

 Prestrobilurin A, formed via benzoate and cinnamate, has been identified as the first enzyme free intermediate in strobilurin biosynthesis.
Prestrobilurin A, formed via benzoate and cinnamate, has been identified as the first enzyme free intermediate in strobilurin biosynthesis.
Abstract
The strobilurins are important antifungal metabolites isolated from a number of basidiomycetes and have been valuable leads for the development of commercially important fungicides. Isotopic labelling studies with early and advanced intermediates confirm for the first time that they are produced via a linear tetraketide, primed with the rare benzoate starter unit, itself derived from phenylalanine via cinnamate. Isolation of a novel biphenyl metabolite, pseudostrobilurin B, provides evidence for the involvement of an epoxide in the key rearrangement to form the β-methoxyacrylate moiety essential for biological activity. Formation of two bolineol related metabolites, strobilurins Y and Z, also probably involves epoxide intermediates. Time course studies indicate a likely biosynthetic pathway from strobilurin A, with the simplest non-subsubstituted benzoate ring, to strobilurin G with a complex dioxepin terpenoid-derived substituent. Precursor-directed biosynthetic studies allow production of a number of novel ring-halogenated analogues as well as a new pyridyl strobilurin. These studies also provide evidence for a non-linear biosynthetic relationship between strobilurin A and strobilurin B.
Introduction
Strobilurins are a group of bioactive metabolites produced by various fungi.1 Mucidin 1 was the first to be isolated in 1965 from the basidiomycete Oudemansiella mucida.2 The triene system was initially assigned the all E,E,E configuration, and its potent antifungal activity led to its commercialisation as “mucidermin” for the treatment of skin infections.3 In 1977, Anke and co-workers isolated two antifungal metabolites, strobilurin A 2 and strobilurin B 3 from Strobilurus tenacellus.4 The former was clearly identical to mucidin which was thus re-assigned the E,Z,E configuration. Many other strobilurin analogues have been isolated from other basidiomycetes. Structural variations include oudemansin A 4 from Oudemansiella mucida in which the central 9/10 double bond of the triene system has formally had methanol added,5 9-methoxystrobilurin A 5 from Flaviolaschia sp. which contains a methyl enol ether6 and 14-hydroxystrobilurin A 6 from a Petrula sp. containing a hydroxymethyl.7 A number of strobilurins have complex dioxepin substituents containing two highly modified prenyl moieties on the phenyl ring, e.g. strobilurin G 7 from Bolinea lutea.8 Bolineol 8 in which the methoxyacrylate substituent of strobilurin A has been replaced by a methyl 3-hydroxy-propionate moiety has also been isolated from B. lutea.9
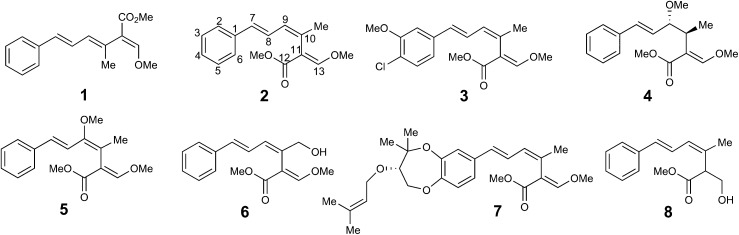 The key β-methoxyacrylate toxophore targets the Qo site of complex III of the mitochondrial electron transport chain and prevents ATP synthesis.10 The major class of β-methoxyacrylate agricultural fungicides were developed from the structures of 2 and 3 with the aim of increasing photo-stability and selectivity. Thus, compounds such as azoxystrobin 9 (Syngenta) and Kresoxim-methyl 10 (BASF) are among the most widely used fungicides worldwide, used as effective treatments against a broad range of destructive fungal plant pathogens.11,12 The strobilurin fungicides are estimated to have been worth $3.4 billion per annum in 2015 and make up 25% of the fungicide market and 6.7% of the total crop protection market.13 Other antifungal natural products containing the β-methoxyacrylate toxophore include myxothiazole 11 and cyrmenin A 12 from the myxobacteria, Myxococcus fulvus and Cystobacter armeniaca respectively.14,15
The key β-methoxyacrylate toxophore targets the Qo site of complex III of the mitochondrial electron transport chain and prevents ATP synthesis.10 The major class of β-methoxyacrylate agricultural fungicides were developed from the structures of 2 and 3 with the aim of increasing photo-stability and selectivity. Thus, compounds such as azoxystrobin 9 (Syngenta) and Kresoxim-methyl 10 (BASF) are among the most widely used fungicides worldwide, used as effective treatments against a broad range of destructive fungal plant pathogens.11,12 The strobilurin fungicides are estimated to have been worth $3.4 billion per annum in 2015 and make up 25% of the fungicide market and 6.7% of the total crop protection market.13 Other antifungal natural products containing the β-methoxyacrylate toxophore include myxothiazole 11 and cyrmenin A 12 from the myxobacteria, Myxococcus fulvus and Cystobacter armeniaca respectively.14,15
 The biosynthesis of the strobilurins, particularly that of the β-methoxyacrylate toxophore, remains obscure despite the fact that the strobilurins and analogues are amongst the most commercially important fungal metabolites known. Early labelling studies on mucidin were consistent with a polyketide produced from a benzoate starter, itself derived via phenylalanine, extended by successive condensations with three malonates, and a C-methylation from S-adenosyl methionine (SAM).16 The labelling of both C-11 and C-12 from [1-13C]-acetate suggested that rearrangement of an originally linear polyketide chain occurs. B. lutea has been described as the only ascomycete to produce the strobilurin/oudemansin family of metabolites.17
The biosynthesis of the strobilurins, particularly that of the β-methoxyacrylate toxophore, remains obscure despite the fact that the strobilurins and analogues are amongst the most commercially important fungal metabolites known. Early labelling studies on mucidin were consistent with a polyketide produced from a benzoate starter, itself derived via phenylalanine, extended by successive condensations with three malonates, and a C-methylation from S-adenosyl methionine (SAM).16 The labelling of both C-11 and C-12 from [1-13C]-acetate suggested that rearrangement of an originally linear polyketide chain occurs. B. lutea has been described as the only ascomycete to produce the strobilurin/oudemansin family of metabolites.17
Results and discussion
Feeding studies using singly and doubly labelled [13C]-acetates, [methyl-13C]-methionine, [2H8]-, [3-13C]- and [2,3-13C2]-phenylalanines, supplemented to cultures of S. tenacellus and B. lutea resulted in the production of isotopically enriched strobilurin A 2 (Scheme 1a). The results are consistent with the pathway summarised in Scheme 1b, in which phenylalanine undergoes loss of ammonia to give cinnamic acid 13,18,19 followed by degradation to benzoate. Although benzoate is a relatively uncommon starter unit in polyketide biosynthesis, benzoyl CoA serves as a component in the formation of a number of important plant metabolites including taxol20 and cocaine.21 The hexaketide moiety in squalestatin (zaragozic acid) biosynthesis in fungi is also formed from a benzoate starter, now known to be produced from phenylalanine via cinnamic acid 13.22,23 The bacterial metabolites enterocin (Streptomyces maritimus)24 and soraphen A (Sorangium cellulosum)25 also have benzoate priming their polyketide synthases.
Scheme 1. (a) Incorporation of 2H and 13C-labelled precursors into strobilurin A 2 in S. tenacellus and B. lutea. (b) Proposed assembly and rearrangement of strobilurin tetraketide.

In strobilurin A 2, the labelling pattern supports a pathway in which benzoyl CoA 14 would undergo three successive chain elongations with one C-methylation via the putative PKS-bound diketide, triketide and tetraketide intermediates 15, 16 and 17 to give prestrobilurin 18 which would then undergo rearrangement of the epoxide 19via20 to the formyl-carboxylic acid 21/22. Finally, two O-methylations using SAM as cofactor complete the biosynthesis of strobilurin A 2. A number of these points are discussed below.
Cinnamate 13 is in the unusual position of potentially acting as the source of the benzoate starter, but also as a polyketide chain elongation intermediate (15, Scheme 1b). Feeding of [2,3-13C2]-phenylanine resulted in only a single 13C label (from C-3) being incorporated, indicating that cinnamate cannot be incorporated intact into the strobilurins. However, precursor-directed biosynthesis studies (discussed in detail below) with 19F-labelled benzoates and cinnamates gave consistently higher incorporation of fluorine label into fluorostrobilurin analogues from cinnamate (78%) than from benzoate (30%) for the 4-fluoro SNAC thiolester analogues. This potential contradiction was re-examined by synthesis (Scheme 2) and feeding of [2,3-13C2]-cinnamate both as the free acid 23 and its SNAC thiol ester 24. SNAC thiol esters have been commonly used in studies of polyketide biosynthesis to mimic the thiol ester linkage of co-enzyme A and acyl carrier protein (ACP) and are usually incorporated into biosynthetic pathways more readily than the parent carboxylic acids.26,27 In this case, however, free cinnamic acid 23 was incorporated significantly more efficiently (85%) than the SNAC thiolester 24 (40%, Fig. S1†). In contrast, [carboxyl-13C]-benzoate is incorporated more efficiently as its SNAC thiol ester compared to the free acid.
Scheme 2. Reagents and conditions: (a) [2-13C]-malonic acid (1.02 equiv.), Na2SO4 (0.15 equiv.), pyridine, piperidine, reflux, 4 h, 91%; (b) DCC (1.13 equiv.), DMAP (0.04 equiv.), HSNAC (1.43 equiv.), DCM, 0 °C, 2 h, 23 °C, 16 h, 58%.

Most fungal PKS belong to the Type I iterative class consisting of a single multi-domain protein encoded by a single gene.28 They have been classified according to the increasing degree of reductive modifications and C-methylations they carry out. One major class, the highly-reducing Type II (HR II) have a trans-acting stand-alone ER domain29 while a minority are Type III systems29 which do not catalyse β-processing reactions.30 The strobilurin PKS is likely to be a Type I PKS and catalyse three iterations of chain-extension and β-processing to produce the three enzyme bound intermediates 15–17 shown in Scheme 1b. In order to investigate the intermediacy of these compounds we decided to feed analogues of 16 and 17 to cultures of B. lutea. To aid metabolite analysis they were synthesised with a fluorinated phenyl ring as fluorine has been demonstrated to be a useful tracer for biosynthetic studies.31 Accordingly, 3-fluorobenzaldehyde was converted to the 2Z,4E isomer of 2-methyl-5-phenylpenta-2,4-dienoic acid 26 and its SNAC thiol ester 27 (Scheme 3A). Key synthetic steps included methylation of ethyl acetoacetate followed by condensation with 3-fluorobenzaldehyde and cyclisation to give the 3-hydroxy-lactone 25. Following reductive removal of the hydroxyl group, ring opening of the resulting unsaturated lactone using TBAF gave acid 26 which was esterified to the corresponding thiol ester 27 in 94% yield. The Z-geometry of the 2,3-double bond was confirmed by nOe studies.
Scheme 3. (a) NaH (60%, 1.08 equiv.), THF, 23 °C, 0.5 h, 50 °C, 20 h, 56%; (b) DIPA (2.5 equiv.), n-BuLi (2.5 equiv.), HMPA (1 equiv.), 3-fluorobenzaldehyde (1.1 equiv.), THF, –78 °C, 3 h; (c) 1 M KOH(aq), 23 °C, 14 h, then 6 M HCl(aq), 0 °C, 65% (over 2 steps); (d) DIPEA (1.5 equiv.), Tf2O (1.1 equiv.), CH2Cl2, –78 °C, 1 h, 97%; (e) Pd(PPh3)4 (0.01 equiv.), Et3SiH (2.0 equiv.), DMF, 60 °C, 2 h, 99%; (f) TBAF (1 M, 5 equiv.), THF, 23 °C, 2 h, 97%; (g) EDCI·HCl (1.6 equiv.), DMAP (1.2 equiv.), HSNAC (1.5 equiv.), DCM, 0 °C, 2 h, 23 °C, 14 h, 94%; (h) Et3N (2 equiv.), EtOCOCl (1.3 equiv.), THF, 0 °C, 0.5 h; (i) NaBH4 (2.5 equiv.), MeOH, –78 °C, 4 h, 60% (over 2 steps); (j) Dess–Martin periodinane (0.3 M, 1.3 equiv.), CH2Cl2, 23 °C, 1.5 h; (k) NaH (60%, 1.4 equiv.), (EtO)2P(O)CH2CO2Et (1.7 equiv.), THF, 0 °C, 10 min, 23 °C, 16 h, 67% (over 2 steps); (l) (CF3CH2O)2P(O)CH2CO2Et (2.1 equiv.), KHMDS (15 wt%, 2 equiv.), 18-crown-6 (2.4 equiv.), THF, –78 °C, 6.5 h, 64% (2Z : 2E::5 : 1, over 2 steps); (m) 1 M NaOH(aq), THF, 23 °C, 91%; (n) EDCI·HCl (2 equiv.), DMAP (2.4 equiv.), HSNAC (8.9 equiv.), DCM, 23 °C, 16 h, 60%; (o) 1 M LiOH(aq), MeOH, 23 °C, 16 h, 72%; (p) DIPA (2 equiv.), n-BuLi (2 equiv.), InCl3 (0.6 equiv.), THF, –78 °C, 2 h, 23 °C, 1 h, 34 (65%, over 2 steps), 35 (67%, over 2 steps); (q) NaH (60%, 1.4 equiv.), (EtO)2P(O)CH2CO2Et (1.7 equiv.), THF, 0 °C, 10 min, 23 °C, 16 h, 81% (over 2 steps).

Both precursors 26 and 27 were pulse fed to cultures of B. lutea after 24, 36 and 48 hours according to the protocol used for previous feeding studies. LCMS analysis of the culture extract, however, failed to detect any fluorinated strobilurin analogues but the peaks corresponding to 26 and 27 remained with no obvious degradation of triketide to benzoate occurring as had been observed to occur for the free acid and SNAC thiolester of the diketide (cinnamate) 15.
Tetraketide 18 (Scheme 1) is proposed to be the first enzyme-free product of the PKS and its fluoro-analogue was also synthesised as both the free acid 30 and SNAC thiol ester 31 (Scheme 3B). Acid 26 was reduced to primary alcohol 28 and, following oxidation to an aldehyde was chain extended under Horner–Wadsworth–Emmons conditions to ester 29 with the 2E double bond. Hydrolysis of ester 29 gave acid 30 which was converted to thiol ester 31. The isomeric acid 33 with the 2Z-double bond was prepared using a similar strategy but with a Still–Gennari reaction for the chain extension.
Acid 30 and thiol ester 31 were pulse fed to B. lutea in separate experiments. On extraction, and LCMS analysis (Fig. 1), a new metabolite was detected at 23.7 min, between the peaks for strobilurin A (23.3 min) and strobilurin B (24.4 min) with a molecular ion [M + Na]+ 299 Da, 18 units heavier than strobilurin A 2. Interestingly, no fluorinated analogue of strobilurin B 3 was detected in either experiment, suggesting that fluorine in the phenyl ring inhibits hydroxylation. The yield of the new metabolite was ca. three times higher from the free acid 30 than from the SNAC ester 31 (Fig. 1). The metabolite 3-fluorostrobilurin 36 was purified and its structure confirmed by 1H and 19F NMR analysis. The proton resonances for the trienoic acid side chain are essentially identical to those of strobilurin A 2 as expected, confirming the 7E,9Z,11E configuration, but the aromatic ring shows the coupling pattern predicted for a 3-fluoro substituent, in particular vicinal H–F couplings of 10.1 and 8.8 Hz respectively to H-4 and H-2, and a 4 bond H–F coupling (5.8 Hz) to H-5. The 19F NMR spectrum shows a signal at –114 ppm as a double, double, doublet with the same coupling constants (Fig. S2b†).
Fig. 1. LCMS analysis shows that the tetraketide 30 fed to culture of B. lutea yields more of the enriched metabolite than the corresponding SNAC thiol ester 31.

As the trienoic acid 18 has not been detected in extracts of B. lutea, it suggests that on release of 17 from the PKS, this first enzyme-free intermediate is rapidly converted to strobilurins (Scheme 1b) as discussed in more detail below. At this stage we could not rule out that the observed incorporation of fluorine label was occurring via degradation of 30 or 31 to 3-fluorobenzoate and reincorporation, as observed for cinnamate. We thus synthesised an isotopomer of 30 (Scheme 3B) with a trideuteriomethyl at C-14 in addition to the original C-3 fluorine label. Incubation of the isotopomer with B. lutea and LCMS analysis of the extract showed a new peak at 24.3 min (strobilurin A 23.9 min) with the correct molecular ion ([M + H]+ 280 Da) for the trideuterio-labelled analogue of 3-fluorostrobilurin 36 to indicate intact incorporation of 30. The structure was again confirmed by full NMR analysis. The 19F NMR spectrum was identical to that previously obtained for 36, as was the 1H NMR spectrum except for the absence of the signal at δ 1.98 ppm for the three protons of the 14-methyl. This confirms that tetraketide 17 is indeed the final enzyme-bound product of the strobilurin PKS.‖
Conversion of tetraketide 18 (thus named prestrobilurin A) to strobilurin A is proposed to proceed via epoxidation and rearrangement to give aldehyde 21, enolisation to 22 and methylation forming the β-methoxyacrylate moiety (Scheme 1b). To further investigate this process we synthesised the epimeric epoxides 34 and 35 with a fluorine label (Scheme 3C). The allylic alcohol 29 was oxidized by Dess–Martin periodinane, and then the unstable aldehyde was treated with the methyl ester or SNAC thiolester of α-bromoacetate under Darzens reaction conditions. However, on feeding to B. lutea no incorporation into strobilurins, direct or indirect could be detected. In addition, fermentation of B. lutea in the presence of varying concentrations of the known cytochrome P450 monooxygenase inhibitor ancymidol32 had no effect on strobilurin production. Thus no direct evidence for an epoxide-mediated arrangement is available.
However, on investigating minor components of a second strain of B. lutea (strain F23523, which in our hands produced strobilurin B 3 as its major metabolite) we observed a minor peak eluting after strobilurin B 3 with a molecular ion [M + H]+ 291 Da with a 37Cl isotopic peak at 293 Da. We purified this metabolite by preparative HPLC, and isolated 0.8 mg of pure compound for NMR characterisation. Signals for a 3-methoxy-4-chloro-phenyl ring as in strobilurin B 3 were evident, but those for the triene side chain were absent, being replaced by signals consistent with the presence of a second 1,2,4-trisubstituted aromatic ring. COSY and HMBC correlations (Fig. S3†) were consistent with the biphenyl structure 39 (Scheme 4). We propose that this novel compound is produced by an alternative rearrangement of the key epoxide intermediate 37 (cf.19 in Scheme 1b) where the initial carbocation intermediate from epoxide ring opening undergoes electrophilic addition to the C-6/C-7 double bond, to give the cyclohexenyl cation 38, which is then aromatised by proton loss and dehydration to give 39 which we have named pseudostrobilurin B. In support of this hypothesis is the fact that 37 must possess the predicted 4Z,6E geometry for the cyclisation to occur.
Scheme 4. Conversion of epoxide 37 to prestrobilurin B 39.

Further detailed analysis of LCMS chromatograms of B. lutea extracts revealed the presence of two new minor compounds (each ca. 1 mg L–1) with UV characteristics typical of strobilurin A and B. These were isolated and their structures 41 and 42 determined from their mass spectra and full NMR analysis (Fig. S4†). They were named strobilurin Y 41 and strobilurin Z 42 respectively. Their formation can be rationalised by methanolysis of the epoxide 40 derived by a P450 or similar mono-oxygenase-mediated oxidation of strobilurins A and B (Scheme 5). In order to rule out formation of these compounds from methanol used as solvent in the original LCMS purification, we repeated the isolation using acetonitrile in place of methanol. Their continued production confirmed that they are the result of enzymatic transformations.
Scheme 5. Formation of strobilurins Y 42 and Z 43via methanolysis of epoxide 40.

In addition, a number of other strobilurins previously isolated from B. lutea were observed after optimisation of fermentation conditions (Fig. S5†). Besides the major metabolites, strobilurins A 2 and B 3, these include strobilurins F1 43, F2 44, G 7, H 45 and bolineol 8. We also observed a number of very minor compounds which were identified by NMR and mass spectrometric characteristics. These are strobilurin C 46 and strobilurin I 47 previously isolated from Xerula longipes33 and an agaricus species34 respectively. Strobilurin C 46 appears to be the earliest prenylated strobilurin, presumably formed from strobilurin F1 43, whereas strobilurin I 47 is likely to be an intermediate to strobilurin G 7. The likely overall biosynthetic interrelationships are summarised in Scheme 6.
Scheme 6. Proposed biosynthetic inter-relationships among strobilurin related metabolites in B. lutea.

Precursor directed biosynthesis, in which analogues of natural substrates are fed to either WT or mutant producing cultures has proved an effective method for producing analogues of microbial natural products, particularly for producing halogenated analogues.35 The presence of halogens often has a beneficial effect for enhancing biological activities. For example, the presence of a halogen is required for bioactivity in the case of salinosporamide, neomangicols and rebeccamycin,36,37 their non-halogenated analogues being inactive.
As indicated above, fluorinated precursors can be efficiently incorporated into strobilurins. With this in mind we made a more systematic study aimed at producing novel fluorinated, chlorinated and brominated analogues. The outcomes of these experiments were dependent on the producing strain used. Our initial studies used B. lutea strain F24510. A series of fluorinated cinnamates and benzoates were fed both as the free acid and SNAC thiol esters. The products were observed by mass spectrometric and 1H and 19F NMR analysis of the extracts after partial purification by thin layer chromatography and the results are summarised in Fig. 2. Mono- and di-fluorinated analogues were incorporated and the structures confirmed by NMR analysis, but individual analogues were not always isolated. Similar results were obtained with both B. lutea F23523 and S. tenacellus, but variations were observed in that relative efficiencies of benzoate and cinnamate incorporations were sometimes reversed with benzoates being preferred by S. tenacellus.
Fig. 2. Strobilurin analogues produced by precursor-directed biosynthesis.

However, feeding of 2-, 3- and 4-fluorobenzoate/cinnamate gave the corresponding fluorostrobilurins 48, 36 and 49 in S. tenacellus in relative yields (compared to levels of strobilurin A) of 8–30% with 2-fluorostrobilurin 48 consistently the poorest yield. The 3,4 and 3,5-difluoro-analogues 50 and 51 were formed from the corresponding benzoates, and feeding 3-fluoro-4-methyl- and 3-fluoro-4-methoxybenzoates gave the corresponding fluorostrobilurins 52 and 53. Feeding of nicotinic acid gave the novel 3-aza-strobilurin A analogue 60 and supplementation of the medium with KBr produced the bromo-analogue 4-dechloro-4-bromostrobilurin B 56. Other attempts to introduce halogens, e.g. by supplementation with chloride, bromide or iodide, or by adding chloro- or bromobenzoates gave only trace amounts (by LCMS), at best, of alternatively halogenated compounds. Similar results were observed in feeding a range of aromatic analogues to squalestatin-producing cultures of Phoma sp.38 In this study a similar range of mono- and difluorinated benzoates were incorporated but no other halogens were accepted.
With B. lutea F23523, feeding of 2-fluorocinnamate gave an interesting result. Production of strobilurin A 2 was completely inhibited, the fermentation giving strobilurin B 3 along with about 50% 2-fluorostrobilurin B 48, 10% 2-fluorostrobilurin G 59, and about 1% 2-fluorostrobilurin H 57 (Fig. S6†). All structures were confirmed by NMR and mass spectrometric analysis. 3-Fluorocinnamate again produced 3-fluorostrobilurin A 36, 4-fluorostrobilurin B 55 and 4-fluorostrobilurin C 58. Finally 4-fluorocinnamate gave strobilurin A 2 along with 4-fluorostrobilurin A 49 only with no strobilurin B, presumably fluorine at C-4 inhibiting oxidative hydroxylation.
The inhibition of strobilurin A production with 2-fluorocinnamate reflected the result of another experiment. The simple triketide analogue 61 was chemically synthesised (Scheme S1†) as a potential substrate analogue for the epoxidase believed to be responsible for the rearrangement to give the methoxyacrylate toxiphore. On feeding to B. lutea it was rapidly metabolised (Fig. S7†) to give a new compound shown to be diol 62. The formation of 62 can be rationalised as summarised in Scheme 7 by a cycle of polyketide condensation, methylation and keto-reduction to give 63. Concerted decarboxylation and dehydration to triene 64 followed by epoxidation and hydrolysis gives diol 62, whose structure was established by 1D and 2D NMR analysis (Fig. S8†).
Scheme 7. Proposed biosynthetic conversion of acid 61 to diol 62.

In this experiment, strobilurin B 3 production remains unaffected but no strobilurin A 2 is detected. Time course experiments (Fig. S9†) clearly indicate that the triketide analogue 61 is rapidly converted to the diol and that the diol itself is responsible for inhibiting strobilurin A production. In both these experiments, feeding acid 61 and 2-fluorocinnamate, strobilurin B production is unaffected. This strongly suggests that the pathways to strobilurin A 2 and strobilurin B 3 are different and that strobilurin A 2 is not converted directly to strobilurin B 3, so that the pathways must diverge at an early stage. On feeding [2,3-13C2]-cinnamate to cultures inhibited as above, no restoration of strobilurin A 2 production is observed, but strobilurin B 3 is produced and labelled from the precursor. This suggest that in these experiments, incorporation post-benzoyl CoA into strobilurin A is being inhibited. This suggests that benzoyl CoA may first be hydroxylated to 3-hydroxybenzoyl CoA before elaboration into the strobilurin B pathway (Scheme 6).
Conclusions
The strobilurins and analogues are amongst the most commercially important fungal metabolites known. Despite this their biosynthesis has been relatively little studied since the initial work of Nerud et al.16 In this study we have demonstrated that phenylalanine is converted to cinnamate, and then onwards to benzoate which forms the starter unit for formation of a linear tetraketide, prestrobilurin A 18. The potential role of cinnamate as both the precursor to the starter unit and an assembly intermediate in its own right has been clarified. Prestobilurin A 18 undergoes oxidative rearrangement to form the core β-methoxyacrylate moiety, but no direct evidence for the process has yet been observed. However, indirect evidence for the intermediacy of an epoxide in the key rearrangement to form the methoxyacrylate toxiphore has been obtained here via the isolation pseudostrobilurin B 39 which is likely formed via the common epoxide intermediate 37. Precursor-directed biosynthesis experiments demonstrated that a wide range of mainly fluorinated starter units can be tolerated by the PKS to provide several halogenated strobilurin analogues. Finally and unexpectedly, the production of strobilurin A 2 can be selectively inhibited with no effect on strobilurin B 3 biosynthesis. This suggests that they are produced by parallel pathways and that strobilurin A 2 is not an intermediate to strobilurin B 3. Further work on the genetic and molecular basis for strobilurin biosynthesis is underway and will be reported elsewhere.
Experimental
Full experimental details are supplies in the ESI.†
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
AMS-S thanks the Commonwealth Scholarship Commission for a postgraduate scholarship. L-CH thanks BrisSynBio, the Centre for Synthetic Biology (BB/L01386X/1) for fellowship support. RN thanks the Directorate General of Resources for Science and Higher Education (Beasiswa Pendidikan Pascasarjana Luar Negeri Direktorat Jenderal Sumber Daya Ilmu Pengetahuan dan Pendidikan Tinggi) (BPP-LN Ditjen SD Iptek-Dikti), Republic of Indonesia, for an Overseas Postgraduate Scholarship and the Alumni foundation of the University of Bristol for financial support. ZI thanks the Higher Education Commission of Pakistan for a post-graduate scholarship. RJC thanks EPSRC (EP/F066104/1) and DFG (INST 187/621-1) for LCMS equipment. GT and AZ are grateful to Prof. Tim Anke for providing us with Strobilurus tenacellus. Dr Y. O'Connell is thanked for synthesis of labelled cinnamate.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob00608c
‖The 2Z isomer 33 was also synthesised and fed as a 5 : 1 mixture with the 2E isomer. A small incorporation into 36 was observed, but this is likely to have been due to the small amount of E-isomer present in the fed mixture.
References
- Anke T. Can. J. Bot. 1995;73:940–945. [Google Scholar]
- Musílek V., Černá J., Šašek V., SemerdŽieva M., Vondráček M. Folia Microbiol. 1969;14:377–388. doi: 10.1007/BF02872707. [DOI] [PubMed] [Google Scholar]
- Sauter H., Steglich W., Anke T. Angew. Chem. 1999;38:1328–1349. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1328::AID-ANIE1328>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Anke T., Oberwinkler F., Steglich W., Schramm G. J. Antibiot. 1977;10:806–810. doi: 10.7164/antibiotics.30.806. [DOI] [PubMed] [Google Scholar]
- Anke T., Hecht H. J., Schramm G., Steglich W. J. Antibiot. 1979;32:1112–1117. doi: 10.7164/antibiotics.32.1112. [DOI] [PubMed] [Google Scholar]
- Schramm G., Steglich W., Anke T., Oberwinkler F. Chem. Ber. 1978;111:2779–2784. [Google Scholar]
- Engler M., Anke T., Klostermeyer D., Steglich W. J. Antibiot. 1995;48:884–885. doi: 10.7164/antibiotics.48.884. [DOI] [PubMed] [Google Scholar]
- Fredenhagen A., Hug P., Peter H. H. J. Antibiot. 1990;43:661–666. doi: 10.7164/antibiotics.43.661. [DOI] [PubMed] [Google Scholar]
- Bedford C. T., Perry D., Sharma R. K. Nat. Prod. Res. 2008;22:1535–1539. doi: 10.1080/14786410802176737. [DOI] [PubMed] [Google Scholar]
- Becker W. F., Von Jagow G., Anke T., Steglich W. FEBS Lett. 1981;132:329–333. doi: 10.1016/0014-5793(81)81190-8. [DOI] [PubMed] [Google Scholar]
- Clough J. M. Nat. Prod. Rep. 1993;10:565–574. doi: 10.1039/np9931000565. [DOI] [PubMed] [Google Scholar]
- Bartlett D. W., Clough J. M., Godwin J. R., Hall A. A., Hamer M., Parr-Dobrzanski B. Pest Manage. Sci. 2002;58:649–662. doi: 10.1002/ps.520. [DOI] [PubMed] [Google Scholar]
- PhillipsMcDougall Product Directory, 2015
- Trowitzsch W., Reifenstahl G., Wray V., Gerth K. J. Antibiot. 1980;23:1480–1490. doi: 10.7164/antibiotics.33.1480. [DOI] [PubMed] [Google Scholar]
- Sasse F., Leibold T., Kunze B., Höfle G., Reichenbach H. J. Antibiot. 2003;56:827–831. doi: 10.7164/antibiotics.56.827. [DOI] [PubMed] [Google Scholar]
- Nerud P., Sedmera P., Zouchova Z., Musílek V., Vondráček M. Collect. Czech. Chem. Commun. 1982;47:1020–1025. [Google Scholar]
- Fredenhagen A., Kuhn A., Peter H. H., Cuomo V., Giuliano U. J. Antibiot. 1990;43:655–660. doi: 10.7164/antibiotics.43.655. [DOI] [PubMed] [Google Scholar]
- Hertweck C., Moore B. S. Tetrahedron. 2000;56:9115–9120. [Google Scholar]
- Xian L., Moore B. S. J. Bacteriol. 2003;185:399–404. doi: 10.1128/JB.185.2.399-404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloglu E., Kingston D. G. I. J. Nat. Prod. 1999;62:1448–1472. doi: 10.1021/np990176i. [DOI] [PubMed] [Google Scholar]
- Bjorklund A. J., Leete E. Phytochemistry. 1992;31:3883–3887. [Google Scholar]
- Bonsch B., Belt V., Bartel C., Duensing N., Koziol M., Lazarus C. M., Bailey A. M., Simpson T. J., Cox R. J. Chem. Commun. 2016;52:6777–6780. doi: 10.1039/c6cc02130a. [DOI] [PubMed] [Google Scholar]
- Liu N., Hung Y.-S., Gao S.-S., Hang L., Zou Y., Chooi Y.-H., Tang Y. Org. Lett. 2017;19:3560–3563. doi: 10.1021/acs.orglett.7b01534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel J., Hertweck C., Shipley P. R., Hunt D. M., Newman M. S., Moore B. S. Chem. Biol. 2000;7:943–955. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- Hill A. M., Thompson B. L., Harris J. P., Segret R. Chem. Commun. 2003:1358–1359. doi: 10.1039/b303542p. [DOI] [PubMed] [Google Scholar]
- Yue S., Duncan J. S., Yamamoto Y., Hutchinson C. R. J. Am. Chem. Soc. 1987;109:1253–1255. [Google Scholar]
- Xian L., Moore B. S. J. Biol. Chem. 2002;277:32505–32509. doi: 10.1074/jbc.M204171200. [DOI] [PubMed] [Google Scholar]
- Cox R. J., Chooi Y.-H., Tang Y. Org. Biomol. Chem. J. Org. Chem. 2007;2012;577:2010–2026. 9933–9953. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- Simpson T. J. Nat. Prod. Rep. 2014;31:1247–1252. doi: 10.1039/c4np00065j. [DOI] [PubMed] [Google Scholar]
- Hashimoto M., Nonaka T., Fujii I. Nat. Prod. Rep. 2104;31:1306–1317. doi: 10.1039/c4np00096j. [DOI] [PubMed] [Google Scholar]
- McKeown D. S. J., McNicholas C., Simpson T. J., Willett N. J. J. Chem. Soc., Chem. Commun. 1996:301–302. [Google Scholar]
- Desjardins A. E., Plattner R. D., Beremand M. N. Appl. Environ. Microbiol. 1987;53:1860–1865. doi: 10.1128/aem.53.8.1860-1865.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anke T., Besl H., Mocek U., Steglich W. J. Antibiot. 1983;143:661–666. doi: 10.7164/antibiotics.36.661. [DOI] [PubMed] [Google Scholar]
- Zapf S., Anke T., Dasenbrock H., Steglich W. Bioengineering. 1993;1:92. [Google Scholar]
- Portmann C., Prestinari C., Myers T., Scharte J., Gademann K. Org. Biomol. Chem. 2009;7:644–646. [Google Scholar]
- Rodrigues P. E., Belin L., Sancelme M., Prudhomme M., Ollier M., Rapp M., Severe D., Riou J., Fabbro D., Mayer T. J. Med. Chem. 1996;39:4471–4477. doi: 10.1021/jm9603779. [DOI] [PubMed] [Google Scholar]
- Renner M. K., Jensen P. R., Fenical W. J. Org. Chem. 1998;63:8346–8354. doi: 10.1021/jo000081h. [DOI] [PubMed] [Google Scholar]
- Cannell R. J. P., Dawson M. J., Hale R. S., Hall R. M., Noble D., Lynn S., Taylor N. L. J. Antibiot. 1993;46:1381–1389. doi: 10.7164/antibiotics.46.1381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.