

Abstract
A unified synthetic strategy leading to the total synthesis of (−)-nodulisporic acids D, C, and B is described. Key synthetic transformations include a nickel-chromium mediated cyclization, an aromatic ring functionalization employing a novel copper-promoted alkylation, a palladium-catalyzed cross coupling cascade/indole ring construction, and a palladium-mediated regio- and diastereoselective allylic substitution/cyclization reaction, the latter to construct ring-D.
Graphical Abstract
The nodulisporic acids A-F (1-6)1 (Scheme 1), reported by the Merck Research Laboratories, comprise an architecturally intriguing family of indole terpenes,2 found to possess potent insecticidal activity.3 Subsequent SAR studies at Merck revealed that the highly substituted indole core and secondary C(24) hydroxyl group are the key structural elements required for the insecticidal activity.4 These two functionalities, in conjugation with the dienoate side chain, also lead to significant instability both in vitro and in vivo;1,4 for example the C(24) hydroxyl group undergoes facile dehydration mediated by the carboxylic acid,1 while exposure to air leads to oxidative ring-opening of the indole core.1a,c This sensitivity pattern clearly conspires to add significant chemical challenge vis-a-vis structural modifications and/or synthetic strategies towards the nodulisporic acids.5,6
Scheme 1.
The Nodulisporic Acids Indole Terpenes (A-F)
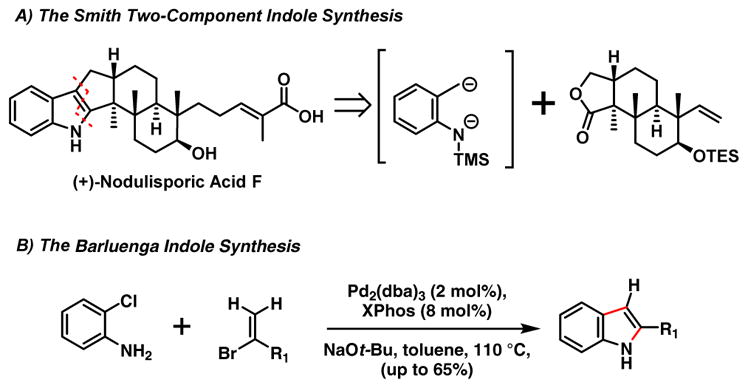
Shortly after the Merck structural/medicinal chemistry program,4,5 we launched synthetic studies towards the total synthesis of members of the nodulisporic acid family. From the outset, this program had two major goals: 1) construct the highly strained CDE tricyclic indole-indoline scaffold found only in these natural products; and 2) develop a unified synthetic strategy not only to the naturally occurring nodulisporic acids, but also to unnatural analogues. This effort initially cumulated in the first synthesis of the simpler, more stable nodulisporic acid (+)-F exploiting at the time a new two-component indole ring construction (Scheme 2A) developed in our laboratory.7 To construct the more advanced nodulisporic acids (1-4), we developed a new union strategy exploiting a palladium-mediated cross coupling/indole construction tactic base on the chemistry of Barluenga and coworkers8 (Scheme 2B).
Scheme 2.

Synthetic Strategy for the Nodulisporic Acids
Initial success of the new strategy was first demonstrated by the total synthesis of (−)-nodulisporic acid D (4)9 again via union of two advanced intermediates. Evolution of that synthetic venture has now led to the first total synthesis of (−)-nodulisporic acid C (3) and (−)-nodulisporic acid B (2), along with generation of an unnatural analogue, 2′-epi-nodulisporic acid B, all as their sodium salts. Herein we report a full account of the development and evolution of this unified strategy for the synthesis of acids D (4), C (3), and B (2).
For construction of (−)-nodulisporic acid D (4), the multisubstituted indole was dissected retrosynthetically into the western and eastern hemispheres 7 and 8 (Scheme 3). While the former was envisioned to be constructed via an Enders alkylation10/Stille-Kelly cyclization11 sequence employing hydrazone (+)-9 and an iodide derived from commercially available benzoic acid 10,12 the latter (8) was to arise via a difunctionalization/cyclization sequence utilizing advanced intermediate (+)-11. We note that we had developed a process-scale synthesis of (+)-11 during our earlier total synthesis of (+)-nodulisporic acid F (6).13
Scheme 3.
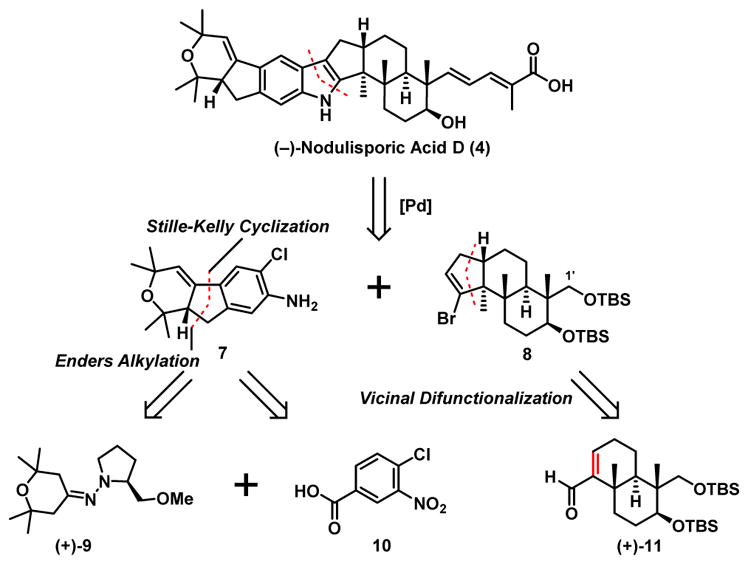
Synthetic Analysis of (−)-Nodulisporic Acid D (4)
Construction of the western hemisphere 7 (Scheme 4) thus began with benzoic acid 10. Borane reduction followed by iodination14 led to benzyl iodide 12. Hydrazone (+)-9 was then employed for the Enders asymmetric alkylation10 with 12 to furnish (+)-13, a single enantiomer with the requisite S stereogenicity (vide infra) at C(23). The chiral auxiliary was next removed via ozonolysis to produce ketone (+)-14 with a Comins triflation protocol15 employed to produce the corresponding vinyl triflate. In turn, reduction of the nitro group using iron/NH4Cl16 delivered aniline (+)-15 in near quantitative yield. A mild electrophilic iodination reagent (BTMA-ICl2)17 was then applied to aniline (+)-15 to furnish iodide (+)-16 in 80% yield, which upon a Stille-Kelly reaction,11 completed construction of the western hemisphere aniline (−)-7, the absolute configuration of which was confirmed by X-ray analysis. The overall yield for the 8-step sequence to (−)-7 was 25%.
Scheme 4.
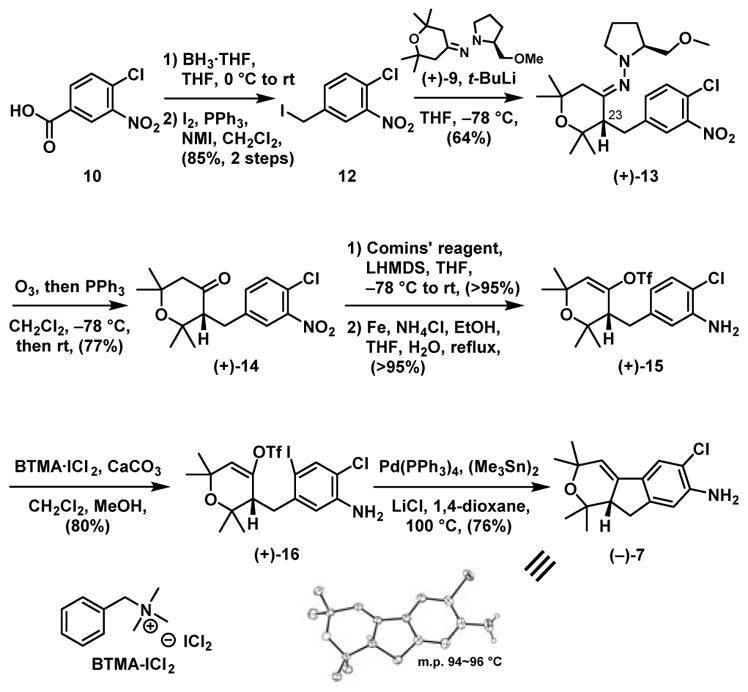
Synthesis of Western Hemisphere (−)-7
Construction of the eastern hemisphere 8 began with aldehyde (+)-11 (Scheme 5). Stereoselective conjugate addition with in situ generated vinyl cuprate18 led to silyl enol ether (−)-17 as a single diastereomer. Surprisingly, (−)-17 could be purified via silica column chromatography, likely due to the steric hindrance which protects the silyl enol ether from hydrolysis. The stereoselectivity of this reaction presumably results from limited accessibility of the top face that is blocked by the C(29) quaternary methyl group. Given the same reasoning, alkylation of the enolate derived via treatment with MeLi was expected to produce the same facial selectivity. To this end, treatment with MeLi in THF followed by alkylation generated the quaternary stereocenter at C(3), albeit with unsatisfactory selectivity (Condition 1; dr=1.4:1). Reasoning that use of a crown ether could chelate the lithium cation thereby generating a more reactive anion species, the alkylation might take place at a lower temperature with improved diastereoselectivity.19 As such, in the presence of 12-crown-4 and a finely-tuned solvent system (THF: Et2O=1:1), alkylation at −40 °C provided a 6:1 diastereomer selectivity (Condition 2). With aldehyde (−)-20 in hand, we moved to construct ring F. Our initially established metathesis cyclization sequence (Scheme 5, Protocol 1)9 proved effective; albeit, 10 mol% of the 2nd generation Grubbs catalyst20 was required, with a large amount of toxic PCC for the oxidation. Upon scale- up these issues could fortunately be addressed by utilizing the 2nd generation Hoveyda-Grubbs catalyst21 (1 mol%) to effect the ring-closing metathesis and a Ley22 oxidation (Protocol 2). The solvent cyclopentyl methyl ether (CPME)23 proved to be optimal for the metathesis, which permitted a higher reaction temperature. Enone (+)-21 was thus produced with a comparable yield (71%) over the three steps. Hydrogenolysis of (+)-21 then led to (−)-22 in 90% yield. The structure of (−)-22 was secured by X-ray analysis.
Scheme 5.

Synthesis of the Eastern Hemisphere (−)-8
Upon further consideration, a possible one-step cyclization of aldehyde (−)-20 to ketone (−)-22 (Protocol 3), which comprise constitutional isomers, was explored employing an intramolecular hydroacylation reaction24 utilizing Rh[NBD]2BF4/(±)-BINAP.25 Pleasingly, (−)-22 was generated in 77% yield. Unfortunately, use of less than a stoichiometric amount of the [Rh] reagent and/or a lower reaction temperature led to a significant decrease in reaction rate, likely due to the steric hindrance of the neopentyl aldehyde (−)-20. Nevertheless, this cyclization comprises a rare example of a rhodium-mediated hydroacylation in a complex structural setting.
Continuing toward vinyl bromide (−)-8, triflation of (−)-22 led to (−)-23, which upon bromination via a Buchwald protocol,26 modified slightly by employing the Pd-GIV dimer precatalyst,27 in combination with CPME as solvent, yielded (−)-8 in excellent yield (84%).
With both western hemisphere aniline (−)-7 and eastern hemisphere bromide (−)-8 in hand, we explored the proposed cascade cross coupling union/indole construction tactic (Scheme 6). Initial attempts applying the Barluenga conditions8 led to a complex mixture with no desired product. Reasoning that the strong basicity of NaOt-Bu may be detrimental to the reaction, a weaker inorganic base (Cs2CO3) was applied in combination with the Buchwald 3rd generation palladacycle RuPhos precatalyst.28 Pleasingly, the desired indole product [(−)-26] was obtained not only using vinyl bromide (−)-8 but also with the more readily available vinyl triflate (−)-23 [Scheme 5; one step from (−)-22] both in similar yields. Mechanistically, this union/cyclization cascade involves a Buchwald-Hartwig reaction via aniline (−)-7 with (−)-8 or (−)-23 to enamine 24, which then undergoes a palladium-mediated enamine cyclization29 via 25 with tautomerization to generate the desired indole core (−)-26. Isolation of indole (−)-26 however proved challenging! Significant decomposition (>50%) was observed after normal silica gel column purification. Analysis of the decomposition mixture led to amide (−)-27 (Scheme 7), as the major component. Further experiments suggested that indole (−)-26 undergoes slow oxidation to (−)-27 in air (days) even in the absence of silica gel. Similar stability issues were observed both during our previous synthetic venture leading to (−)-21-isopentenylpaxilline30 and in Merck reports.1a,c To resolve this purification issue, we developed a nitrogen-purged vacuum silica gel column chromatography purification method (see Supporting Information) to afforded (−)-26 in 60% and 56% yield respectively from bromide (−)-8 and triflate (−)-23.
Scheme 6.
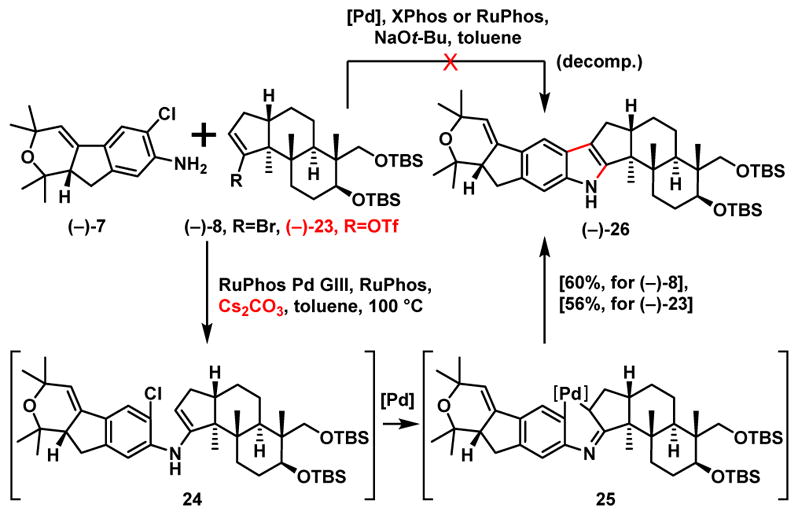
Development of a Barluenga Cross Coupling Union
Scheme 7.
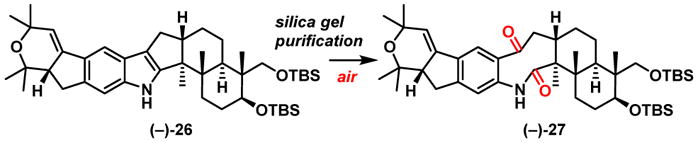
Air Oxidation of the Nodulisporic Indole Core
At this stage of the synthesis, considering the significant oxygen sensitivity of the indole core, we revised our synthetic plan to avoid any late-stage oxidations. Specifically, the C(1′) position of the new eastern hemisphere (Scheme 8) was now planned to be oxidized prior to construction of the indole core. Towards this end, removal of both TBS groups in ketone (−)-22 led to diol (−)-28, which was oxidized chemoselectively to aldehyde (−)-29 employing a TEMPO mediated protocol.31 The reported oxidation conditions led to an epimeric mixture at C(7) likely via an intramolecular aldol/retro-aldol process. This issue was resolved by employing a biphasic solvent system (CH2Cl2/H2O, 1:1). Protection of the secondary alcohol followed by triflation then furnished the new eastern hemisphere aldehyde (−)-33. The refined 4-step sequence to (−)-33 proceeded in an overall yield of 65%.
Scheme 8.
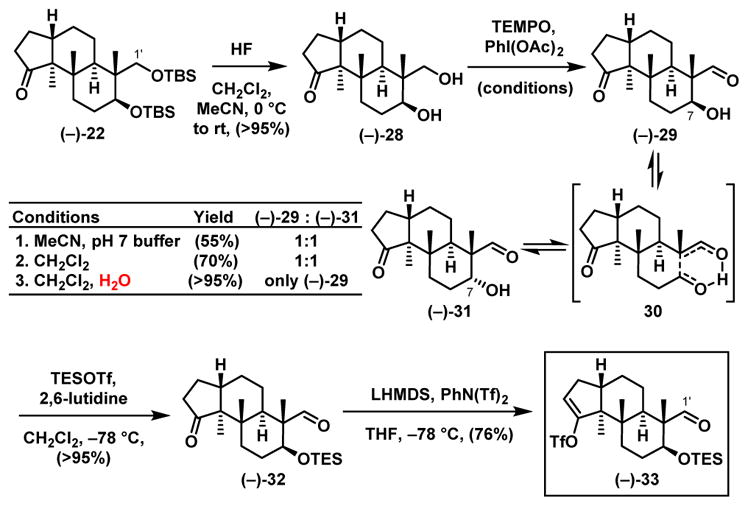
Construction of the Revised Eastern Hemisphere (−)-33
Applying our union conditions to western hemisphere (−)-7 and the revised eastern hemisphere (−)-33 (Scheme 9) now at a lower temperature (70 °C) and higher concentration led to the desired indole (−)-34 in 69% yield, importantly with the aldehyde group tolerated. Surprisingly however, the aldehyde functionality proved inert toward the envisioned Horner–Wadsworth–Emmons reaction32 to install the dienoate side chain. We reasoned this lack of reactivity was likely due to the steric hindrance of the C(8) neopentyl position. The TES group was therefore removed to alleviate the steric constraint. The derived alcohol (−)-37 again only led to a complex mixture with the same olefination protocol. This observation led us to envision that an acetyl group at C(7) might serve not only as a temporary protecting group but also as an electrophilic anion directing group. Alcohol (−)-37 was therefore acetylated. Pleasingly, the Horner–Wadsworth–Emmons reaction with acetate (−)-40 and phosphonate 3533 yielded the desired dienoate (−)-41 in 60% yield. Hydrolysis of the latter (LiOH) completed the first total synthesis of (−)-nodulisporic acid D (4), identical in all respects with the published spectroscopic data.1e
Scheme 9.

End Game of (−)-Nodulisporic Acid D
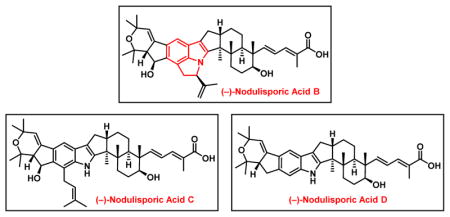
With the total synthesis of (−)-nodulisporic acid D (4) achieved, we were encouraged to continue our synthetic drive to orchestrate a unified strategy to access the architecturally more complex nodulisporic acids (C, B, and A; Scheme 10). Noteworthy here in each is a C(24) hydroxy group known to undergo readily elimination, as well as more complex indole cores.1 Nodulisporic acid C (3) for example possesses a C(26) prenyl unit, while for nodulisporic acids B (2) and A (1) a strained1a,1c,6b CDE tricyclic indole-indoline motif, including a stereogenic center at C(2′) in B(2) and A(1), distal form the other stereogenicity. Finally A(1) presents a carbonyl at C(1′)!
Scheme 10.

A Unified Synthetic Strategy to the Advanced Nodulisporic Acids
To access the more complex nodulisporic acids, we first required a protocol that would permit functionalization of the western hemisphere 43 at C(26) with a prenyl unit (Scheme 10), followed by union with eastern hemisphere fragment 44, leading to an advanced indole intermediate (42). Depending on the specific prenyl unit selected, such a strategy would hold the promise not only to access (−)-nodulisporic acid C (3), but potentially to (−)-B (2) and eventually (+)-A (1) via a late-stage cyclization/D-ring constructions.
Towards this end, the indole core (45, Scheme 11) of (−)- nodulisporic acid C (3) was disconnected retrosynthetically to yield the western hemisphere 46 and now the C(7)-acetate eastern hemisphere 47 to avoid a late-stage two-step protecting group interchange as required in the acid D (4) synthesis (vide supra). While 47 would derive from advanced FGH-intermediate (−)-33, employed in our (−)-nodulisporic D synthesis (Scheme 9),9 the critical C(26) functionalized western hemisphere 46 was envisioned to be generated via an N-Boc directed ortho-lithiation/alkylation34 utilizing tricyclic intermediate 48. Construction of 48 in turn would entail union of the aldehyde derived from commercially available benzoic acid 49 with hydrazone (+)-9, now via an Enders asymmetric addition35 to establish both the relative and absolute configurations, as employed in our earlier nodulisporic acid synthetic ventures.12 Then instead of the previously established Stille-Kelly cyclization protocol employed in the acid D(4) synthesis that required use of the toxic and volatile reagent hexamethylditin36 which could be a significant issue upon scale-up, a Nozaki-Hiyama-Kishi cyclization37 was envisioned.
Scheme 11.

Synthetic Analysis of (−)-Nodulisporic Acid C (3)
Towards this end (Scheme 12A), borane reduction of 49 followed by electrophilic iodination led to benzyl alcohol 51. Oxidation (MnO2) then provided benzyl aldehyde 52, which was submitted to the Enders asymmetric addition35 with (+)-9 to deliver hydrazone (+)-53, again as a single enantiomer. Next, employing the oxidation protocol established previously in our laboratory,6c β-hydroxyl ketone (−)-54 was obtained, albeit with enone 55 as a significant side product lacking the benzylic hydroxy group. The structure of 55 was assigned by X-ray analysis, while the relative configuration of (−)-54 was confirmed by NOESY analysis of (−)-56, derived from (−)-54 via a reduction/acetalization sequence.
Scheme 12.

A) Substrate Construction; and B) The Nozaki-Hiyama-Kishi Reaction
At this stage, given the previously cited toxicity issues related to the Stille-Kelly cyclization, we turned to the Nozaki-Hiyama-Kishi protocol.37 Treatment of (−)-54 (Scheme 12B) with chromium (II) chloride in the presence of a catalytic amount of nickel (II) chloride smoothly led to tricyclic diol (+)-57 in 79% yield. Notably this transformation was carried out in the presence of a free benzylic-hydroxyl group. Having constructed the tricyclic system, the secondary hydroxyl of (+)-57 was protected chemoselectively as the TBS ether and the tertiary hydroxyl group eliminated to deliver olefin (−)-58, along with a minor side product (−)-59, wherein the double bond had migrated.
With (−)-58 secure (ca. > 1g), we turned to explore the critical ortho-lithiation/alkylation. Initial attempts at deprotonation of the C(26) position of (−)-58 proved unrewarding (Scheme 13A), only minimal lithiation was observed in commonly employed ethereal solvents (THF or Et2O) with or without an additive (i.e., HMPA or TMEDA).34 We postulated that the steric congestion of the TBS group at the C(26) of (−)-58 was the issue. We therefore turned to the use of a -OPiv group at C(24), introduced by a two-step deprotection/pivalation sequence (Scheme 13). Although the steric environment of the derivative pivalate (−)-60 may be similar to TBS ether (−)-58, as suggested by the X-ray structure of (−)-60, we reasoned that the pivalate group might facilitate deprotonation at C(26) as an additional directing group. As such, (−)-60 was subjected to direct lithiation employing t-BuLi/HMPA/CPME. Upon addition of MeOD, we observed considerable deuterium incorporation at C(26) (D% = 80%, by 1H-NMR), however the pivalate group was gone. Nevertheless, this observation encouraged us to explore the alcohol (−)-48 for the directed lithiation.
Scheme 13.

Synthesis of Western Hemisphere 46 and Eastern Hemisphere 47
Pleasingly, successful deprotonation of (−)-48 at C(26) was observed with t-BuLi (6 eq.)/HMPA/CPME (Scheme 13B). A MeOD quench experiment revealed 82% D-incorporation at C(26). However, upon addition of prenyl bromide, C-alkylation did not occur, only O- and N-alkylation to furnish (−)-63 in 49% yield after removal of the Boc group. Presumably strong chelation from the neighboring alkoxide and/or carbamate groups reduces the desired reactivity of the aryl anion 62. We reasoned that addition of a copper (I) salt might decrease the reactivity of the N- and O- anions, while increasing the reactivity of the C-anion, possibly via generation of a cuprate species.38 After investigation of a number of different copper (I) salts (see Supporting Information), we discovered that addition of CuCN permitted exclusive C-alkylation (72%). Protection of the derived alcohol (−)-64 as the silyl ether and removal of the Boc group, both achieved in “one-pot”, completed construction of western hemisphere (−)-46. The requisite eastern hemisphere 47 in turn was obtained via a single flask deprotection/acetylation operation employing the advanced intermediate (−)-33 used in the acid D (4) synthetic venture.
With both hemispheres (−)-46 and (−)-47 in hand, we moved to the key union tactic exploited for nodulisporic acid D (4). Disappointedly, conditions used in the (−)-nodulisporic acid D (4) synthesis (i.e., RuPhos, Pd2dba3 or the palladacycle precatalyst), as well as the use of other biaryl phosphine ligands, solvents, and bases led only to either recovery of both starting materials or complex mixtures.39 It appeared that the desired cross coupling was inhibited due to the additional steric hindrance of the prenyl side chain now present in the western hemisphere (−)-46. This lack of productive cross coupling provides another example where palladium-mediated amination of two highly encumbered substrates is often challenging.40
Reasoning that different phosphine ligands may alter the behavior of the palladium catalyst, we turned to a screen to identify possible phosphine ligands (see Supporting Information). Eventually we discovered that a combination of Pd(OAc)2 and APhos41 with triflate (−)-47 (Scheme 14) delivered the desired union product (+)-72, while tolerating the multiple sensitive functionalities, in particular the C(8) aldehyde and the base sensitive C(7) acetate. It is likely that the specific combination of both steric and electronic effects of APhos facilitated the union sequence,41b as suggested by the superior reactivity of Aphos towards Suzuki-Miyaura Coupling.41
Scheme 14.

End Game for (−)-Nodulisporic Acid C (3)
Initially however the reproducibility and scalability of the union reaction proved troublesome. Experimentally, we observed that the employed base (K3PO4) often deposited on the inner surface of the reaction flask as a thick residual, especially on prolonged heating. As a result, either incomplete conversion or decomposition occurred. Pleasingly addition of sand (ca. 50 mg/1 mL solvent) to the reaction mixture prevented deposition of the base via agitation, thereby furnishing a constant yield (51%) upon scale-up. With the critical union achieved, the Horner–Wadsworth–Emmons reaction32 with phosphonate 359 on aldehyde (+)-72 installed the dienoate side chain in near quantitative yield. Hydrolysis of the derived dienoate (+)-73 with LiOH/MeOH/H2O, followed by salt exchange pleasingly completed the first total synthesis of (−)-nodulisporic acid C (3), stabilized as the sodium salt, identical in all respects upon spectral comparison with the published Merck data.1d
With the total synthesis of both (−)-nodulisporic acid D (4) and C (3) achieved, we proceeded to construction of the highly-strained tricyclic indole-indoline core with the embedded D-ring present in nodulisporic acid B (2) (Scheme 15). Here we envisioned a Tsuji-Trost palladium-promoted allylic cyclization reaction,42 employing 75 as the appropriately C(26) functionalized advanced indole to construct ring-D.
Scheme 15.
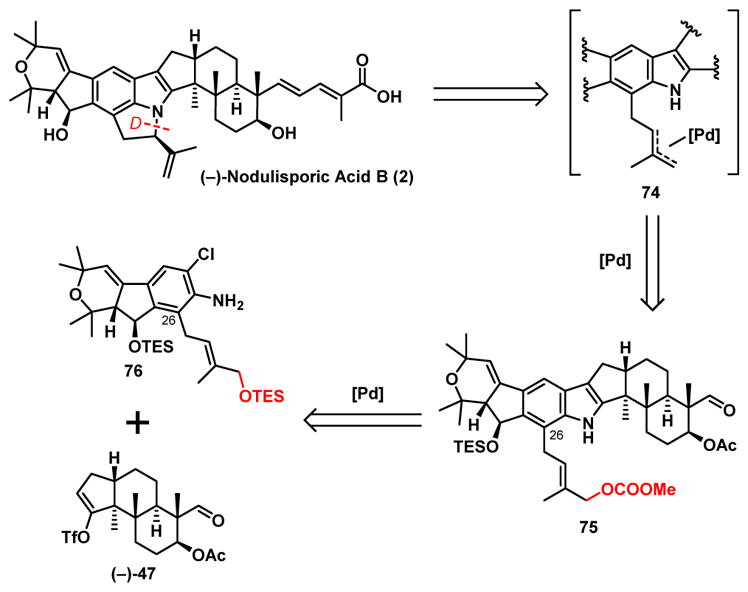
Synthetic Analysis of (−)-Nodulisporic Acid B (2)
At the outset of this venture, and with a critical need for more overall efficacy for scale-up, we devised a second-generation route to the advanced tricyclic intermediate (−)-48 that was employed in the nodulisporic acid C (3) synthesis (Scheme 13). To this end, the hydroxy group of hydrazone (+)-53 (Scheme 16) was first protected as TES ether, and then submitted to oxidation (SeO2, H2O2), applying a biphasic solvent system (water/cyclohexane), to deliver ketone (−)-77 in 60% yield over the two step, thereby avoiding the previous issue of facile hydroxy elimination. Next, application of the Nozaki-Hiyama-Kishi cyclization followed by elimination provided (−)-79, now without producing the double bond isomer (see Scheme 12B). Removal of the TES group employing the refined conditions of H2SiF6/t-BuOH43 completed a second-generation synthesis of (−)-48 with an overall yield of 34%, compared to 14% of the first-generation, over the five steps from (+)-53.
Scheme 16.
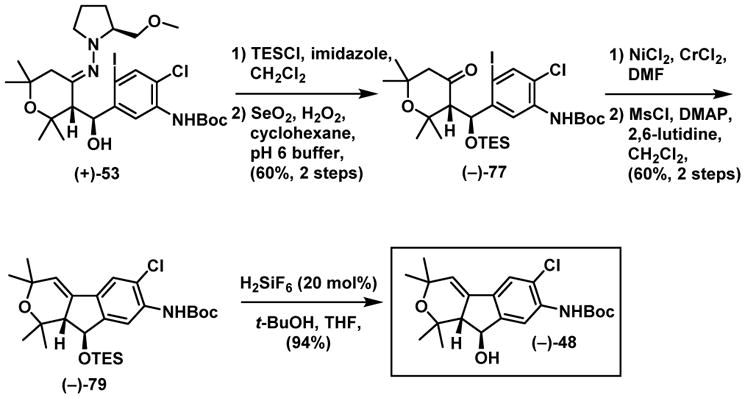
Second-Generation Western Hemisphere Synthesis
Application of our ortho lithiation/alkylation tactic from the nodulisporic acid C synthesis with iodide 80 (Scheme 17),44 now possessing the requisite allylic OTES substituent for ring-D generation led to (−)-81, albeit in modest yield (45%), with 37% starting material (−)-48 recovered, that fortunately could be recycled. Alcohol (−)-81 was next converted in a single flask operation to (−)-76 in 77% yield via silylation and Boc group removal. Union with triflate (−)-47 employed in the acid C (3) synthesis via our now established cross coupling protocol then led to the corresponding indole (+)-82 in 35% yield (vide infra), which in turn was converted to carbonate (−)-75 via a two-step chemoselective deprotection/carbonation sequence.
Scheme 17.
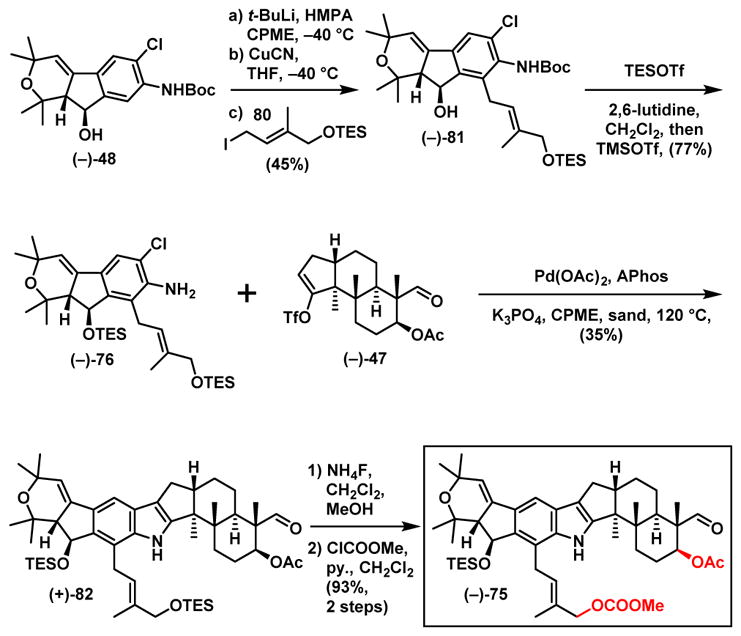
Synthesis of the Cyclization Precursor
With (−)-75 in hand, we explored the proposed Tsuji-Trost cyclization. Initial attempts at generating ring-D utilizing either a palladium or iridium catalytic system,45 with or without an external base such as K3PO4 or DBU in various solvent, proved unrewarding; only recovery of starting materials was achieved. Employing a stronger base such as NaOt-Bu also only led to decomposition, likely due to the base sensitivity of the -OAc group. We therefore adjusted the synthetic scheme revisiting the use of triflate (−)-33 employed in nodulisporic acid D (4) synthesis (Scheme 18).
Scheme 18.
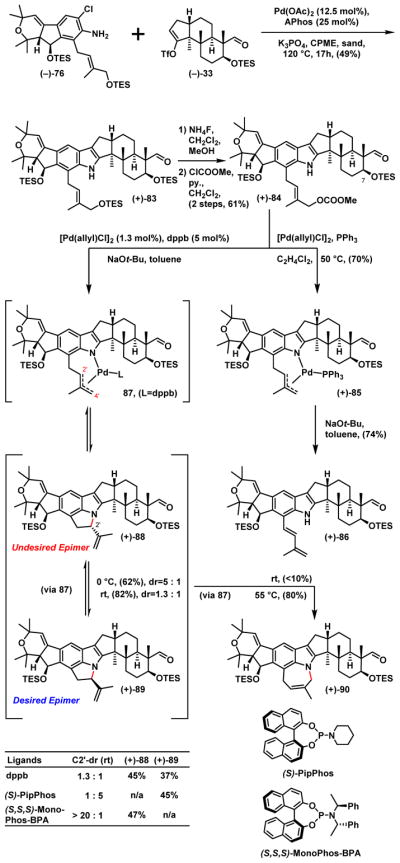
The Key Cyclization Reaction
To this end, union of aniline (−)-76 with triflate (−)-33 led to the corresponding indole (+)-83 now in 49% yield. Next employing the same synthetic sequence as describe in Scheme 17, carbonate (+)-84 was acquired in 61% over the 2 steps. With the hydroxyl group at C(7) now protected as the TES ether, the issue of the base sensitivity of the acetyl group had been eliminated.
Use of [Pd(allyl)Cl]2/PPh3 again however resulted in none of the desired cyclization, but now producing diene (+)-86, with isolation of a small amount of the palladium species (+)-85, which did not undergo the desired ring-D cyclization under either neutral or basic conditions, but instead provided diene (+)-86 again, likely via beta-hydride elimination. Reasoning that the beta-hydride elimination might be suppressed by a specific ligand that could occupy the empty coordination site on palladium, we investigated different ligands (see Supporting Information). After extensive optimization, we eventually discovered that employing the bidentate ligand 1,4-bis(diphenylphosphino)butane (dppb) at room temperature for 17 hours resulted in 82% combined yield of (+)-88 and (+)-89, albeit with a dr of 1.3:1. However, if the reaction was conducted at 55 °C for 17 hours, (+)-90 containing a seven- instead of a five-membered ring, was produced as a major product (80%). The ring expansion is likely driven by the high strain energy1a of (+)-88 and (+)-89 inherent in the nodulisporic tricyclic indole-indoline core. Fortunately (+)-88 and (+)-89 proved readily separable via silica gel chromatography, and importantly the structures could be assigned by extensive NMR analysis.
From the mechanistic perspective, we envisioned that the initially formed palladium π-allyl species 87 undergoes cyclization at the C(2′) position to produce the kinetic product (+)-88. Importantly, this cyclization is reversible, specifically with dppb as the ligand;46 at room temperature, a thermodynamic mixture [(+)-88:(+)-89 = 1.3:1] is produced. Again for the same reason, but now at 55 °C, the thermodynamically more stable product (+)-90 dominates.47
At this stage, we reasoned that introduction of external stereogenicity might influence the cyclization transition state. We therefore turned to the chiral phosphoramidite ligands developed by Feringa.48 Pleasingly, treatment of (−)-84 as before, but now with addition of (S)-PipPhos as the ligand (5 mol%) led predominately to the desired epimer (+)-89 (dr=5:1) albeit in 45% yield, whereas the (S,S,S)-MonoPhos-BPA49 ligand (4 mol%) gave almost exclusively (+)-88. Notwithstanding the modest yield of (+)-89, a regio- and diastereoselective cyclization had been achieved that successfully led to the construction of the highly strained tricyclic indole-indoline core of (−)-nodulisporic acid B (2).
Having arrived at the tricyclic indole-indoline core (−)-89 for (−)-nodulisporic acid B, not unexpectedly the acid and oxygen sensitivities of such advanced intermediates became an even greater issue;1a,c exposure to either air or to standard silica gel chromatographic protocols led to significant decomposition. Notwithstanding the stability issues, careful elaboration of (+)-89 and (+)-88 individually in the absence of oxygen, followed by removal of the silyl groups and “one-pot” acetylation, exploiting our earlier developed Horner–Wadsworth–Emmons directed olefination tactic (Scheme 19) led, after careful hydrolysis protocols, to the first total synthesis of (−)-nodulisporic acid B (2) and the epimer (−)- 2′-epi-nodulisporic acid B (94), both stabilized as their sodium salts; the former identical in all respects to the published Merck NMR date,1c the latter assigned upon extensive NMR and HRMS analysis.
Scheme 19.

The End Game
In summary, the first total syntheses of (−)-nodulisporic acids D, C, and B as well as the unnatural analogue (−)-2′-epi-nodulisporic acid B, each stabilized and fully characterized as their sodium salts, have been achieved in a stereocontrolled fashion via a unified synthetic strategy. Synthetic studies towards the more complex nodulisporic acid A, as well as analogs thereof continue in our laboratory.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health (National Institute of General Medical Sciences) through Grant GM-29028 and National Cancer Institute through Grant CA-19033.
Footnotes
The Supporting Information is available free of charge on the ACS Publication website at DOI:
Experimental procedures, as well as spectroscopic and analytical data for all new compounds (PDF)
References
- 1.(a) Ondeyka JG, Helms GL, Hensens OD, Goetz MA, Zink DL, Tsipouras A, Shoop WL, Slayton L, Dombrowski AW, Polishook JD, Ostlind DA, Tsou NN, Ball RG, Singh SB. Nodulisporic Acid A, a Novel and Potent Insecticide from a Nodulisporium Sp. Isolation, Structure Determination, and Chemical Transformations J Am Chem Soc. 1997;119:8809. [Google Scholar]; (b) Hensens OD, Ondeyka JG, Dombrowski AW, Ostlind DA, Zink DL. Isolation and Structure of Nodulisporic Acid A1 and A2, Novel Insecticides from a Nodulisporium Sp Tetrahedron Lett. 1999;40:5455. [Google Scholar]; (c) Ondeyka JG, Dahl-Roshak AM, Tkacz JS, Zink DL, Zakson-Aiken M, Shoop WL, Goetz MA, Singh SB. Nodulisporic Acid B, B1, and B2: A Series of 1′-Deoxy-Nodulisporic Acids from Nodulisporium Sp. Bioorg Med Chem Lett. 2002;12:2941. doi: 10.1016/s0960-894x(02)00621-2. [DOI] [PubMed] [Google Scholar]; (d) Ondeyka JG, Byrne K, Vesey D, Zink DL, Shoop WL, Goetz MA, Singh SB. Nodulisporic Acids C, C1, and C2: A Series of D-Ring-Opened Nodulisporic Acids from the Fungus Nodulisporium Sp. J Nat Prod. 2003;66:121. doi: 10.1021/np020339u. [DOI] [PubMed] [Google Scholar]; (e) Singh SB, Ondeyka JG, Jayasuriya H, Zink DL, Ha SN, Dahl-Roshak A, Greene J, Kim JA, Smith MM, Shoop W, Tkacz JS. Nodulisporic Acids D–F: Structure, Biological Activities, and Biogenetic Relationships. J Nat Prod. 2004;67:1496. doi: 10.1021/np0498455. [DOI] [PubMed] [Google Scholar]
- 2.Other examples of syntheses of indole terpenes see: Smith AB, Mewshaw R. Total Synthesis of (−)-Paspaline J Am Chem Soc. 1985;107:1769.Smith AB, Sunazuka T, Leenay TL, Kingery-Wood J. Total Syntheses of (+)-Paspalicine and (+)-Paspalinine J Am Chem Soc. 1990;112:8197.Smith AB, Kanoh N, Ishiyama H, Hartz RA. Total Synthesis of (−)-Penitrem D. J Am Chem Soc. 2000;122:11254. doi: 10.1021/ja034842k.Zou Y, Smith AB. Total Synthesis of Architecturally Complex Indole Terpenoids: Strategic and Tactical Evolution. J Antibiot. 2017;30:94. doi: 10.1038/ja.2017.94.Corsello MA, Kim J, Garg NK. Indole Diterpenoid Natural Products as the Inspiration for New Synthetic Methods and Strategies. Chemical Science. 2017;8:5836. doi: 10.1039/c7sc01248a.Enomoto M, Morita A, Kuwahara S. Total Synthesis of the Tremorgenic Indole Diterpene Paspalinine. Angew Chem Int Ed. 2012;51:12833. doi: 10.1002/anie.201206299.Lu Z, Li H, Bian M, Li A. Total Synthesis of Epoxyeujindole A. J Am Chem Soc. 2015;137:13764. doi: 10.1021/jacs.5b09198.Li H, Chen Q, Lu Z, Li A. Total Syntheses of Aflavazole and 14-Hydroxyaflavinine. J Am Chem Soc. 2016;138:15555. doi: 10.1021/jacs.6b10880.
- 3.(a) Shoop WL, Gregory LM, Zakson-Aiken M, Michael BF, Haines HW, Ondeyka JG, Meinke PT, Schmatz DM. Systemic Efficacy of Nodulisporic Acid against Fleas on Dogs. J Parasitol. 2001;87:419. doi: 10.1645/0022-3395(2001)087[0419:SEONAA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]; (b) Felcetto T, Ondeyka J, Colletti SL, Meinke PT, Shoop WL. Comparison of Nodulisporic Acid Analogs in a Lucilia Sericata in Vitro Assay and a Ctenocephalides Felis Membrane Feeding System. J Parasitol. 2002;88:223. doi: 10.1645/0022-3395(2002)088[0223:CONAAI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 4.(a) Meinke PT, Ayer MB, Colletti SL, Li C, Lim J, Ok D, Salva S, Schmatz DM, Shih TL, Shoop WL, Warmke LM, Wyvratt MJ, Zakson-Aiken M, Fisher MH. Chemical Modification of Nodulisporic Acid A: Preliminary Structure-Activity Relationships. Bioorg Med Chem Lett. 2000;10:2371. doi: 10.1016/s0960-894x(00)00469-8. [DOI] [PubMed] [Google Scholar]; (b) Meinke PT, Colletti SL, Fisher MH, Wyvratt MJ, Shih TL, Ayer MB, Li C, Lim J, Ok D, Salva S, Warmke LM, Zakson M, Michael BF, de Montigny P, Ostlind DA, Fink D, Drag M, Schmatz DM, Shoop WL. Discovery of the Development Candidate N-Tert-Butyl Nodulisporamide: A Safe and Efficacious Once Monthly Oral Agent for the Control of Fleas and Ticks on Companion Animals. J Med Chem. 2009;52:3505. doi: 10.1021/jm801334v. [DOI] [PubMed] [Google Scholar]
- 5.(a) Ok D, Li C, Shih TL, Salva S, Ayer MB, Colletti SL, Chakravarty PK, Wyvratt MJ, Fisher MH, Gregory L, Zakson-Aiken M, Shoop WL, Schmatz DM, Meinke PT. Side-Chain Homologation of Nodulisporic Acid: Synthesis of Potent New Dienyl Derivatives. Bioorg Med Chem Lett. 2002;12:1751. doi: 10.1016/s0960-894x(02)00284-6. [DOI] [PubMed] [Google Scholar]; (b) Chakravarty PK, Shih TL, Colletti SL, Ayer MB, Snedden C, Kuo H, Tyagarajan S, Gregory L, Zakson-Aiken M, Shoop WL, Schmatz DM, Wyvratt M, Fisher MH, Meinke PT. Nodulisporic Acid Side-Chain Modifications: Access to the 2″, 3″, 4″, and 6″ Registers. Bioorg Med Chem Lett. 2003;13:147. doi: 10.1016/s0960-894x(02)00826-0. [DOI] [PubMed] [Google Scholar]
- 6.(a) Smith AB, Davulcu AH, Cho YS, Ohmoto K, Kürti L, Ishiyama H. Indole Diterpene Synthetic Studies. Total Synthesis of (+)-Nodulisporic Acid F and Construction of the Heptacyclic Cores of (+)-Nodulisporic Acids a and B and (−)-Nodulisporic Acid D. J Org Chem. 2007;72:4596. doi: 10.1021/jo062422i. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, Kürti L, Davulcu AH, Cho YS, Ohmoto K. Indole Diterpene Synthetic Studies: Development of a Second-Generation Synthetic Strategy for (+)-Nodulisporic Acids A and B. J Org Chem. 2007;72:4611. doi: 10.1021/jo062423a. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, Liu Z, Simov V. An Efficient Protocol for the Oxidative Hydrolysis of Ketone Samp Hydrazones Employing SeO2 and H2O2 under Buffered (Ph 7) Conditions. Synlett. 2009;19:3131. doi: 10.1055/S-0029-1218352. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Magnus P, Mansley TE. Synthesis of the Abcd-Rings of the Insecticidal Indole Alkaloid Nodulisporic Acid Tetrahedron Lett. 1999;40:6909. [Google Scholar]
- 7.(a) Smith AB, Visnick M. An Expedient Synthesis of Substituted Indoles Tetrahedron Lett. 1985;26:3757. [Google Scholar]; (b) Smith AB, Visnick M, Haseltine JN, Sprengeler PA. Organometallic Reagents in Synthesis: A New Protocol for Construction of the Indole Nucleus Tetrahedron. 1986;42:2957. [Google Scholar]; (c) Smith AB, Davulcu AH, Kürti L. Indole Diterpenoid Synthetic Studies. The Total Synthesis of (+)-Nodulisporic Acid F. Org Lett. 2006;8:1665. doi: 10.1021/ol060290+. [DOI] [PubMed] [Google Scholar]
- 8.(a) Barluenga J, Valdes C. Palladium Catalyzed Alkenyl Amination: From Enamines to Heterocyclic Synthesis. Chem Commun. 2005:4891. doi: 10.1039/b509311b. [DOI] [PubMed] [Google Scholar]; (b) Barluenga J, Fernandez MA, Aznar F, Valdes C. Cascade Alkenyl Amination/Heck Reaction Promoted by a Bifunctional Palladium Catalyst: A Novel One-Pot Synthesis of Indoles from O-Haloanilines and Alkenyl Halides. Chem - Eur J. 2005;11:2276. doi: 10.1002/chem.200401274. [DOI] [PubMed] [Google Scholar]
- 9.Zou Y, Melvin JE, Gonzales SS, Spafford MJ, Smith AB. Total Synthesis of (−)-Nodulisporic Acid D. J Am Chem Soc. 2015;137:7095. doi: 10.1021/jacs.5b04728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Enders D, Eichenauer H. Asymmetric Synthesis of A-Substituted Ketones by Metalation and Alkylation of Chiral Hydrazones Angewandte Chemie International Edition in English. 1976;15:549. [Google Scholar]; (b) Enders D, Zamponi A, Raabe G, Runsink J. Enantioselective Synthesis of 2-Alkyl-2-Cyanocycloalkanones with a Quaternary Stereogenic Center Synthesis. 1993;1993:725. [Google Scholar]
- 11.(a) Ross Kelly T, Li Q, Bhushan V. Intramolecular Biaryl Coupling: Asymmetric Synthesis of the Chiral B-Ring Diol Unit of Pradimicinone Tetrahedron Lett. 1990;31:161. [Google Scholar]; (b) Sheffy FK, Godschalx JP, Stille JK. Palladium-Catalyzed Cross Coupling of Allyl Halides with Organotin Reagents: A Method of Joining Highly Functionalized Partners Regioselectively and Stereospecifically J Am Chem Soc. 1984;106:4833. [Google Scholar]; (c) Azarian D, Dua SS, Eaborn C, Walton DRM. Reactions of Organic Halides with R3 MMR3 Compounds (M = Si, Ge, Sn) in the Presence of Tetrakis(Triarylphosphine)Palladium J Organomet Chem. 1976;117:C55. [Google Scholar]
- 12.Smith AB, Ishiyama H, Cho YS, Ohmoto K. Nodulisporic Acid a Synthetic Studies. 1. Overall Strategy and Construction of a Western Hemisphere Subtarget. Org Lett. 2001;3:3967. doi: 10.1021/ol0168871. [DOI] [PubMed] [Google Scholar]
- 13.Smith AB, Kürti L, Davulcu AH, Cho YS. Development of a Scalable Synthesis of a Common Eastern Tricyclic Lactone for Construction of the Nodulisporic Acids Org Process Res Dev. 2007;11:19. [Google Scholar]
- 14.Garegg PJ, Samuelsson B. Novel Reagent System for Converting a Hydroxy-Group into an Iodo-Group in Carbohydrates with Inversion of Configuration J Chem Soc, Chem Commun. 1979:978. [Google Scholar]
- 15.Comins DL, Dehghani A. Pyridine-Derived Triflating Reagents: An Improved Preparation of Vinyl Triflates from Metallo Enolates Tetrahedron Lett. 1992;33:6299. [Google Scholar]
- 16.Ramadas K, Srinivasan N. Iron-Ammonium Chloride - a Convenient and Inexpensive Reductant Synth Commun. 1992;22:3189. [Google Scholar]
- 17.Kajigaeshi S, Kakinami T, Yamasaki H, Fujisaki S, Kondo M, Okamoto T. Iodination of Phenols by Use of Benzyltrimethylammonium Dichloroiodate Chem Lett. 1987;16:2109. [Google Scholar]
- 18.Normant JF, Bourgain M. Synthese Stereospecifique and Reactivite D′ Organocuivreux Vinyliques Tetrahedron Lett. 1971;12:2583. [Google Scholar]
- 19.Cram DJ, Roitman JN. Electrophilic Substitution at Saturated Carbon. XLVI. Crown Ethers’ Ability to Alter Role of Metal Cations in Control of Stereochemical Fate of Carbanions J Am Chem Soc. 1971;93:2231. [Google Scholar]
- 20.Scholl M, Ding S, Lee CW, Grubbs RH. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-Dihydroimidazol-2-Ylidene Ligands. Org Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 21.(a) Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts J Am Chem Soc. 2000;122:8168. [Google Scholar]; (b) Gessler S, Randl S, Blechert S. Synthesis and Metathesis Reactions of a Phosphine-Free Dihydroimidazole Carbene Ruthenium Complex Tetrahedron Lett. 2000;41:9973. [Google Scholar]
- 22.Griffith WP, Ley SV, Whitcombe GP, White AD. Preparation and Use of Tetra-N-Butylammonium Per-Ruthenate (Tbap Reagent) and Tetra-N-Propylammonium Per-Ruthenate (Tpap Reagent) as New Catalytic Oxidants for Alcohols J Chem Soc, Chem Commun. 1987:1625. [Google Scholar]
- 23.Watanabe K, Yamagiwa N, Torisawa Y. Cyclopentyl Methyl Ether as a New and Alternative Process Solvent Organic Process Research & Development. 2007;11:251. [Google Scholar]
- 24.Willis MC. Transition Metal Catalyzed Alkene and Alkyne Hydroacylation. Chem Rev. 2010;110:725. doi: 10.1021/cr900096x. [DOI] [PubMed] [Google Scholar]
- 25.Wu XM, Funakoshi K, Sakai K. Highly Enantioselective Cyclization Using Cationic Rh(I) with Chiral Ligand Tetrahedron Lett. 1992;33:6331. [Google Scholar]
- 26.(a) Shen X, Hyde AM, Buchwald SL. Palladium-Catalyzed Conversion of Aryl and Vinyl Triflates to Bromides and Chlorides. J Am Chem Soc. 2010;132:14076. doi: 10.1021/ja107481a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pan J, Wang X, Zhang Y, Buchwald SL. An Improved Palladium-Catalyzed Conversion of Aryl and Vinyl Triflates to Bromides and Chlorides. Org Lett. 2011;13:4974. doi: 10.1021/ol202098h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruno NC, Niljianskul N, Buchwald SL. N-Substituted 2-Aminobiphenylpalladium Methanesulfonate Precatalysts and Their Use in C–C and C–N Cross-Couplings. J Org Chem. 2014;79:4161. doi: 10.1021/jo500355k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Bruno NC, Tudge MT, Buchwald SL. Design and Preparation of New Palladium Precatalysts for C-C and C-N Cross-Coupling Reactions. Chemical Science. 2013;4:916. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kinzel T, Zhang Y, Buchwald SL. A New Palladium Precatalyst Allows for the Fast Suzuki–Miyaura Coupling Reactions of Unstable Polyfluorophenyl and 2-Heteroaryl Boronic Acids. J Am Chem Soc. 2010;132:14073. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Biscoe MR, Fors BP, Buchwald SL. A New Class of Easily Activated Palladium Precatalysts for Facile C–N Cross-Coupling Reactions and the Low Temperature Oxidative Addition of Aryl Chlorides. J Am Chem Soc. 2008;130:6686. doi: 10.1021/ja801137k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knapp JM, Zhu JS, Tantillo DJ, Kurth MJ. Multicomponent Assembly of Highly Substituted Indoles by Dual Palladium-Catalyzed Coupling Reactions. Angew Chem Int Ed. 2012;51:10588. doi: 10.1002/anie.201204633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith AB, Cui H. Indole-Diterpene Synthetic Studies: Total Synthesis of (−)-21-Isopentenylpaxilline Helv Chim Acta. 2003;86:3908. [Google Scholar]
- 31.De Mico A, Margarita R, Parlanti L, Vescovi A, Piancatelli G. A Versatile and Highly Selective Hypervalent Iodine (III)/2,2,6,6-Tetramethyl-1-Piperidinyloxyl-Mediated Oxidation of Alcohols to Carbonyl Compounds J Org Chem. 1997;62:6974. [Google Scholar]
- 32.(a) Horner L, Hoffmann H, Wippel HG. Phosphororganische Verbindungen, XII. Phosphinoxyde Als Olefinierungsreagenzien Chem Ber. 1958;91:61. [Google Scholar]; (b) Wadsworth WS, Emmons WD. The Utility of Phosphonate Carbanions in Olefin Synthesis J Am Chem Soc. 1961;83:1733. [Google Scholar]; (c) Wadsworth WS. Organic Reactions. John Wiley & Sons, Inc; 2004. Synthetic Applications of Phosphoryl-Stabilized Anions [Google Scholar]; Wadsworth WS. Organic Reactions. 1977;25:73. [Google Scholar]
- 33.Trost BM, Thiel OR, Tsui H-C. Total Syntheses of Furaquinocin a, B, and E. J Am Chem Soc. 2003;125:13155. doi: 10.1021/ja0364118. [DOI] [PubMed] [Google Scholar]
- 34.(a) Muchowski JM, Venuti MC. Ortho Functionalization of N-(Tert-Butoxycarbonyl)Aniline J Org Chem. 1980;45:4798. [Google Scholar]; (b) Stanetty P, Koller H, Mihovilovic M. Directed Ortho Lithiation of Phenylcarbamic Acid 1,1-Dimethylethyl Ester (N-Boc-Aniline). Revision and Improvements J Org Chem. 1992;57:6833. [Google Scholar]; (c) Mulhern TA, Davis M, Krikke JJ, Thomas JA. A Practical Ortholithiation-Based Synthesis of 2-Chloro-6-Methylaniline J Org Chem. 1993;58:5537. [Google Scholar]
- 35.Enders D, Kipphardt H, Fey P, Guzmán B, Hall SS, Saucy G. Asymmetric Syntheses Using the SAMP-/RAMP-Hydrazone Method: (S)-(+)-4-Methyl-3-Heptanone Organic Syntheses. 2003 [Google Scholar]
- 36.Mitchell TN, Platonov A, Nikonov G. Encyclopedia of Reagents for Organic Synthesis. Hexamethyldistannane; 2010. [Google Scholar]
- 37.(a) Okude Y, Hirano S, Hiyama T, Nozaki H. Grignard-Type Carbonyl Addition of Allyl Halides by Means of Chromous Salt. A Chemospecific Synthesis of Homoallyl Alcohols J Am Chem Soc. 1977;99:3179. [Google Scholar]; (b) Takai K, Kimura K, Kuroda T, Hiyama T, Nozaki H. Selective Grignard-Type Carbonyl Addition of Alkenyl Halides Mediated by Chromium(II) Chloride Tetrahedron Lett. 1983;24:5281. [Google Scholar]; (c) Jin H, Uenishi J, Christ WJ, Kishi Y. Catalytic Effect of Nickel(II) Chloride and Palladium(II) Acetate on Chromium(II)-Mediated Coupling Reaction of Iodo Olefins with Aldehydes J Am Chem Soc. 1986;108:5644. [Google Scholar]
- 38.(a) Normant JF. Organocopper(I) Compounds and Organocuprates in Synthesis Synthesis. 1972;1972:63. [Google Scholar]; (b) Krause N, editor. Modern Organocopper Chemistry. Wiley-VCH; 2002. [Google Scholar]
- 39.see Supporting Information for the detailed investigation
- 40.(a) Park NH, Vinogradova EV, Surry DS, Buchwald SL. Design of New Ligands for the Palladium-Catalyzed Arylation of A-Branched Secondary Amines. Angew Chem Int Ed. 2015;54:8259. doi: 10.1002/anie.201502626. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stefan R, Oliver K, Hubert SJ, NB Synthesis of Sterically Congested Triarylamines by Palladium-Catalyzed Amination Eur J Org Chem. 2014;2014:1391. [Google Scholar]
- 41.(a) Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ, Reider PJ. New Air-Stable Catalysts for General and Efficient Suzuki–Miyaura Cross-Coupling Reactions of Heteroaryl Chlorides. Org Lett. 2006;8:1787. doi: 10.1021/ol060268g. [DOI] [PubMed] [Google Scholar]; (b) Guram AS, Wang X, Bunel EE, Faul MM, Larsen RD, Martinelli MJ. New Catalysts for Suzuki–Miyaura Coupling Reactions of Heteroatom-Substituted Heteroaryl Chlorides. J Org Chem. 2007;72:5104. doi: 10.1021/jo070341w. [DOI] [PubMed] [Google Scholar]
- 42.(a) Tsuji J, Takahashi H, Morikawa M. Organic Syntheses by Means of Noble Metal Compounds XVII. Reaction of π-Allylpalladium Chloride with Nucleophiles Tetrahedron Lett. 1965;6:4387. [Google Scholar]; (b) Trost BM, Fullerton TJ. New Synthetic Reactions. Allylic Alkylation J Am Chem Soc. 1973;95:292. [Google Scholar]; (c) Trost BM. Metal Catalyzed Allylic Alkylation: Its Development in the Trost Laboratories. Tetrahedron. 2015;71:5708. doi: 10.1016/j.tet.2015.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pilcher AS, DeShong P. Improved Protocols for the Selective Deprotection of Trialkylsilyl Ethers Using Fluorosilicic Acid J Org Chem. 1993;58:5130. [Google Scholar]
- 44.See Supporting Information for the preparation of 80
- 45.Selected examples of palladium or iridium catalyzed indole N-alkylation see: Trost BM, Krische MJ, Berl V, Grenzer EM. Chemo-, Regio-, and Enantioselective Pd-Catalyzed Allylic Alkylation of Indolocarbazole Pro-Aglycons. Org Lett. 2002;4:2005. doi: 10.1021/ol020046s.Bandini M, Melloni A, Umani-Ronchi A. New Versatile Pd-Catalyzed Alkylation of Indoles Via Nucleophilic Allylic Substitution: Controlling the Regioselectivity. Org Lett. 2004;6:3199. doi: 10.1021/ol048663z.Ming C, Hartwig JF. Iridium-Catalyzed Regio- and Enantioselective N-Allylation of Indoles. Angew Chem Int Ed. 2009;48:7841. doi: 10.1002/anie.200904338.Ke-Yin Y, Qiang C, Chun-Xiang Z, Li-Xin D, Shu-Li Y. An Iridium(I) N-Heterocyclic Carbene Complex Catalyzes Asymmetric Intramolecular Allylic Amination Reactions. Angew Chem Int Ed. 2016;55:8113. doi: 10.1002/anie.201603266.
- 46.Detailed study on the reversibility of palladium catalyzed allylic aminations see: Amatore C, Génin E, Jutand A, Mensah L. Palladium(0)-Catalyzed Allylic Aminations: Kinetics and Mechanism of the Reaction of Secondary Amines with Cationic [(η3-Allyl)PdL2]+ Complexes Organometallics. 2007;26:1875.Caminiti NS, Goodstein MB, Leibler INM, Holtzman BS, Jia ZB, Martini ML, Nelson NC, Bunt RC. Reversible Nucleophilic Addition Can Lower the Observed Enantioselectivity in Palladium-Catalyzed Allylic Amination Reactions with a Variety of Chiral Ligands Tetrahedron Lett. 2015;56:5445.
- 47.See Supporting Information for the detailed experiments
- 48.Teichert JF, Feringa BL. Phosphoramidites: Privileged Ligands in Asymmetric Catalysis. Angew Chem Int Ed. 2010;49:2486. doi: 10.1002/anie.200904948. [DOI] [PubMed] [Google Scholar]
- 49.(S,S,S)-(+)-(3,5-dioxa-4-phosphacyclohepta[2,1-a:3,4-a′]dinaphthalen-4-yl)bis(1-phenylethyl)amine
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.