

Abstract
Calcium-triggered exocytosis is key to many physiological processes, including neurotransmitter and hormone release by neurons and endocrine cells. Dozens of proteins regulate exocytosis, yet the temporal and spatial dynamics of these factors during vesicle fusion remain unclear. Here we use total internal reflection fluorescence microscopy to visualize local protein dynamics at single sites of exocytosis of small synaptic-like microvesicles in live cultured neuroendocrine PC12 cells. We employ two-color imaging to simultaneously observe membrane fusion (using vesicular acetylcholine ACh transporter tagged to pHluorin) and the dynamics of associated proteins at the moments surrounding exocytosis. Our experiments show that many proteins, including the SNAREs syntaxin1 and VAMP2, the SNARE modulator tomosyn, and Rab proteins, are preclustered at fusion sites and rapidly lost at fusion. The ATPase N-ethylmaleimide–sensitive factor is locally recruited at fusion. Interestingly, the endocytic Bin-Amphiphysin-Rvs domain–containing proteins amphiphysin1, syndapin2, and endophilins are dynamically recruited to fusion sites and slow the loss of vesicle membrane-bound cargo from fusion sites. A similar effect on vesicle membrane protein dynamics was seen with the overexpression of the GTPases dynamin1 and dynamin2. These results suggest that proteins involved in classical clathrin-mediated endocytosis can regulate exocytosis of synaptic-like microvesicles. Our findings provide insights into the dynamics, assembly, and mechanistic roles of many key factors of exocytosis and endocytosis at single sites of microvesicle fusion in live cells.
INTRODUCTION
Exocytosis is the cellular process in which cytoplasmic membrane-bound vesicles fuse with the plasma membrane and release their contents into the extracellular space. During synaptic transmission, action potentials depolarize the presynaptic terminal triggering Ca2+ influx into the cell. Local elevations in intracellular Ca2+ cause synaptic vesicles (SVs) in the terminal to fuse with the plasma membrane, releasing neurotransmitters into the synaptic cleft (Jahn and Fasshauer, 2012). SV exocytosis is a carefully orchestrated process that involves multiple steps and dozens of proteins. SVs are small, ∼50 nm in diameter, and contain a repertoire of proteins on their membranes (Takamori et al., 2006). These vesicular membrane proteins and several cytoplasmic and plasma membrane-associated proteins play important roles in regulating SV exocytosis (Sudhof, 2013d).
Specifically, SNARE proteins are thought to drive SV fusion with the plasma membrane (Sudhof and Rothman, 2009). The vesicular SNARE, synaptobrevin (VAMP), and the plasma membrane SNAREs, syntaxin and synaptosomal–associated protein 25 (SNAP25), form a four-helical zippered complex that pulls the two lipid bilayers together resulting in fusion. The SNAREs are sufficient for fusion in vitro (van den Bogaart et al., 2010). However, under physiological conditions, several proteins, such as Rabs and their effector molecules (Fukuda, 2008), complexin (Trimbuch and Rosenmund, 2016), the Ca2+ sensor synaptotagmin (Sudhof, 2013a), tomosyn (Ashery et al., 2009; Bielopolski et al., 2014), Ca2+-dependent activator protein for secretion (CAPS) (Stevens and Rettig, 2009), Munc18, and Munc13 (Sudhof and Rothman, 2009), have been proposed to regulate the steps leading to fusion, including docking (attaching the SV to the active zone) and priming (preparing the SV for fusion) (Sudhof, 2013d). While much is known about the biochemical properties of these proteins and their physiological effects, a comprehensive understanding of their spatial and temporal dynamics during SV exocytosis in live cells is lacking, partly due to the small size of the SVs and the challenges associated with labeling and imaging single vesicles in synaptic terminals (Kavalali and Jorgensen, 2014). Understanding the dynamics of these key mediators of SV exocytosis will provide direct insights into their regulatory functions, biological mechanisms, and roles in disease.
Neuroendocrine PC12 cells have two distinct Ca2+-triggered exocytic vesicle pools, ∼50-nm-diameter synaptic-like microvesicles (SLMVs) and the larger ∼150-nm-diameter dense core vesicles (DCVs) (Thomas-Reetz and De Camilli, 1994). Both vesicle populations fuse with the plasma membrane with a rise in intracellular Ca2+. However, each vesicle-type exhibits distinct Ca2+ sensitivities and fusion kinetics and is responsible for the release of different signals (Ninomiya et al., 1997). Specifically, DCVs release peptide neurotransmitters, proteins, and amines while microvesicles release the chemical neurotransmitter acetylcholine (ACh) or γ-aminobutyric acid. Furthermore, SLMVs are stimulated at much lower intracellular Ca2+ concentrations, allowing for a more rapid burst of fusion during depolarization (Ninomiya et al., 1997). SLMVs are structurally and functionally similar to neuronal SVs (Thomas-Reetz and De Camilli, 1994). They maintain an acidic pH, accumulate neurotransmitters, and fuse with the plasma membrane to release their luminal contents in a Ca2+-dependent manner. Furthermore, they contain many of the same proteins required for cargo packaging, transport, exocytosis, and recycling, but unlike SVs, SLMVs do not appear clustered at active zones via synapsins (Thomas-Reetz and De Camilli, 1994). Aside from the interest in their signaling functions in endocrine cells, these vesicles have been used as experimentally tractable surrogates for the study of SV behavior (de Wit et al., 2001; Brauchi et al., 2008; Sochacki et al., 2012). Here we used total internal reflection fluorescence (TIRF) microscopy to monitor the dynamics of over two dozen proteins during Ca2+-triggered exocytosis of single SLMVs in PC12 cells (Sochacki et al., 2012; Trexler et al., 2016). In an imaging-based screen examining key exocytic and endocytic proteins, we show that many proteins, including syntaxin, VAMP, tomosyn, and Rab GTPases, are preclustered at fusion sites and rapidly diffuse away following exocytosis. Interestingly, Bin-Amphiphysin-Rvs (BAR) domain–containing proteins, and dynamin, known to be important in clathrin-mediated endocytosis, are recruited during fusion and influence the loss of vesicle membrane proteins from the fusion sites. Our study provides insights into the local dynamics, assembly, and function of key regulators of exocytosis and endocytosis during microvesicle fusion in live cells.
RESULTS
To image microvesicle fusion in PC12 cells, we expressed the vesicular ACh transporter (VAChT) tagged on its luminal side to pHluorin, a pH-sensitive variant of green fluorescent protein (GFP) (Miesenbock et al., 1998). VAChT is targeted specifically to SLMVs in PC12 cells (Liu and Edwards, 1997), and VAChT-pHluorin has been used to track SLMV exocytosis (Brauchi et al., 2008; Sochacki et al., 2012). pHluorin fluorescence is quenched in the acidic lumen of the vesicle, but on fusion the lumenal pH is neutralized by the extracellular buffer causing pHluorin signal to dramatically increase, enabling the detection of exocytic events. To stimulate exocytosis, we depolarized cells by applying buffer containing high extracellular KCl using a superfusion pipette positioned close to the cell (Trexler et al., 2016; Trexler and Taraska, 2017). Membrane depolarization caused a substantial increase in the frequency of fusion events as shown in Supplemental Figure 1A. Fusion events were detected as sudden bright flashes of green fluorescence (Figure 1A). This is observed as a local sharp increase in signal that decays with time as VAChT-pH diffuses away from the sites of exocytosis (Figure 1A, bottom, and Supplemental Figure 1B). In the red fluorescent channel, we monitored coexpressed proteins fused to mCherry, mRFP, or mKate2 (Supplemental Figure 1D table). In the example shown in Figure 1, VAChT-pH was coexpressed with mRFP-Rab3A (Figure 1B), where measurable changes in signal were detected during fusion (Figure 1B, bottom). Background-subtracted fluorescence intensities from hundreds of individual fusion events were extracted, normalized, and averaged in the green and red channels for every protein examined in the study to produce the average time-dependent changes in local protein signals at fusion sites (Figure 1, C and D). We analyzed ∼8000 fusion events from over 300 cells to track, quantitate, and characterize the dynamics of dozens of proteins (Supplemental Figure 1D table). To verify that fusion events predominantly occurred from microvesicles in our experimental system, we stimulated PC12 cells coexpressing VAChT-pHuji and neuropeptide Y (NPY) tagged to GFP (to label large dense core vesicles [LDCVs]) (Shen et al., 2014; Martineau et al., 2017). As expected, VAChT-labeled fusion events were detected in the pHuji channel (23 events in four cells), but not in the NPY-GFP channel, and no specific increase in GFP signal was measured in regions corresponding to VAChT-labeled events (Supplemental Figure 1C). These results further confirm that VAChT is a specific marker of exocytic SLMVs in PC12 cells.
FIGURE 1:

Analysis of protein dynamics at SLMV fusion sites in PC12 cells. (A, B) Images of a PC12 cell transfected with VAChT-pH (A) and mRFP-Rab3A (B) imaged using TIRF microscopy. Arrows (yellow) show a fusion event in the green channel and the corresponding region in the red channel, after application of stimulation buffer. Bar, 5 μm. Middle, snapshots of the fusion event shown above at the indicated time points. Time-point “0 s” indicates the manually identified first frame of brightening in the green channel. Circles (∼1 µm diameter) represent regions used for intensity analysis. Bar, 1 μm. Bottom, time-lapse traces of normalized fluorescence intensities for the event shown above in the green and red channels. (C, D) Average time-lapse traces of normalized fluorescence intensities for VAChT-pH (green) and mRFP-Rab3A (magenta) (196 events, five cells). Individual event traces were time aligned to 0 s, which corresponds to the fusion frame in the green channel. Standard errors are plotted as shaded areas around the average traces. ***p ≤ 0.0001 when compared with baseline.
Rab GTPases and effectors are rapidly lost from sites of SLMV exocytosis
We first examined the dynamics of the Rab family of GTPases and Rab effector molecules, which play important roles in targeting and docking synaptic vesicles to the plasma membrane (Fukuda, 2008). In PC12 cells, Rab27A and Rab3A were localized at exocytic sites before fusion and diffused away rapidly following exocytosis, consistent with their vesicle membrane-anchored nature (Figure 2, A and B, and Supplemental Figure 2). The cytosolic Rab3A effector molecule, Rabphilin3A, also displayed similar localization and behavior (Figure 2C and Supplemental Figure 2). An early endosomal Rab, Rab5A (Woodman, 2000), showed some enrichment at fusion sites that slowly decreased following fusion (Figure 2D, Supplemental Figure 2). Rab27B did not exhibit specific localization at SLMVs or a change in intensity following fusion (Figure 2E and Supplemental Figure 2). These results demonstrate that the Rab proteins, Rab27A and Rab3A, and the effector Rabphilin3A, are targeted to microvesicle sites before exocytosis and are dynamically lost into the cytosol or plasma membrane following fusion.
FIGURE 2:
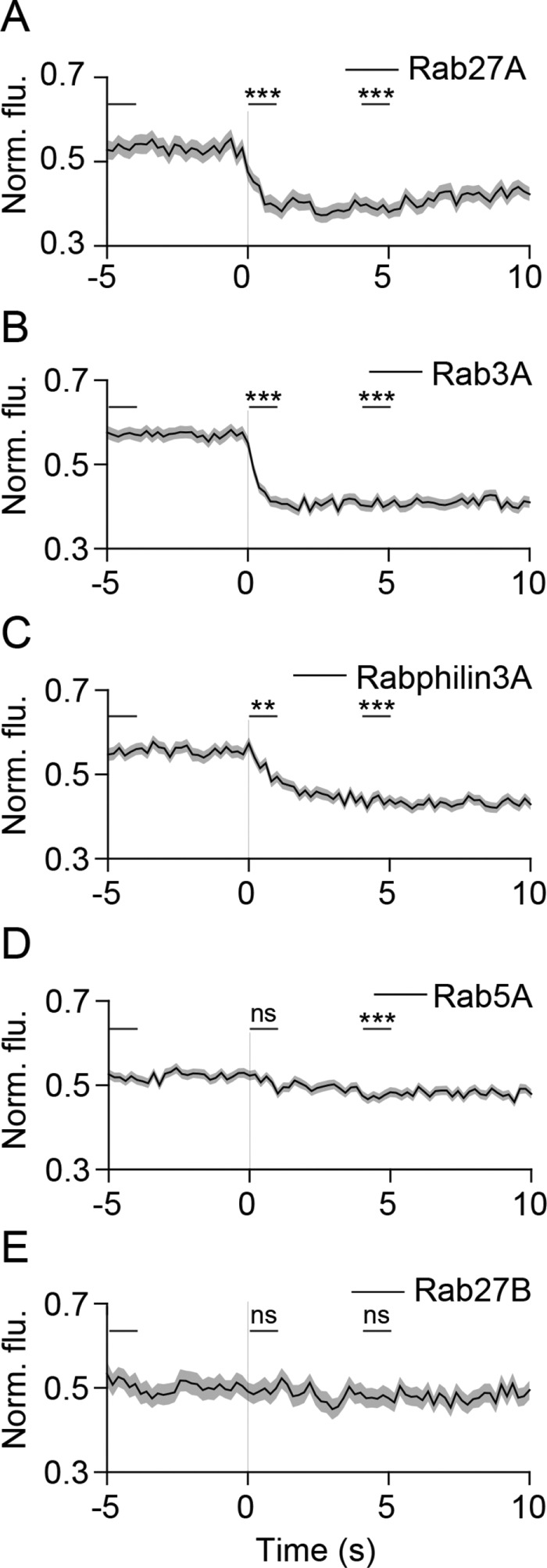
Dynamics of Rab proteins during SLMV fusion. (A–E) Average time-lapse traces of normalized fluorescence intensities for (A) mCherry-Rab27A (103 events, four cells), (B) mRFP-Rab3A (196 events, five cells), (C) mCherry-Rabphilin3A (198 events, six cells), (D) mCherry-Rab27B (73 events, three cells), and (E) mCherry-Rab5A (276 events, six cells). Individual event traces were time aligned to 0 s (vertical black line), which corresponds to the fusion frame in the green channel. Standard errors are plotted as shaded areas around the average traces. **p ≤ 0.01, ***p ≤ 0.0001, ns, not significant, when compared with baseline.
SNAREs, syntaxin1 and VAMP2, are clustered at fusion sites and lost following fusion
Docked vesicles fuse with the plasma membrane by the concerted action of the SNARE proteins syntaxin, SNAP25, and VAMP2 (Sudhof and Rothman, 2009). We found that the plasma membrane SNARE, syntaxin1, decreased in intensity at fusion sites during exocytosis (Figure 3A). Similar to the distribution seen in LDCVs (Lang et al., 2001; Barg et al., 2010; Gandasi and Barg, 2014; Ullrich et al., 2015), radial scan analysis of syntaxin1 fluorescence revealed locally elevated syntaxin1 that diffused away following fusion, indicating that syntaxin1 is clustered on the plasma membrane at sites of microvesicle exocytosis (Supplemental Figure 3). We did not observe reclustering of syntaxin1 at the original fusion sites (Supplemental Figure 3). The plasma membrane-attached SNARE, SNAP25, did not exhibit increased localization at fusion sites or a substantial change in signal or distribution during fusion (Figure 3B and Supplemental Figure 3). VAMP2 showed some concentration at fusion sites, consistent with its expression on the vesicular membrane, and diffused away following fusion (Figure 3C and Supplemental Figure 3). To rule out potential artifacts induced by the red fluorescent tag, we examined the distribution and dynamics of cytosolic mCherry and membrane-attached farnesyl-mCherry during fusion. Cytosolic mCherry was diffusely distributed both before and during fusion and showed a small increase in signal following fusion (Figure 3D and Supplemental Figure 3) likely due to mCherry diffusing into cytoplasmic space previously occupied by microvesicles (Taraska et al., 2003). Farnesyl-mCherry signal increased slightly during fusion, perhaps due to the delivery of a small amount of probe contained on the vesicle to the plasma membrane (Figure 3E and Supplemental Figure 3). Thus, the control mCherry proteins exhibited small changes in signal during fusion that are markedly different from the dynamics of syntaxin1 and VAMP2 described above. In conclusion, the SNAREs syntaxin1 and VAMP2 are locally preassembled at microvesicle fusion sites and diffuse away from the sites of release.
FIGURE 3:
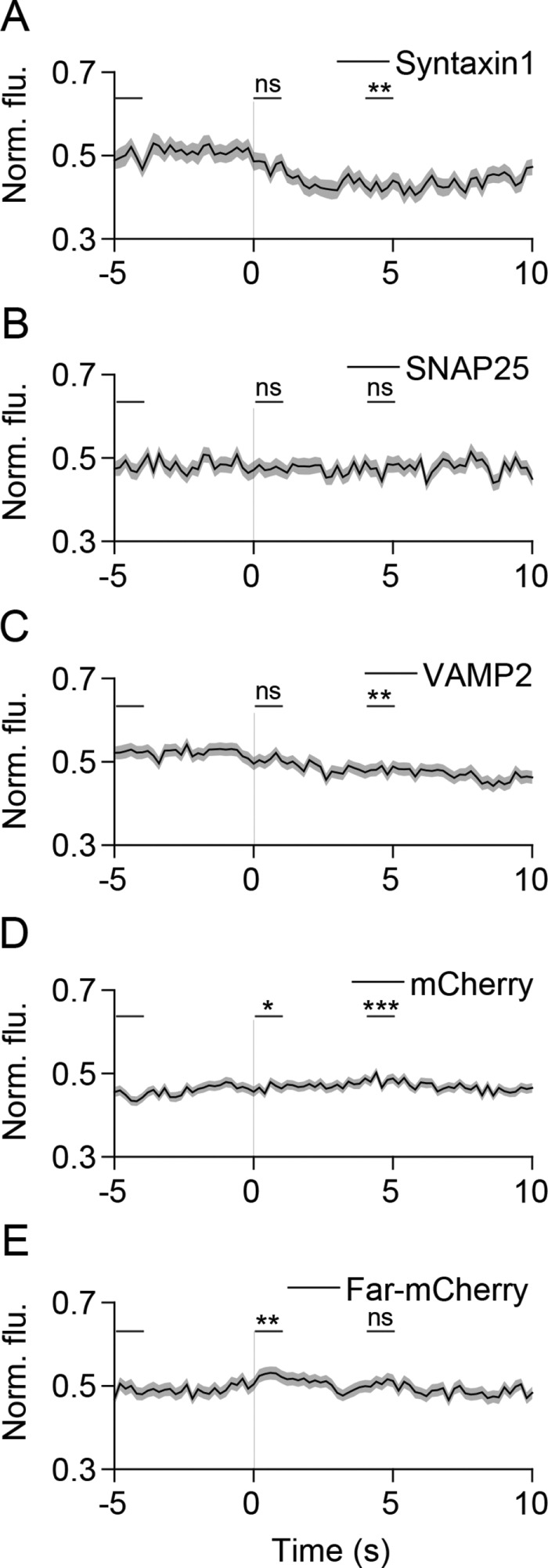
SNAREs syntaxin1 and VAMP2 diffuse away from sites of SLMV fusion. (A–E) Average time-lapse traces of normalized fluorescence intensities for (A) dCMV-mCherry-syntaxin1 (76 events, four cells), (B) dCMV-mCherry-SNAP25 (97 events, eight cells), (C) VAMP2-mCherry (172 events, 7 cells), (D) mCherry (274 events, eight cells), and (E) farnesyl-mCherry (166 events, nine cells). Individual event traces were time-aligned to 0 s (vertical black line), which corresponds to the fusion frame in the green channel. Standard errors are plotted as shaded areas around the average traces. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.0001, ns, not significant, when compared with baseline.
SNARE modulators exhibit diverse behaviors during SLMV fusion
SNARE-mediated fusion is regulated by several proteins (Sudhof, 2013d). To gain insights into the function of these SNARE modulators in live cells, we next examined their dynamics during SLMV fusion in PC12 cells. Multiple roles have been proposed for the small protein complexin, which is thought to either clamp the SNARE complex in a partially zippered state, or facilitate fusion (Brose, 2008; Yoon et al., 2008; An et al., 2010; Wragg et al., 2013). Complexin2 signal decreases slightly during fusion (Figure 4A); however, most of the protein appeared diffusely distributed before and after fusion (Supplemental Figure 5). A small decrease in signal was also seen with CAPS (Figure 4B), a protein essential for SV priming (Jockusch et al., 2007). Unlike complexin2, CAPS displayed some preferential localization at fusion sites at rest (Supplemental Figure 5). The Ca2+ sensor synaptotagmin1 (Sudhof, 2013a) was concentrated at fusion sites but, surprisingly, did not diffuse away following fusion (Supplemental Figures 4A and 5). Tomosyn, thought to play a largely inhibitory role in SV priming (Gracheva et al., 2006; McEwen et al., 2006; Ashery et al., 2009; Bielopolski et al., 2014; Cazares et al., 2016), was located as clusters at vesicles and diffused away following fusion (Figure 4C and Supplemental Figure 5). Munc18a and Munc13, proteins proposed to bind the SNAREs and have essential roles in SV fusion (Sudhof and Rothman, 2009), did not exhibit significant changes in dynamics during fusion (Supplemental Figures 4, B and C, and 5). Interestingly, the ATPase N-ethylmaleimide–sensitive factor (NSF) was transiently recruited to fusion sites during exocytosis (Figure 4D and Supplemental Figure 5), consistent with its role in disassembling SNARE complexes (Ryu et al., 2016). Overall, SNARE modulators exhibited diverse and subtle behaviors, with some preassembled at fusion sites and lost following fusion (CAPS and tomosyn), some recruited to fusion sites (NSF), and others localized at fusion sites throughout fusion (synaptotagmin1). These data likely reflect the diverse functions of these distinct classes of proteins along with the transient and stepwise nature of their activities during microvesicle fusion.
FIGURE 4:
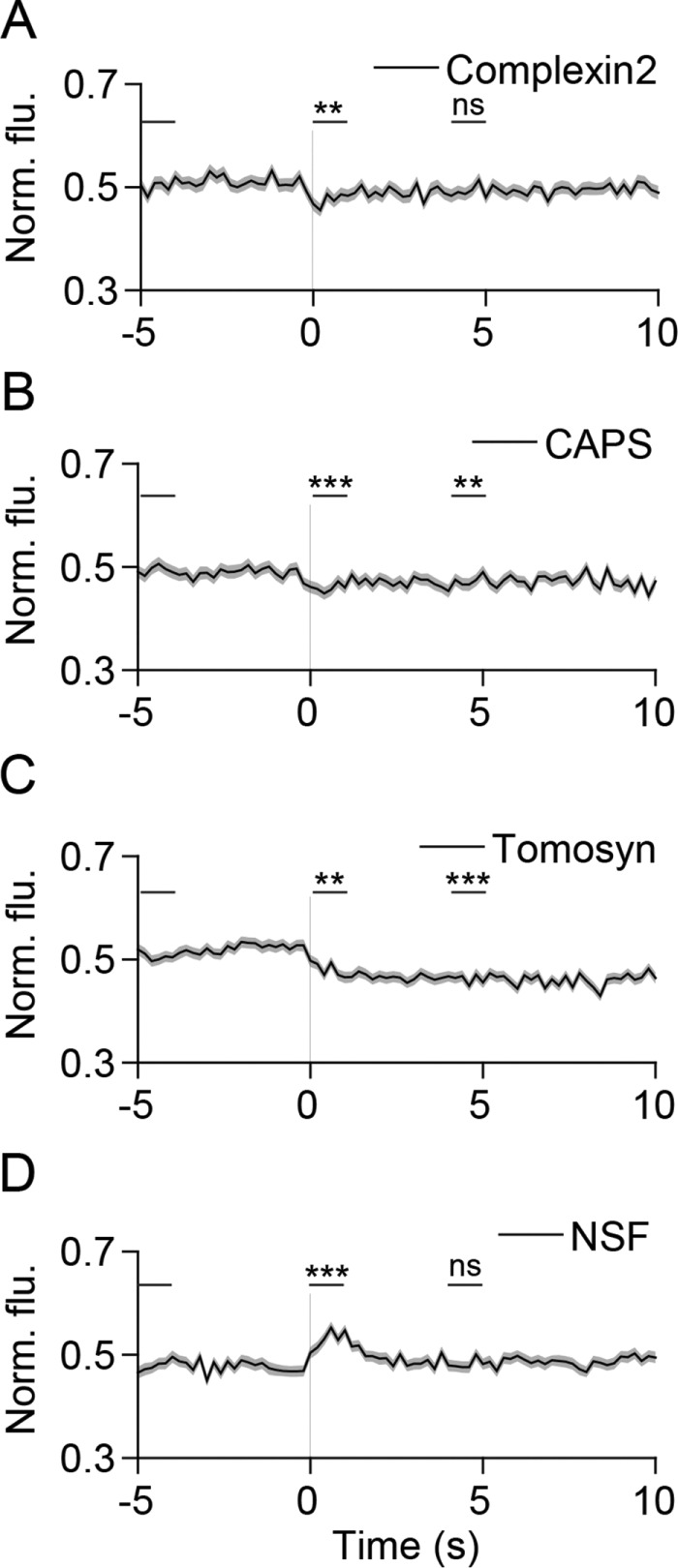
SNARE modulators exhibit diverse behaviors during SLMV fusion. (A–C) Average time-lapse traces of normalized fluorescence intensities for (A) complexin2-mCherry (202 events, seven cells), (B) CAPS-mKate2 (232 events, five cells), (C) mCherry-tomosyn (289 events, nine cells), and (D) NSF-mCherry (274 events, five cells). Individual event traces were time aligned to 0 s (vertical black line), which corresponds to the fusion frame in the green channel. Standard errors are plotted as shaded areas around the average traces. **p ≤ 0.01, ***p ≤ 0.0001, ns, not significant, when compared with baseline.
BAR domain proteins are recruited to sites of SLMV exocytosis
Previous studies have shown that the proteins amphiphysin, syndapin, and dynamin, involved in the maturation and scission of clathrin-coated endocytic structures (McMahon and Boucrot, 2011; Daumke et al., 2014), are recruited to exocytic sites (Tsuboi et al., 2004; Jaiswal et al., 2009; Trexler et al., 2016) and regulate Ca2+-dependent cargo release from LDCVs in endocrine cells (Graham et al., 2002; Holroyd et al., 2002; Tsuboi et al., 2004; Min et al., 2007; Fulop et al., 2008; Llobet et al., 2008; Anantharam et al., 2010, 2011; Samasilp et al., 2012; Trexler et al., 2016) and constitutive secretion from post-Golgi vesicles (Jaiswal et al., 2009). Because SLMVs are approximately fivefold smaller than LDCVs and likely have different curvatures, we hypothesized that a distinct set of proteins might be recruited (Xu and Xu, 2008; Zhang and Jackson, 2010). We first imaged the dynamics of the curvature sensing/inducing BAR domain proteins amphiphysin1 and syndapin2. Both proteins were specifically recruited to microvesicles during exocytosis (Figure 5 and Supplemental Figure 6A). BAR domain proteins endophilinA1 and endophilinA2 were also recruited during fusion (Supplemental Figure 7, A and B). EndophilinB1, however, appeared localized at fusion sites even at rest and did not exhibit significant changes in dynamics during fusion (Supplemental Figure 7D). Overall, these results indicate that specific BAR domain proteins are recruited to SLMVs at exocytosis in neuroendocrine cells.
FIGURE 5:

Amphiphysin1 and syndapin2 are transiently recruited to SLMV fusion sites. (A, B) Images of PC12 cells expressing (A) VAChT-pH and amphiphysin1-mCherry or (B) VAChT-pH and syndapin2-mCherry. Arrowheads show fusion events in the green channel and the corresponding regions in the red channel, after application of stimulation buffer. Bar, 5 μm. Middle, snapshots of the fusion event shown above at the indicated time points. Time-point “0 s” indicates the first frame of brightening in the green channel. Bar, 1 μm. Bottom, time-lapse traces of normalized fluorescence intensities in the green and red channels for the fusion event shown above. (C, D) Average time-lapse traces of normalized fluorescence intensities in the red channel for (C) Amph1-WT (371 events, nine cells) and (D) Synd2-WT (228 events, six cells). Individual event traces were time-aligned to 0 s (vertical black line), which corresponds to the fusion frame in the green channel. Standard errors are plotted as shaded areas around the average traces. ***p ≤ 0.0001 when compared with baseline.
To determine whether amphiphysin1 and syndapin2 recruitment to the site of fusion is driven by the curvature-sensitive BAR domains (Daumke et al., 2014), we imaged mutants lacking BAR domains. Unlike the wild-type (WT) proteins, Amph1-ΔBAR and Synd2-ΔBAR did not show specific localization at fusion sites during exocytosis (Figure 6, A–D, and Supplemental Figure 6A), indicating that the BAR domain is required for amphiphysin1 and syndapin2 recruitment. Deletion of the protein–protein interaction domain (SH3) (David et al., 1996; Grabs et al., 1997; Qualmann et al., 1999) in amphiphysin1 (Amph1-ΔSH3) and syndapin2 (Synd2-ΔSH3) did not prevent recruitment (Figure 6, E and F, and Supplemental Figure 6A), indicating that the SH3 domain is not required for their localization to SLMVs during fusion. Moreover, the BAR domain of syndapin2 (Synd2-BAR) showed strong recruitment (Figure 6H and Supplemental Figure 6A), whereas overexpression of the SH3 domain alone of amphiphysin1 (Amph1-SH3) did not show recruitment to fusion sites (Figure 6G and Supplemental Figure 6A). Thus, amphiphysin1 and syndapin2 recruitment is dependent on BAR domains, suggesting that these proteins could be targeted to the highly curved neck of the expanding fusion pore. Interestingly, recruitment appeared to slightly precede fusion (Figure 6), suggesting a possible localization of these proteins to a transient hemifusion state (Zhao et al., 2016). Also, the retention of these proteins at fusion sites seconds after fusion (Figure 6 and Supplemental Figure 6A) suggests either the persistence of a curved neck of the fusion pore or the rapid formation of endocytic structures at or near the fusion sites (Watanabe et al., 2013a, c). Radial scan analysis shows diffusional spread of VAChT-pH following fusion in the presence of Synd2-BAR (Supplemental Figure 6B), suggesting that at least some vesicular proteins can leave the sites of microvesicle exocytosis.
FIGURE 6:

Amphiphysin1 and syndapin2 recruitment to SLMV fusion sites is dependent on the BAR domain. (A–H) Average time-lapse traces of normalized fluorescence intensities in the red channel for (A) Amph1-WT (371 events, nine cells), (B) Synd2-WT (228 events, six cells), (C) Amph1-ΔBAR (169 events, four cells), (D) Synd2-ΔBAR (136 events, 11 cells), (E) Amph1-ΔSH3 (214 events, 13 cells), (F) Synd2-ΔSH3 (184 events, nine cells), (G) Amph1-SH3 (117 events, eight cells), and (H) Synd2-BAR (418 events, 13 cells). Individual event traces were time-aligned to 0 s (vertical black line), which corresponds to the fusion frame in the green channel. Standard errors are plotted as shaded areas around the average traces. **p ≤ 0.01, ***p ≤ 0.0001, ns, not significant, when compared with baseline.
BAR domain proteins modulate loss of SLMV membrane cargo from fusion sites
We next investigated whether the recruitment of BAR domain proteins described above impacts the loss of vesicle membrane cargo from fusion sites by measuring VAChT-pH fluorescence decay (Sochacki et al., 2012). Expression of Amph1-WT did not significantly alter VAChT-pH loss when compared with farnesyl-mCherry (Supplemental Figures 8A and 10G, τ = 2.39 ± 0.04 s vs. τ = 2.33 ± 0.04 s). However, overexpression of Amph1-ΔBAR and Amph1-SH3, proteins that were not recruited to fusion sites (Figure 6 and Supplemental Figure 6), resulted in faster VAChT-pH decay when compared with WT (Figure 7A and Supplemental Figure 10G, τ = 1.36 ± 0.06 s, τ = 1.88 ± 0.08 s). On the other hand, overexpression of Amph1-ΔSH3, which was recruited to fusion sites (Figure 6 and Supplemental Figure 6), resulted in slower VAChT-pH decay (Figure 7A and Supplemental Figure 10G, τ = 3.12 ± 0.05 s). These results suggest that amphiphysin1 recruitment during SLMV fusion slows the loss of vesicle membrane proteins from fusion sites.
FIGURE 7:

Amphiphysin1 and syndapin2 mutants slow the loss of VAChT-pH from fusion sites. (A) Average time-lapse traces of normalized VAChT-pH fluorescence intensities in PC12 cells coexpressing VAChT-pH and (A) WT or mutant amphiphysin1 and (B) WT or mutant syndapin2 constructs. Individual event traces were time aligned to 0 s, which corresponds to the frame of fusion. Standard errors are plotted as shaded areas around the average traces.
We obtained similar results with syndapin2. Like Amph1-WT, expression of Synd2-WT did not alter VAChT-pH decay when compared with farnesyl-mCherry (Supplemental Figures 8B and 10G, τ = 2.28 ± 0.04 s vs. τ = 2.33 ± 0.04 s). Synd2-ΔBAR, which was not recruited to fusion sites (Figure 6 and Supplemental Figure 6), exhibited VAChT decay slightly slower than Synd2-WT (Figure 7B and Supplemental Figure 10G, τ = 2.63 ± 0.05). However, Synd2-ΔSH3 and Synd2-BAR, which showed strong and specific recruitment to fusion sites (Figure 6 and Supplemental Figure 6), substantially slowed down VAChT decay (Figure 7B and Supplemental Figure 10G, τ = 3.43 ± 0.07 s, τ = 2.94 ± 0.04 s). Thus, the lack of the SH3 domain in amphiphysin1 and syndapin2 further slows down the loss of membrane cargo when compared with WT (Figure 7). Furthermore, the BAR domain proteins endophilinA1, endophilinA2 and endophilinB1 that showed recruitment or localization at fusion sites (Supplemental Figure 7), resulted in significantly slower VAChT-pH decay when compared with farnesyl-mCherry (Supplemental Figures 8C and 10G, τ = 3.24 ± 0.04 s, τ = 3.07 ± 0.06 s, τ = 4.16 ± 0.07 s). Intriguingly, the VAChT-pH decay with farnesyl-mCherry was slower than with cytosolic mCherry (Supplemental Figures 8D and 10G, τ = 2.33 ± 0.04 s vs. 2.07 ± 0.10 s). It is possible that the small increase in farnesyl-mCherry seen during exocytosis (Figure 3 and Supplemental Figure 3) alters membrane properties including fluidity or packing to slow down cargo release (Stachowiak et al., 2012). Because we are interested in the impact of membrane-associated BAR domain proteins on fusion dynamics, we chose to compare the resulting VAChT-pH decay profiles with that seen with farnesyl-mCherry (Supplemental Figure 8). Overall, our results indicate that BAR domain proteins slow the loss of vesicle membrane cargo from fusion sites during SLMV exocytosis.
We further examined the effects of knock-down of endogenous amphiphysin1 and syndapin2 on VAChT-pH loss by treating cells with small interfering RNA (siRNA) against these molecules. Western blot analysis revealed syndapin2 expression in PC12 cells, which was reduced with siRNA treatment (Supplemental Figure 9A). We were unsuccessful in our attempts to detect endogenous amphiphysin1 using commercial antibodies (unpublished data). Contrary to our expectation, the VAChT-pH decay in PC12 cells treated with siSyndapin2 was slower than that with control siRNA (Supplemental Figures 9B and 10G, τ = 4.01 ± 0.06 s vs. τ = 3.15 ± 0.07 s). It is possible that syndapin2 knock-down resulted in increased recruitment of other BAR domain proteins to fusion sites, but this complication was not explored further. Nonetheless, our experiments with amphiphysin1 and syndapin2 mutants, and endophilins, provide evidence for the modulation of microvesicle membrane cargo dynamics by BAR domain proteins.
Dynamin is recruited to fusion sites and delays loss of SLMV membrane cargo
Expression of amphiphysin1 and syndapin2 mutants lacking the SH3 domains resulted in slower VAChT-pH decay than that seen with full-length proteins (Figure 7), suggesting a role for SH3 binding partners in hastening the loss of VAChT from fusion sites. To test this hypothesis, we examined the dynamics of the well-studied SH3 binding partner, the GTPase dynamin (David et al., 1996; Qualmann et al., 1999; Ferguson and De Camilli, 2012). During clathrin-mediated endocytosis (CME), dynamin localizes to the curved neck of the invaginating clathrin-coated pit through its interactions with BAR domain proteins and causes scission (Daumke et al., 2014). Dynamin1 and dynamin2 have also been shown to cluster at LDCV fusion sites (Holroyd et al., 2002; Tsuboi et al., 2004; Trexler et al., 2016) and regulate fusion pore expansion (Holroyd et al., 2002; Tsuboi et al., 2004; Min et al., 2007; Fulop et al., 2008; Anantharam et al., 2011; Samasilp et al., 2012; Gonzalez-Jamett et al., 2013; Fan et al., 2015; Trexler et al., 2016). We found that dynamin1 is transiently recruited to SLMVs during fusion (Supplemental Figure 10A) and slows down the loss of VAChT-pH when compared with control (Figure 8A and Supplemental Figure 10G, τ = 3.47 ± 0.05 s vs. τ = 2.33 ± 0.04 s). Dyn1-K44A, a GTPase mutant, was recruited to SLMV fusion sites (Supplemental Figure 10B), suggesting that the GTPase activity of dynamin1 is dispensable for its recruitment. Dyn1-K44A, however, hastened VAChT-pH decay (Figure 8B and Supplemental Figure 10G, τ = 2.09 ± 0.08 s). Thus, the recruitment of functional dynamin1 to SLMV fusion sites slows down loss of membrane cargo from the fusion site.
FIGURE 8:

Dynamin1 and dynamin2 slow the loss of VAChT-pH from fusion sites. (A–D) Average time-lapse traces of normalized VAChT-pH fluorescence intensities in PC12 cells coexpressing VAChT-pH and (A, B) WT or mutant dynamin1 and (C, D) WT or mutant dynamin2 constructs. Individual event traces were time aligned to 0 s, which corresponds to the frame of fusion. Standard errors are plotted as shaded areas around the average traces.
We next examined the effects of disrupting dynamin1’s interactions with BAR domain proteins by mutating residues in its proline-rich domain (PRD) (Okamoto et al., 1997). Dyn1 deficient in binding amphiphysin1 (Dyn1-833-838A, Dyn1-ΔAmph1) (Grabs et al., 1997) showed mild localization at fusion sites, whereas dynamin1 deficient in binding syndapin2 (Dyn1-S774E/S778E, Dyn1-ΔSynd2) (Anggono et al., 2006) exhibited substantial recruitment to SLMVs during fusion (Supplemental Figure 10, C and D). Both mutants, however, resulted in VAChT-pH decay that was comparable to that observed with Dyn1-WT (Figure 8B and Supplemental Figure 10G, τ = 3.68 ± 0.05 s, τ = 3.93 ± 0.07 s) and slower than with farnesyl-mCherry. Thus, our findings suggest that dynamin1 slows the loss of membrane cargo, and disrupting its interactions with either amphiphysin1 or syndapin2 does not lessen its effects.
Unlike dynamin1, dynamin2 (Dyn2-WT) did not show substantial recruitment to fusion sites (Supplemental Figure 10E). PC12 cells expressing Dyn2-WT, however, exhibited slower VAChT-pH decay when compared with farnesyl-mCherry (Figure 8C and Supplemental Figure 10G, τ = 3.56 ± 0.09 s vs. τ = 2.33 ± 0.04 s). This suggests that low levels of Dyn2-WT at fusion sites can affect cargo loss or that dynamin2 acts at a step prior to or distant from exocytosis. Dynamin2 lacking the PRD domain (Dyn2-ΔPRD) (Supplemental Figure 10F) resulted in faster loss of membrane cargo (Figure 8D and Supplemental Figure 10G, τ = 2.69 ± 0.06 s), suggesting that dynamin2’s interaction with BAR domain proteins is essential for this effect. Taken together, these results suggest that dynamin1 and dynamin2 slow the loss of SLMV membrane cargo from fusion sites. This is consistent with the observation that Amph1-SH3, which sequesters SH3 binding partners such as dynamin (Shupliakov et al., 1997; Wigge et al., 1997; Holroyd et al., 2002), results in faster VAChT-pH decay (Figure 7A). These results also suggest that the slower VAChT-pH decay seen in Amph1-ΔSH3 and Synd2-ΔSH3 mutants (Figure 7) lacking dynamin binding is likely due to a lack of interactions with SH3 binding partners other than dynamin.
Examination of the dynamics of neural Wiskott–Aldrich syndrome protein (N-WASP), a protein that binds SH3-domain containing proteins and stimulates actin polymerization during CME (Qualmann et al., 1999; Kessels and Qualmann, 2002; Yamada et al., 2009), did not reveal significant changes in signal or distribution during fusion (Supplemental Figure 11A) or a substantial change in the VAChT-pH decay when compared with control (Supplemental Figure 10G, τ = 2.64 ± 0.05 s, vs. τ = 2.33 ± 0.04 s). Interestingly, we observed a slow, but significant, increase in clathrin around fusion sites several seconds after fusion (Supplemental Figure 11B). Moreover, apart from the specific and transient recruitment of amphiphysin1 to fusion sites described earlier (Figure 5), in five of nine cells we observed a marked increase in amphiphysin1 clusters across the plasma membrane that peaked tens of seconds after stimulation and then disappeared (Supplemental Figure 12), suggesting compensatory endocytosis of released cargo or membrane, controlled by amphiphysin. This additional slow recruitment did not seem correlated to fusion sites. Last, the adaptor protein AP-2 and the scaffolding protein intersectin showed mild or no recruitment to fusion sites (Supplemental Figure 11, C and D), suggesting perhaps the presence of sufficient amounts of these proteins to effect endocytosis. Overall, our results support the model that BAR domain proteins and dynamin play important roles in modulating microvesicle membrane cargo dynamics in neuroendocrine cells.
DISCUSSION
Calcium-triggered exocytosis of SVs is a highly coordinated process involving dozens of proteins. The dynamics and assembly of these factors in live cells is not fully understood. Here we systematically analyzed the spatial and temporal dynamics of two dozen proteins around the moment of fusion of single SLMVs in living cells. Our experiments reveal distinct local dynamics of exocytic and endocytic proteins and a key role for BAR domain-containing proteins, along with dynamin, in modulating membrane cargo dynamics at microvesicle fusion sites.
First, we show that many proteins involved in SV exocytosis are concentrated at fusion sites several seconds before fusion (Lang et al., 2001; Tsuboi and Rutter, 2003; Barg et al., 2010; Gandasi and Barg, 2014; Ullrich et al., 2015; Trexler et al., 2016; Geerts et al., 2017). These include the SNAREs, VAMP2 and syntaxin1 (Figure 3); the Ca sensor synaptotagmin1 (Supplemental Figure 4); the SNARE modulators tomosyn and CAPS (Figure 4); Rab proteins Rab3A and Rab27A; and the Rab effector molecule Rabphilin3A (Figure 2) (Supplemental Figures 2, 3, and 5). Because Rab proteins and VAMP2 are known to be associated with the vesicle membrane, their presence before fusion indicates that many SLMVs are docked at the plasma membrane prior to exocytosis. We did not measure additional recruitment of these molecules before fusion, suggesting that many proteins needed in exocytosis are preassembled at the vesicle. All these factors, except for synaptotagmin1, diffused away within seconds of fusion (Figures 2–4 and Supplemental Figures 2–5), indicating the highly dynamic and transient nature of the docking and fusion complex.
Synaptotagmin1 remained localized at fusion sites after fusion (Supplemental Figure 5), raising the possibility that SLMV fusion is incomplete. The complete release of VAChT-pH (Figure 1) suggests that classic kiss-and-run (Alabi and Tsien, 2013) is not the predominant mode of SLMV exocytosis in PC12 cells (Sochacki et al., 2012). The residual VAChT-pH signal results from VAChT trapped in neighboring clathrin structures (Sochacki et al., 2012). We cannot, however, rule out a cavicapture-type fusion mechanism (Holroyd et al., 2002; Taraska et al., 2003; Tsuboi et al., 2004), where VAChT-pH is lost, but other components such as synaptotagmin1 are retained. Previous reports have suggested that synaptotagmin stays clustered after SV exocytosis (Willig et al., 2006). Given that synaptotagmin1 is an important cargo for CME, it is possible that it is internalized very close to fusion sites through interactions with local adaptor proteins (Haucke and De Camilli, 1999; Martina et al., 2001; McMahon and Boucrot, 2011). This is supported by the increase in clathrin at the plasma membrane seconds after fusion (Supplemental Figure 11).
We did not see clusters, or changes in the dynamics of SNAP25 at fusion sites (Figure 3 and Supplemental Figure 3). It is possible that high levels of endogenous SNAP25 prevent the concentration of overexpressed or labeled protein at fusion sites (Knowles et al., 2010). Surprisingly, we also did not see significant changes in signal for Munc18a and Munc13 (Supplemental Figures 4 and 5), molecules thought to be critical for vesicle docking and priming (Sudhof and Rothman, 2009; Gandasi and Barg, 2014), though Munc18a showed mild clustering at fusion sites (Supplemental Figure 5). This is different from the dynamics of these proteins observed with LDCVs in endocrine cells (Gandasi and Barg, 2014; Trexler et al., 2016). It is possible that overexpression of these proteins masked small changes in signal. But a Munc18a construct with diminished promoter activity and reduced background signal also failed to show changes during fusion (unpublished data). The position and nature of the fluorescent tag could also interfere with its function (Crivat and Taraska, 2012). Detecting small transient dynamics of all proteins during fusion in this system will require future technical developments.
Of note, we found that the ATPase NSF is recruited at exocytosis (Figure 4 and Supplemental Figure 5). NSF and its binding partner α-soluble NSF-attachment protein (α-SNAP) (Sollner et al., 1993; Ungermann et al., 1998; Ryu et al., 2016) are thought to disassemble the SNARE complexes into monomeric SNAREs making them available for subsequent rounds of fusion (Ryu et al., 2016). It is unclear whether NSF acts after fusion (Littleton et al., 2001) or immediately prior to fusion (Banerjee et al., 1996; Kuner et al., 2008). Here the NSF signal appears to increase at fusion sites near the beginning of fusion (Figure 4 and Supplemental Figure 5) and stays elevated for ∼2 s, suggesting that NSF activity is coupled to microvesicle exocytosis, both spatially and temporally.
Importantly, we show that curvature sensing BAR domain proteins and the GTPase dynamin are locally recruited to SLMV fusion sites. Specifically, we measure dynamic recruitment of amphiphysin1, syndapin2, endophilinA1, endophilinA2, and dynamin1 to SLMVs during fusion (Figures 5 and 6 and Supplemental Figures 6, 7, and 10). Amphiphysin1 and syndapin2 recruitment was dependent on their curvature sensing BAR domains (Figure 6 and Supplemental Figure 6), suggesting that these proteins are recruited to the curved membrane of the vesicle. EndophilinB1 appeared preclustered at fusion sites (Supplemental Figure 7), consistent with previous findings suggesting association of endophilins with SVs at rest in nerve terminals (Bai et al., 2010).
Furthermore, we show that the presence of BAR domain proteins and dynamin at fusion sites slows the loss of membrane cargo following exocytosis. Supporting this idea, overexpression of endophilinA1, endophilinA2, endophilinB1, dynamin1, and dynamin2 resulted in slower VAChT decay (Supplemental Figure 7 and Figure 8). Second, amphiphysin1 mutants that failed to assemble at fusion sites resulted in faster VAChT decay (Figure 7). Third, amphiphysin1 and syndapin2 mutants that lacked the SH3 domain but showed strong recruitment to fusion sites slowed down VAChT decay (Figure 7). Fourth, dynamin1 lacking its GTPase activity and, fifth, dynamin2 deficient in binding BAR domain proteins resulted in faster loss of VAChT at fusion sites (Figure 8). Amphiphysin1 and syndapin2 mutants lacking the SH3 domain (Figure 7) exhibited slower VAChT decay; however, the prominent SH3 binding proteins dynamin1 and dynamin2 resulted in even further delays (Figure 8), and N-WASP did not show significant recruitment (Supplemental Figure 11) or affect loss of VAChT from fusion sites, suggesting that interactions with other SH3 binding proteins (McPherson et al., 1994, 1996), lipids (Martin, 2015), or rearrangements in the cytoskeleton (Malacombe et al., 2006; Felmy, 2007; Wen et al., 2016; Gonzalez-Jamett et al., 2017) may be involved in modulating vesicle membrane cargo dynamics following fusion.
Because the loss of VAChT from fusion sites depends on both the rate of VAChT release from vesicles and the rate of VAChT diffusion in the plasma membrane, the delayed loss in the presence of BAR domain proteins and dynamins could be attributed to one or more of the following reasons. First, the recruitment of these proteins to the highly curved neck of the fusion pore could alter the structure of the pore and modulate cargo release. Such a model has been described previously for LDCVs (Wang et al., 2001; Llobet et al., 2008; Anantharam et al., 2010, 2011; Trexler et al., 2016). Specifically, in insulin-secreting beta cells, amphiphysin1, syndapin2, endophilinA2, dynamin1, and dynamin2 are recruited to LDCVs, and mutants of dynamin deficient in their interactions with BAR domain proteins hasten cargo release (Trexler et al., 2016). Moreover, in neurons, neurotransmitter release rates have been linked to the molecular machinery of the synaptic vesicle pore (Pawlu et al., 2004; Guzman et al., 2010). However, without a direct measure of microvesicle fusion pore properties in PC12 cells, it is unclear whether the recruitment of BAR domain proteins modulates membrane protein release and ACh release in a similar manner. Second, BAR domain proteins and dynamin could constrict the neck of the vesicle causing endocytosis in a cavicapture-type fusion model as described for LDCVs (Holroyd et al., 2002; Taraska et al., 2003; Taraska and Almers, 2004; Perrais et al., 2004; Tsuboi et al., 2004; Fulop et al., 2008) or alter the merger of the omega-profile with the plasma membrane (Chiang et al., 2014). Examination of radial scans reveals diffusion of VAChT away from fusion sites, even in the presence of the BAR domain (Supplemental Figure 6). It is possible that most of VAChT diffuses away, and the remaining vesicle components are endocytosed, or that there is heterogeneity in the vesicle population. The third possibility is that BAR domain proteins and dynamin are not recruited to the neck of the fusion pore but to sites very close to fusion but below the diffraction limit to effect endocytosis. This would be supported by the prolonged retention of BAR domain proteins at fusion sites (Supplemental Figure 6) and the delayed recruitment of clathrin (Supplemental Figure 11) and is consistent with previous studies demonstrating spatial and temporal proximity of exocytosis and compensatory endocytosis (Roos and Kelly, 1999; Sochacki et al., 2012; Watanabe et al., 2013a,c; Wu et al., 2014). And, last, BAR domain proteins and dynamin may have nonlocal effects on plasma membrane tension (Stachowiak et al., 2012), the cytoskeleton, or membrane fluidity, and affect fusion dynamics and/or diffusion of membrane cargo without directly modulating the fusion pore or endocytosis.
At the neuronal synapse, the association of endocytic proteins with SVs is thought to ensure the availability of these proteins for compensatory endocytosis (Shupliakov, 2009; Bai et al., 2010). Our findings that endocytic proteins are dynamically recruited to sites of fusing SLMVs, which are structurally and functionally similar to SVs (Thomas-Reetz and De Camilli, 1994), suggests that these proteins could play more active roles in modulating exocytosis, endocytosis, and the coupling between these two processes. Direct evaluation of the effects of endocytic protein recruitment on fusion pore dynamics and rapid compensatory endocytosis in neurons could provide insights into the complex regulation of synaptic membrane trafficking.
The protein dynamics observed at SLMV fusion sites is like that seen with the much larger LDCVs in endocrine cells. While SVs and LDCVs have broadly similar molecular requirements for fusion (Xu and Xu, 2008), differences in their size (∼50 vs. ∼150 nm diameter), Ca2+ dependencies (Verhage et al., 1991; Heidelberger et al., 1994; Ninomiya et al., 1997; Sudhof, 2013a), and latencies to fusion (Chow et al., 1992; Sabatini and Regehr, 1996) suggest differential regulation of the exocytic fusion machinery. Furthermore, their different curvatures, pore sizes (19 vs. 213 pS conductance) (Klyachko and Jackson, 2002), and pore stabilities (Zhang and Jackson, 2010) suggest distinct structures of the pore. However, in both SLMVs and LDCVs, syntaxin1 (Lang et al., 2001; Barg et al., 2010; Gandasi and Barg, 2014), VAMP (Tsuboi and Rutter, 2003; Trexler et al., 2016), Rab3A (Gandasi and Barg, 2014; Trexler et al., 2016), Rab27A, Rabphilin3A, CAPS, and tomosyn (Trexler et al., 2016) are concentrated at fusion sites and diffuse away following fusion, supporting parallels in the molecular assembly and disassembly of key components during LDCV and microvesicle exocytosis. Moreover, BAR domain proteins and dynamin are recruited to fusion sites of both SLMVs and LDCVs sites (Holroyd et al., 2002; Tsuboi et al., 2004; Trexler et al., 2016). Given the different sensitivities to membrane curvature among the various BAR domain proteins (Daumke et al., 2014) and dynamin isoforms (Yoshida et al., 2004; Liu et al., 2011), the differences in their assembly and effects on LDCV and SLMV exocytosis may be nuanced. For example, all three endophilins associated with fusing SLMVs, but only endophilinA2 localizes with LDCVs (Trexler et al., 2016). Future work at higher temporal or spatial resolutions comparing LDCV and microvesicle fusion in the same cells, under expression-controlled conditions, will help identify key differences in protein dynamics needed to regulate different pools of vesicles, their fusion kinetics, release of their unique cargos, and their roles in signaling and cellular communication.
MATERIALS AND METHODS
Cells and solutions
PC12-GR5 cells were grown in DMEM containing 4 mM l-glutamine, supplemented with 5% fetal bovine serum (FBS), 5% horse serum, and 1% penicillin/streptomycin at 37°C in 5% CO2. The cell line was originally obtained from Rae Nishi (Marine Biological Laboratory), expanded from low passage frozen stocks, and was not further authenticated. The cells tested negative for mycoplasma contamination. Cells were plated onto 25-mm, #1.5, round, poly-d-lysine–coated glass cover-slips and transfected ∼24–48 h later with 1 µg each of the indicated plasmids, or 25 nmol of siRNA (Dharmacon), using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. The list of plasmids used and the n values for all the experiments are listed in Supplemental Figure 1D. Cells were imaged ∼24–48 h after transfection. The imaging buffer contained (in mM): 130 NaCl, 2.8 KCl, 5 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose. The pH was adjusted to 7.4 with 1 N NaOH. The stimulation buffer contained (in mM): 50 NaCl, 105 KCl, 5 CaCl2, 1 MgCl2, 10 HEPES, and 1 NaH2PO4. The pH was adjusted to 7.4 with 5 M KOH.
TIRF microscopy
TIRF microscopy was done as previously described (Trexler et al, 2016; Trexler and Taraska, 2017). Cells were imaged on an inverted fluorescent microscope (IX-81, Olympus), equipped with a ×100, 1.45 NA objective (Olympus). Combined green (488 nm) and red (561 nm) lasers (Melles Griot) were controlled with an acousto-optic tunable filter (Andor) and passed through a LF405/488/561/635 dichroic mirror. Emitted light was filtered using a 565 DCXR dichroic mirror on the image splitter (Photometrics), passed through 525Q/50 and 605Q/55 filters and projected onto the chip of an electron-multiplying charge-coupled device (EMCCD) camera. Images were acquired using the Andor IQ2 software. Cells were excited with alternate green and red excitation light, and images in each channel were acquired at 100-ms exposure at 5 Hz. To trigger exocytosis, stimulation buffer was applied for 30–40 s using a µFlow perfusion system (ALA Scientific Instruments) with a 100-µm pipette positioned close to the surface of the cell. Each day before experiments, 100-nm yellow-green fluorescent beads (Invitrogen) were imaged in the green and red channels and superimposed by mapping corresponding bead positions. The green and red cell images were aligned postacquisition using projective image transformation as described before (Sochacki et al, 2012; Trexler et al, 2016). All experiments were carried out at 25°C.
Image analysis
Image analysis was performed using Metamorph (Molecular Devices), ImageJ (National Institutes of Health), and custom scripts on MATLAB (Mathworks). The coordinates of the brightest pixel in the first frame of brightening for individual fusion events in the green channel were identified by eye and assigned as the center of the fusion event. All events were time aligned to the first frame of fusion (0 s). A circular region of interest (ROI) of 6 pixels (∼990 nm) diameter and a square of 21 pixels (∼3.5 µm) were drawn around the center, and the mean intensities in the surrounding square, excluding the circular region, were subtracted from the mean intensities in the center for each fusion event. This analysis was done for every frame from –10 to +50 s relative to the first frame of fusion. The background subtracted time-lapse intensities in the green channel were normalized 0–1 for each event, with “0” being the mean prefusion intensity, obtained from –5 to –3 s, and “1” being the peak intensity. The background subtracted time-lapse intensities for each event in the red channel were also normalized 0–1, with “0” being the minimum and “1” the maximum value, within –10 to +50 s. The resulting normalized traces were averaged across all events and truncated to show data from –5 to +10 s to better represent the protein dynamics around the moment of fusion. For the radial scan analyses, the intensities along 32 overlapping lines of 3.5 µm length arranged in a star pattern were averaged for each frame. The resulting kymograph through time for each event was normalized from 0–1000 and averaged across all events. Local background subtraction was not performed for radial scan analyses. The VAChT decay constants were obtained by single exponential fits of average VAChT fluorescence from 0.2 s to 5 s in the time-lapse traces, with y-offset constrained to 0, using OriginPro. Fusion events were excluded from time-lapse intensity analysis if one or more additional fusion events occurred in the circular ROI within 5 s before or after fusion (–5 to 5 s in the time-lapse traces). If additional events occurred >5 s after fusion, the original fusion event was not excluded from analysis. Therefore, the average intensities of VAChT and red proteins after 5 s in traces and radial scan images should be interpreted with this caveat in mind.
Western blots
PC12 cells were transfected with Syndapin2-GFP, or siRNA (Dharmacon) using Lipofectamine 2000, and protein was isolated ∼24 h later. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (1% Anatrace, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM EDTA and 1 mM phenylmethylsulfonyl fluoride [PMSF] in PBS) on ice for 30 min. Lysates were centrifuged at 12,000 × g at 4°C for 10 min. Supernatants were boiled in lithium dodecyl sulfate (LDS) sample buffer containing 62.5 mM dithiothreitol (DTT) for 10 min and loaded onto 4–12% Tris-Bis gels (NuPAGE). Protein was transferred onto nitrocellulose membrane using the iBlot dry transfer system (ThermoFisher Scientific), and syndapin2 detected with monoclonal antibody (ThermoFisher Scientific) and peroxidase-labeled secondary antibody. Blots were stripped with stripping buffer (ThermoFisher Scientific) and reprobed with α-actin antibody (Abcam).
Statistical tests
All data are expressed as mean ± SEM. Statistical analysis was done using two-tailed paired Student’s t test to compare average normalized red protein fluorescence intensities at baseline (–5 to –4 s) with average intensities during (0 to 1 s), and after (4 to 5 s) fusion for the same set of events, as indicated in the figure legends.
Supplementary Material
Acknowledgments
We thank Marie-Paule Strub (National Institutes of Health) for assistance with molecular biology; J. Silver, K. Sochacki, and A. Trexler for critical reading of the manuscript; and J. Silver, S. Kale, and members of the Taraska laboratory for discussions and input on data analysis methods. J.W.T. is supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health.
Abbreviations used:
- CME
clathrin-mediated endocytosis
- LDCV
large dense core vesicle
- SLMV
synaptic-like microvesicle
- SV
synaptic vesicle
- TIRF
total internal reflection fluorescence
- VAChT
vesicular acetylcholine transporter.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-12-0716) on June 6, 2018.
REFERENCES
- Alabi AA, Tsien RW. (2013). Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu Rev Physiol , 393–422. [DOI] [PubMed] [Google Scholar]
- An SJ, Grabner CP, Zenisek D. (2010). Real-time visualization of complexin during single exocytic events. Nat Neurosci , 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam A, Bittner MA, Aikman RL, Stuenkel EL, Schmid SL, Axelrod D, Holz RW. (2011). A new role for the dynamin GTPase in the regulation of fusion pore expansion. Mol Biol Cell , 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam A, Onoa B, Edwards RH, Holz RW, Axelrod D. (2010). Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J Cell Biol , 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Smillie KJ, Graham ME, Valova VA, Cousin MA, Robinson PJ. (2006). Syndapin I is the phosphorylation-regulated dynamin I partner in synaptic vesicle endocytosis. Nat Neurosci , 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery U, Bielopolski N, Barak B, Yizhar O. (2009). Friends and foes in synaptic transmission: the role of tomosyn in vesicle priming. Trends Neurosci , 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Hu Z, Dittman JS, Pym EC, Kaplan JM. (2010). Endophilin functions as a membrane-bending molecule and is delivered to endocytic zones by exocytosis. Cell , 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Barry VA, DasGupta BR, Martin TF. (1996). N-Ethylmaleimide-sensitive factor acts at a prefusion ATP-dependent step in Ca2+-activated exocytosis. J Biol Chem , 20223–20226. [DOI] [PubMed] [Google Scholar]
- Barg S, Knowles MK, Chen X, Midorikawa M, Almers W. (2010). Syntaxin clusters assemble reversibly at sites of secretory granules in live cells. Proc Natl Acad Sci USA , 20804–20809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielopolski N, Lam AD, Bar-On D, Sauer M, Stuenkel EL, Ashery U. (2014). Differential interaction of tomosyn with syntaxin and SNAP25 depends on domains in the WD40 beta-propeller core and determines its inhibitory activity. J Biol Chem , 17087–17099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauchi S, Krapivinsky G, Krapivinsky L, Clapham DE. (2008). TRPM7 facilitates cholinergic vesicle fusion with the plasma membrane. Proc Natl Acad Sci USA , 8304–8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N. (2008). For better or for worse: complexins regulate SNARE function and vesicle fusion. Traffic , 1403–1413. [DOI] [PubMed] [Google Scholar]
- Cazares VA, Njus MM, Manly A, Saldate JJ, Subramani A, Ben-Simon Y, Sutton MA, Ashery U, Stuenkel EL. (2016). Dynamic partitioning of synaptic vesicle pools by the SNARE-binding protein Tomosyn. J Neurosci , 11208–11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HC, Shin W, Zhao WD, Hamid E, Sheng J, Baydyuk M, Wen PJ, Jin A, Momboisse F, Wu LG. (2014). Post-fusion structural changes and their roles in exocytosis and endocytosis of dense-core vesicles. Nat Commun , 3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow RH, von Ruden L, Neher E. (1992). Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature , 60–63. [DOI] [PubMed] [Google Scholar]
- Crivat G, Taraska JW. (2012). Imaging proteins inside cells with fluorescent tags. Trends Biotechnol , 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daumke O, Roux A, Haucke V. (2014). BAR domain scaffolds in dynamin-mediated membrane fission. Cell , 882–892. [DOI] [PubMed] [Google Scholar]
- David C, McPherson PS, Mundigl O, de Camilli P. (1996). A role of amphiphysin in synaptic vesicle endocytosis suggested by its binding to dynamin in nerve terminals. Proc Natl Acad Sci USA , 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Lichtenstein Y, Kelly RB, Geuze HJ, Klumperman J, van der Sluijs P. (2001). Rab4 regulates formation of synaptic-like microvesicles from early endosomes in PC12 cells. Mol Biol Cell , 3703–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Ji C, Wu Y, Ferguson SM, Tamarina N, Philipson LH, Lou X. (2015). Dynamin 2 regulates biphasic insulin secretion and plasma glucose homeostasis. J Clin Invest , 4026–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmy F. (2007). Modulation of cargo release from dense core granules by size and actin network. Traffic , 983–997. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, De Camilli P. (2012). Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol , 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M. (2008). Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol Life Sci , 2801–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Doreian B, Smith C. (2008). Dynamin I plays dual roles in the activity-dependent shift in exocytic mode in mouse adrenal chromaffin cells. Arch Biochem Biophys , 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandasi NR, Barg S. (2014). Contact-induced clustering of syntaxin and munc18 docks secretory granules at the exocytosis site. Nat Commun , 3914. [DOI] [PubMed] [Google Scholar]
- Geerts CJ, Mancini R, Chen N, Koopmans FTW, Li KW, Smit AB, van Weering JRT, Verhage M, Groffen AJA. (2017). Tomosyn associates with secretory vesicles in neurons through its N- and C-terminal domains. PLoS One , e0180912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Jamett AM, Guerra MJ, Olivares MJ, Haro-Acuna V, Baez-Matus X, Vasquez-Navarrete J, Momboisse F, Martinez-Quiles N, Cardenas AM. (2017). The F-actin binding protein cortactin regulates the dynamics of the exocytotic fusion pore through its SH3 domain. Front Cell Neurosci , 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Jamett AM, Momboisse F, Guerra MJ, Ory S, Baez-Matus X, Barraza N, Calco V, Houy S, Couve E, Neely A, et al (2013). Dynamin-2 regulates fusion pore expansion and quantal release through a mechanism that involves actin dynamics in neuroendocrine chromaffin cells. PLoS One , e70638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabs D, Slepnev VI, Songyang Z, David C, Lynch M, Cantley LC, De Camilli P. (1997). The SH3 domain of amphiphysin binds the proline-rich domain of dynamin at a single site that defines a new SH3 binding consensus sequence. J Biol Chem , 13419–13425. [DOI] [PubMed] [Google Scholar]
- Gracheva EO, Burdina AO, Holgado AM, Berthelot-Grosjean M, Ackley BD, Hadwiger G, Nonet ML, Weimer RM, Richmond JE. (2006). Tomosyn inhibits synaptic vesicle priming in Caenorhabditis elegans. PLoS Biol , e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham ME, O’Callaghan DW, McMahon HT, Burgoyne RD. (2002). Dynamin-dependent and dynamin-independent processes contribute to the regulation of single vesicle release kinetics and quantal size. Proc Natl Acad Sci USA , 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman RE, Schwarz YN, Rettig J, Bruns D. (2010). SNARE force synchronizes synaptic vesicle fusion and controls the kinetics of quantal synaptic transmission. J Neurosci , 10272–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haucke V, De Camilli P. (1999). AP-2 recruitment to synaptotagmin stimulated by tyrosine-based endocytic motifs. Science , 1268–1271. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. (1994). Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature , 513–515. [DOI] [PubMed] [Google Scholar]
- Holroyd P, Lang T, Wenzel D, De Camilli P, Jahn R. (2002). Imaging direct, dynamin-dependent recapture of fusing secretory granules on plasma membrane lawns from PC12 cells. Proc Natl Acad Sci USA , 16806–16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. (2012). Molecular machines governing exocytosis of synaptic vesicles. Nature , 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal JK, Rivera VM, Simon SM. (2009). Exocytosis of post-Golgi vesicles is regulated by components of the endocytic machinery. Cell , 1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockusch WJ, Speidel D, Sigler A, Sorensen JB, Varoqueaux F, Rhee JS, Brose N. (2007). CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell , 796–808. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Jorgensen EM. (2014). Visualizing presynaptic function. Nat Neurosci , 10–16. [DOI] [PubMed] [Google Scholar]
- Kessels MM, Qualmann B. (2002). Syndapins integrate N-WASP in receptor-mediated endocytosis. EMBO J , 6083–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyachko VA, Jackson MB. (2002). Capacitance steps and fusion pores of small and large-dense-core vesicles in nerve terminals. Nature , 89–92. [DOI] [PubMed] [Google Scholar]
- Knowles MK, Barg S, Wan L, Midorikawa M, Chen X, Almers W. (2010). Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc Natl Acad Sci USA , 20810–20815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner T, Li Y, Gee KR, Bonewald LF, Augustine GJ. (2008). Photolysis of a caged peptide reveals rapid action of N-ethylmaleimide sensitive factor before neurotransmitter release. Proc Natl Acad Sci USA , 347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C, Jahn R. (2001). SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J , 2202–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton JT, Barnard RJ, Titus SA, Slind J, Chapman ER, Ganetzky B. (2001). SNARE-complex disassembly by NSF follows synaptic-vesicle fusion. Proc Natl Acad Sci USA , 12233–12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Edwards RH. (1997). Differential localization of vesicular acetylcholine and monoamine transporters in PC12 cells but not CHO cells. J Cell Biol , 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YW, Neumann S, Ramachandran R, Ferguson SM, Pucadyil TJ, Schmid SL. (2011). Differential curvature sensing and generating activities of dynamin isoforms provide opportunities for tissue-specific regulation. Proc Natl Acad Sci USA , E234–E242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llobet A, Wu M, Lagnado L. (2008). The mouth of a dense-core vesicle opens and closes in a concerted action regulated by calcium and amphiphysin. J Cell Biol , 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malacombe M, Bader MF, Gasman S. (2006). Exocytosis in neuroendocrine cells: new tasks for actin. Biochim Biophys Acta , 1175–1183. [DOI] [PubMed] [Google Scholar]
- Martin TF. (2015). PI(4,5)P(2)-binding effector proteins for vesicle exocytosis. Biochim Biophys Acta , 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Bonangelino CJ, Aguilar RC, Bonifacino JS. (2001). Stonin 2: an adaptor-like protein that interacts with components of the endocytic machinery. J Cell Biol , 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau M, Somasundaram A, Grimm JB, Gruber TD, Choquet D, Taraska JW, Lavis LD, Perrais D. (2017). Semisynthetic fluorescent pH sensors for imaging exocytosis and endocytosis. Nat Commun , 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen JM, Madison JM, Dybbs M, Kaplan JM. (2006). Antagonistic regulation of synaptic vesicle priming by Tomosyn and UNC-13. Neuron , 303–315. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. (2011). Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol , 517–533. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Czernik AJ, Chilcote TJ, Onofri F, Benfenati F, Greengard P, Schlessinger J, De Camilli P. (1994). Interaction of Grb2 via its Src homology 3 domains with synaptic proteins including synapsin I. Proc Natl Acad Sci USA , 6486–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson PS, Garcia EP, Slepnev VI, David C, Zhang X, Grabs D, Sossin WS, Bauerfeind R, Nemoto Y, De Camilli P. (1996). A presynaptic inositol-5-phosphatase. Nature , 353–357. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature , 192–195. [DOI] [PubMed] [Google Scholar]
- Min L, Leung YM, Tomas A, Watson RT, Gaisano HY, Halban PA, Pessin JE, Hou JC. (2007). Dynamin is functionally coupled to insulin granule exocytosis. J Biol Chem , 33530–33536. [DOI] [PubMed] [Google Scholar]
- Ninomiya Y, Kishimoto T, Yamazawa T, Ikeda H, Miyashita Y, Kasai H. (1997). Kinetic diversity in the fusion of exocytotic vesicles. EMBO J , 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto PM, Herskovits JS, Vallee RB. (1997). Role of the basic, proline-rich region of dynamin in Src homology 3 domain binding and endocytosis. J Biol Chem , 11629–11635. [DOI] [PubMed] [Google Scholar]
- Pawlu C, DiAntonio A, Heckmann M. (2004). Postfusional control of quantal current shape. Neuron , 607–618. [DOI] [PubMed] [Google Scholar]
- Perrais D, Kleppe IC, Taraska JW, Almers W. (2004). Recapture after exocytosis causes differential retention of protein in granules of bovine chromaffin cells. J Physiol , 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualmann B, Roos J, DiGregorio PJ, Kelly RB. (1999). Syndapin I, a synaptic dynamin-binding protein that associates with the neural Wiskott-Aldrich syndrome protein. Mol Biol Cell , 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, Kelly RB. (1999). The endocytic machinery in nerve terminals surrounds sites of exocytosis. Curr Biol , 1411–1414. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Jahn R, Yoon TY. (2016). Review: Progresses in understanding N-ethylmaleimide sensitive factor (NSF) mediated disassembly of SNARE complexes. Biopolymers , 518–531. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. (1996). Timing of neurotransmission at fast synapses in the mammalian brain. Nature , 170–172. [DOI] [PubMed] [Google Scholar]
- Samasilp P, Chan SA, Smith C. (2012). Activity-dependent fusion pore expansion regulated by a calcineurin-dependent dynamin-syndapin pathway in mouse adrenal chromaffin cells. J Neurosci , 10438–10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Rosendale M, Campbell RE, Perrais D. (2014). pHuji, a pH-sensitive red fluorescent protein for imaging of exo- and endocytosis. J Cell Biol , 419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O. (2009). The synaptic vesicle cluster: a source of endocytic proteins during neurotransmitter release. Neuroscience , 204–210. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Low P, Grabs D, Gad H, Chen H, David C, Takei K, De Camilli P, Brodin L. (1997). Synaptic vesicle endocytosis impaired by disruption of dynamin-SH3 domain interactions. Science , 259–263. [DOI] [PubMed] [Google Scholar]
- Sochacki KA, Larson BT, Sengupta DC, Daniels MP, Shtengel G, Hess HF, Taraska JW. (2012). Imaging the post-fusion release and capture of a vesicle membrane protein. Nat Commun , 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature , 318–324. [DOI] [PubMed] [Google Scholar]
- Stachowiak JC, Schmid EM, Ryan CJ, Ann HS, Sasaki DY, Sherman MB, Geissler PL, Fletcher DA, Hayden CC. (2012). Membrane bending by protein-protein crowding. Nat Cell Biol , 944–949. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Rettig J. (2009). The Ca(2+)-dependent activator protein for secretion CAPS: do I dock or do I prime? Mol Neurobiol , 62–72. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. (2013a). A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med , 1227–1231. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. (2013d). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron , 675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. (2009). Membrane fusion: grappling with SNARE and SM proteins. Science , 474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, et al (2006). Molecular anatomy of a trafficking organelle. Cell , 831–846. [DOI] [PubMed] [Google Scholar]
- Taraska JW, Almers W. (2004). Bilayers merge even when exocytosis is transient. Proc Natl Acad Sci USA , 8780–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraska JW, Perrais D, Ohara-Imaizumi M, Nagamatsu S, Almers W. (2003). Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proc Natl Acad Sci USA , 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Reetz AC, De Camilli P. (1994). A role for synaptic vesicles in non-neuronal cells: clues from pancreatic beta cells and from chromaffin cells. FASEB J , 209–216. [DOI] [PubMed] [Google Scholar]
- Trexler AJ, Sochacki KA, Taraska JW. (2016). Imaging the recruitment and loss of proteins and lipids at single sites of calcium-triggered exocytosis. Mol Biol Cell , 2423–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trexler AJ, Taraska JW. (2017). Two-color total internal reflection fluorescence microscopy of exocytosis in endocrine cells. Methods Mol Biol , 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimbuch T, Rosenmund C. (2016). Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci , 118–125. [DOI] [PubMed] [Google Scholar]
- Tsuboi T, McMahon HT, Rutter GA. (2004). Mechanisms of dense core vesicle recapture following “kiss and run” (“cavicapture”) exocytosis in insulin-secreting cells. J Biol Chem , 47115–47124. [DOI] [PubMed] [Google Scholar]
- Tsuboi T, Rutter GA. (2003). Multiple forms of “kiss-and-run” exocytosis revealed by evanescent wave microscopy. Curr Biol , 563–567. [DOI] [PubMed] [Google Scholar]
- Ullrich A, Bohme MA, Schoneberg J, Depner H, Sigrist SJ, Noe F. (2015). Dynamical organization of syntaxin-1A at the presynaptic active zone. PLoS Comput Biol , e1004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C, Nichols BJ, Pelham HR, Wickner W. (1998). A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol , 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R. (2010). One SNARE complex is sufficient for membrane fusion. Nat Struct Mol Biol , 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, McMahon HT, Ghijsen WE, Boomsma F, Scholten G, Wiegant VM, Nicholls DG. (1991). Differential release of amino acids, neuropeptides, and catecholamines from isolated nerve terminals. Neuron , 517–524. [DOI] [PubMed] [Google Scholar]
- Wang CT, Grishanin R, Earles CA, Chang PY, Martin TF, Chapman ER, Jackson MB. (2001). Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science , 1111–1115. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Liu Q, Davis MW, Hollopeter G, Thomas N, Jorgensen NB, Jorgensen EM. (2013a). Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. Elife , e00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Rost BR, Camacho-Perez M, Davis MW, Sohl-Kielczynski B, Rosenmund C, Jorgensen EM. (2013c). Ultrafast endocytosis at mouse hippocampal synapses. Nature , 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PJ, Grenklo S, Arpino G, Tan X, Liao HS, Heureaux J, Peng SY, Chiang HC, Hamid E, Zhao WD, et al (2016). Actin dynamics provides membrane tension to merge fusing vesicles into the plasma membrane. Nat Commun , 12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge P, Vallis Y, McMahon HT. (1997). Inhibition of receptor-mediated endocytosis by the amphiphysin SH3 domain. Curr Biol , 554–560. [DOI] [PubMed] [Google Scholar]
- Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. (2006). STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature , 935–939. [DOI] [PubMed] [Google Scholar]
- Woodman PG. (2000). Biogenesis of the sorting endosome: the role of Rab5. Traffic , 695–701. [DOI] [PubMed] [Google Scholar]
- Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, Bai J, Eliezer D, Dittman JS. (2013). Synaptic vesicles position complexin to block spontaneous fusion. Neuron , 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Hamid E, Shin W, Chiang HC. (2014). Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu Rev Physiol , 301–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Xu P. (2008). Searching for molecular players differentially involved in neurotransmitter and neuropeptide release. Neurochem Res , 1915–1919. [DOI] [PubMed] [Google Scholar]
- Yamada H, Padilla-Parra S, Park SJ, Itoh T, Chaineau M, Monaldi I, Cremona O, Benfenati F, De Camilli P, Coppey-Moisan M, et al (2009). Dynamic interaction of amphiphysin with N-WASP regulates actin assembly. J Biol Chem , 34244–34256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon TY, Lu X, Diao J, Lee SM, Ha T, Shin YK. (2008). Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat Struct Mol Biol , 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Kinuta M, Abe T, Liang S, Araki K, Cremona O, Di Paolo G, Moriyama Y, Yasuda T, De Camilli P, Takei K. (2004). The stimulatory action of amphiphysin on dynamin function is dependent on lipid bilayer curvature. EMBO J , 3483–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jackson MB. (2010). Membrane bending energy and fusion pore kinetics in Ca(2+)-triggered exocytosis. Biophys J , 2524–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WD, Hamid E, Shin W, Wen PJ, Krystofiak ES, Villarreal SA, Chiang HC, Kachar B, Wu LG. (2016). Hemi-fused structure mediates and controls fusion and fission in live cells. Nature , 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.