- 16.Chu H.W., Balzar S., Seedorf G.J., Westcott J.Y., Trudeau J.B., Silkoff P. Transforming growth factor-beta2 induces bronchial epithelial mucin expression in asthma. Am J Pathol. 2004;165(4):1097–1106. doi: 10.1016/s0002-9440(10)63371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrop C.A., Gore R.B., Evans C.M., Thornton D.J., Herrick S.E. TGF-beta(2) decreases baseline and IL-13-stimulated mucin production by primary human bronchial epithelial cells. Exp Lung Res. 2013;39(1):39–47. doi: 10.3109/01902148.2012.748854. [DOI] [PubMed] [Google Scholar]
- 18.Tham R., Vicendese D., Dharmage S.C., Hyndman R.J., Newbigin E., Lewis E. Associations between outdoor fungal spores and childhood and adolescent asthma hospitalizations. J Allergy Clin Immunol. 2017;139(4):1140–1147. doi: 10.1016/j.jaci.2016.06.046. [e4] [DOI] [PubMed] [Google Scholar]
- 19.Zhou J.S., Zhao Y., Zhou H.B., Wang Y., Wu Y.F., Li Z.Y. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):L1042–L1052. doi: 10.1152/ajplung.00418.2015. [DOI] [PubMed] [Google Scholar]
- 20.Dickinson J.D., Alevy Y., Malvin N.P., Patel K.K., Gunsten S.P., Holtzman M.J. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. 2016;12(2):397–409. doi: 10.1080/15548627.2015.1056967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poon A., Eidelman D., Laprise C., Hamid Q. ATG5, autophagy and lung function in asthma. Autophagy. 2012;8(4):694–695. doi: 10.4161/auto.19315. [DOI] [PubMed] [Google Scholar]
- 22.Chen Z.H., Wu Y.F., Wang P.L., Wu Y.P., Li Z.Y., Zhao Y. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. 2016;12(2):297–311. doi: 10.1080/15548627.2015.1124224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dickinson J.D., Sweeter J.M., Warren K.J., Ahmad I.M., De Deken X., Zimmerman M.C. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol. 2018;14:272–284. doi: 10.1016/j.redox.2017.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia F., Deng C., Jiang Y., Qu Y., Deng J., Cai Z. IL4 (interleukin 4) induces autophagy in B cells leading to exacerbated asthma. Autophagy. 2018;14(3):450–464. doi: 10.1080/15548627.2017.1421884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghavami S., Yeganeh B., Zeki A.A., Shojaei S., Kenyon N.J., Ott S. Autophagy and the unfolded protein response promote profibrotic effects of TGF-beta1 in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2018;314(3):L493–L504. doi: 10.1152/ajplung.00372.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen J., Zhao D.S., Li M.Z. TGF-beta1 promotes human gastric carcinoma SGC7901 cells invasion by inducing autophagy. Eur Rev Med Pharmacol Sci. 2017;21(5):1013–1019. [PubMed] [Google Scholar]
- 27.Zou M., Zhu W., Wang L., Shi L., Gao R., Ou Y. AEG-1/MTDH-activated autophagy enhances human malignant glioma susceptibility to TGF-beta1-triggered epithelial-mesenchymal transition. Oncotarget. 2016;7(11):13122–13138. doi: 10.18632/oncotarget.7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma C.L., Qiao S., Li Y.C., Wang X.F., Sun R.J., Zhang X. TGF-beta1 promotes human hepatic carcinoma HepG2 cells invasion by upregulating autophagy. Eur Rev Med Pharmacol Sci. 2017;21(11):2604–2610. [PubMed] [Google Scholar]
- 29.Korah J., Canaff L., Lebrun J.J. The retinoblastoma tumor suppressor protein (pRb)/E2 promoter binding factor 1 (E2F1) pathway as a novel mediator of TGFbeta-induced autophagy. J Biol Chem. 2016;291(5):2043–2054. doi: 10.1074/jbc.M115.678557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C., Zhang X., Xu R., Huang B., Chen A.J., Li C. TGF-beta2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells' invasion. J Exp Clin Cancer Res: CR. 2017;36(1):162. doi: 10.1186/s13046-017-0628-8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Wang X., Li Y., Luo D., Wang X., Zhang Y., Liu Z. Lyn regulates mucus secretion and MUC5AC via the STAT6 signaling pathway during allergic airway inflammation. Sci Rep. 2017;7 doi: 10.1038/srep42675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie T., Luo G., Zhang Y., Wang X., Wang X., Wu M. Rho-kinase inhibitor fasudil reduces allergic airway inflammation and mucus hypersecretion by regulating STAT6 and NFkappaB. Clin Exp Allergy: J British Soc Allergy Clin Immunol. 2015;45(12):1812–1822. doi: 10.1111/cea.12606. [DOI] [PubMed] [Google Scholar]
- 33.Wang X., Yang X., Li Y., Wang X., Zhang Y., Dai X. Lyn kinase represses mucus hypersecretion by regulating IL-13-induced endoplasmic reticulum stress in asthma. EBioMedicine. 2017;15:137–149. doi: 10.1016/j.ebiom.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeki A.A., Yeganeh B., Kenyon N.J., Post M., Ghavami S. Autophagy in airway diseases: a new frontier in human asthma? Allergy. 2016;71(1):5–14. doi: 10.1111/all.12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ban G.Y., Pham D.L., Trinh T.H., Lee S.I., Suh D.H., Yang E.M. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: a new therapeutic target. Clin Exp Allergy: J British Soc Allergy Clin Immunol. 2016;46(1):48–59. doi: 10.1111/cea.12585. [DOI] [PubMed] [Google Scholar]
- 36.Wu Y.T., Tan H.L., Shui G., Bauvy C., Huang Q., Wenk M.R. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makinde T., Murphy R.F., Agrawal D.K. The regulatory role of TGF-beta in airway remodeling in asthma. Immunol Cell Biol. 2007;85(5):348–356. doi: 10.1038/sj.icb.7100044. [DOI] [PubMed] [Google Scholar]
- 38.Koeppen M., McNamee E.N., Brodsky K.S., Aherne C.M., Faigle M., Downey G.P. Detrimental role of the airway mucin Muc5ac during ventilator-induced lung injury. Mucosal Immunol. 2013;6(4):762–775. doi: 10.1038/mi.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eferl R., Wagner E.F. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3(11):859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 40.Xu X., Wang H., Liu S., Xing C., Liu Y. Aodengqimuge, et al. TP53-dependent autophagy links the ATR-CHEK1 axis activation to proinflammatory VEGFA production in human bronchial epithelial cells exposed to fine particulate matter (PM2.5) Autophagy. 2016;12(10):1832–1848. doi: 10.1080/15548627.2016.1204496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J., Yeganeh B., Ermini L., Post M. Sphingolipids as cell fate regulators in lung development and disease. Apoptosis: Int J Program Cell Death. 2015;20(5):740–757. doi: 10.1007/s10495-015-1112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu T., Liu Y., Miller M., Cao L., Zhao J., Wu J. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am J Physiol Lung Cell Mol Physiol. 2017;313(1):L27–L40. doi: 10.1152/ajplung.00510.2016. [DOI] [PubMed] [Google Scholar]
- 43.Johnson J.R., Nishioka M., Chakir J., Risse P.A., Almaghlouth I., Bazarbashi A.N. IL-22 contributes to TGF-beta1-mediated epithelial-mesenchymal transition in asthmatic bronchial epithelial cells. Respir Res. 2013;14:118. doi: 10.1186/1465-9921-14-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabeya Y., Kawamata T., Suzuki K., Ohsumi Y. Cis1/Atg31 is required for autophagosome formation in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2007;356(2):405–410. doi: 10.1016/j.bbrc.2007.02.150. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki K., Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007;581(11):2156–2161. doi: 10.1016/j.febslet.2007.01.096. [DOI] [PubMed] [Google Scholar]
- 46.Yue Z., Jin S., Yang C., Levine A.J., Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 48.Bjorkoy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang Y., Woosley A.N., Sivalingam N., Natarajan S., Howe P.H. Cathepsin-B-mediated cleavage of Disabled-2 regulates TGF-beta-induced autophagy. Nat Cell Biol. 2016;18(8):851–863. doi: 10.1038/ncb3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun C., Wang F.J., Zhang H.G., Xu X.Z., Jia R.C., Yao L. miR-34a mediates oxaliplatin resistance of colorectal cancer cells by inhibiting macroautophagy via transforming growth factor-beta/Smad4 pathway. World J Gastroenterol. 2017;23(10):1816–1827. doi: 10.3748/wjg.v23.i10.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu L., Hebert M.C., Zhang Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21(14):3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim S.I., Na H.J., Ding Y., Wang Z., Lee S.J., Choi M.E. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J Biol Chem. 2012;287(15):11677–11688. doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sagara H., Okada T., Okumura K., Ogawa H., Ra C., Fukuda T. Activation of TGF-beta/Smad2 signaling is associated with airway remodeling in asthma. J Allergy Clin Immunol. 2002;110(2):249–254. doi: 10.1067/mai.2002.126078. [DOI] [PubMed] [Google Scholar]
- 54.Rahman I., Gilmour P.S., Jimenez L.A., MacNee W. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002;234–235(1–2):239–248. [PubMed] [Google Scholar]
- 55.Li X.M., Sun S.Z., Wu F.L., Shi T., Fan H.J., Li D.Z. Study on JNK/AP-1 signaling pathway of airway mucus hypersecretion of severe pneumonia under RSV infection. Eur Rev Med Pharmacol Sci. 2016;20(5):853–857. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material