

Abstract
Objective:
Allergic airway diseases (AADs) are a group of heterogeneous disease mediated by T-helper type 2 (Th2) immune response and characterized with airway inflammation and remodeling, including allergic asthma, allergic rhinitis, and chronic rhinosinusitis with allergic background. This review aimed to discuss the abnormal epithelial-mesenchymal crosstalk in the pathogenesis of AADs.
Data Sources:
Articles referred in this review were collected from the database of PubMed published in English up to January 2018.
Study Selection:
We had done a literature search using the following terms “allergic airway disease OR asthma OR allergic rhinitis OR chronic sinusitis AND IL-25 OR IL-33 OR thymic stromal lymphopoietin OR fibrocyte”. Related original or review articles were included and carefully analyzed.
Results:
It is now believed that abnormal epithelial-mesenchymal crosstalk underlies the pathogenesis of AADs. However, the key regulatory factors and molecular events involved in this process still remain unclear. Epithelium-derived triple cytokines, including interleukin (IL)-25, IL-33, and thymic stromal lymphopoietin (TSLP), are shown to act on various target cells and promote the Th2 immune response. Circulating fibrocyte is an important mesenchymal cell that can mediate tissue remodeling. We previously found that IL-25-circulating fibrocyte axis was significantly upregulated in patients with asthma, which may greatly contribute to asthmatic airway inflammation and remodeling.
Conclusions:
In view of the redundancy of cytokines and “united airway” theory, we propose a new concept that IL-25/IL-33/TSLP-fibrocyte axis may play a vital role in the abnormal epithelial-mesenchymal crosstalk in some endotypes of AADs. This novel idea will guide potential new intervention schema for the common treatment of AADs sharing common pathogenesis in the future.
Keywords: Allergic Airway Diseases, Axis, Crosstalk, Fibrocyte, Interleukin-25/Interleukin-33/Thymic Stromal Lymphopoietin
摘要
目的:
过敏性气道疾病(Allergic airway diseases , AADs)是一组Th2型免疫反应介导的慢性气道炎症和异常修复的异质性疾病 群,包括具有过敏背景的某些过敏性哮喘、过敏性鼻炎和慢性鼻窦炎亚型。本综述旨在探讨上皮-间质异常通讯在过敏性气 道疾病发病机制中的作用。
数据来源:
本综述所引用的论文是通过检索2018年1月以来PubMed数据库收录的参考文献而获得。
资料选择:
我们用“(allergic airway disease OR asthma OR allergic rhinitis OR chronic sinusitis) AND (IL-25 OR IL-33 OR thymic stromal lymphopoietin OR fibrocyte)”这一检索式进行文献检索,并仔细阅读和分析相关研究论文或综述。
结果:
目前认为上皮-间质异常通讯是过敏性气道疾病的核心发病机制,但参与其中的关键调控因子和分子事件尚未明确。IL- 25/IL-33/TSLP(胸腺基质淋巴生成素)是上皮来源的促Th2型免疫反应的细胞因子,而循环纤维细胞(circulating fibrocyte)是一 种可介导组织重塑的重要间质细胞。我们前期研究发现IL-25-fibrocyte轴在哮喘患者中表达明显上调,且和哮喘严重程度相关。
结论:
鉴于细胞因子生物学作用的冗余性和“同一气道”理论,我们认为IL-25/IL-33/TSLP-纤维细胞轴可能参与不同亚型的过 敏性气道疾病上皮-间质异常通讯过程。这一新理论将为AADs这类有着共同发病机制的异质性疾病群的共有治疗提供新型潜 在靶点。
INTRODUCTION
Allergic airway diseases (AADs) are a group of heterogeneous disorder characterized by excessive T-helper type 2 (Th2) cytokine responses and abnormal airway inflammation and remodeling, including allergic rhinitis (AR), allergic asthma (AA), and chronic rhinosinusitis (CRS) with atopic nature. Since the world Industrial Revolution, the worldwide incidence of AR, AA, and CRS has significantly increased, resulting in a huge social and economic burden.[1] Recent concept believes that although AADs are distinct in terms of clinical features and the lesion site, they may coexist in one individual epidemiologically and share some common underlying pathophysiological mechanisms, indicating “one/united allergic airway”.[1,2] It is important to elucidate the common key regulatory factors and molecular events underlying AADs because any treatment on these “master switch” is superior to the traditional therapeutic modality. In this review, we would discuss the abnormal communication axis between epithelium-derived triple cytokines (interleukin [IL]-25/IL-33/thymic stromal lymphopoietin [TSLP])[3] and circulating fibrocyte in the common pathogenesis of AADs.
ONE/UNITED ALLERGIC AIRWAY CONCEPT
One/united allergic airway concept argues that the allergic diseases of upper and lower airway are not isolated, but they are a group of similar diseases with common allergic background. First, epidemiological studies showed that AR, AA, and CRS can coexist in the same patient. More than 40% of patients with AR had asthmatic symptoms; on the other hand, more than 80% of patients with AA suffered from symptomatic AR.[4] Furthermore, CRS is often associated with the severity of asthma. Nearly 88% of patients with moderate-to-severe asthma have CRS-like symptoms and paranasal sinus CT scan showed that 100% of patients with hormone- dependent severe asthma combined with CRS.[5,6]
Second, AR, AA, and CRS share common immunopathological mechanisms. Antigen-specific IgE production, mast cell activation/degranulation, and significant eosinophil infiltration are all features present in patients with AADs.[7] More importantly, Th2 cytokine response bias underlies different endotypes of AADs because excessive production of IL-4 (a cytokine that can promote cell surface IgE type conversion and Th2 cell activation), IL-5 (a cytokine that can promote eosinophil activation, recruitment, and differentiation), and IL-13 (a cytokine that can promote allergic inflammatory response and mucus formation) co-exists in patients with AR, AA, and CRS (especially CRS patients with nasal polyps [CRSwNP]).[8,9,10]
Third, chronic inflammatory injury and abnormal remodeling of airway are the basic pathophysiological changes in AR, AA, and CRS. Previous studies have shown that there are some wound-healing events in asthmatic airways, which were characterized by mucosa epithelial apoptosis/erosion, goblet cell hyperplasia/hypertrophy, thickening of basement membrane, epithelial fibrosis, submucosal gland hyperplasia, mucus secretion, hyperplasia of smooth muscle hypertrophy, hyperplasia of blood vessels, etc.[11,12] Studies on the abnormal injury and repair of upper airway allergic diseases are relatively scarce. A previous study showed that abnormal epithelial cell phenotype (epithelial-mesenchymal transition [EMT], characterized by downregulation of E-cadherin and cytokeratin and upregulation of vimentin) exists in nasal mucosal tissue in patients with CRSwNP.[13] In addition, the hyperplasia of goblet cells, excessive mucoprotein secretion, and thickening of basement membrane are all features in upper airways of CRSwNP patients, which significantly correlates with the severity and duration of the disease.[13] Another study showed that increased mucosal/submucosal collagen and proteoglycan deposition and increased numbers of lymphatic vessels and length were significant in patients with mild or severe persistent AR, indicating that abnormal remodeling occurs in upper airways (nose) of patients with AR.[14]
Collectively, we suggest that although AR, AA, and CRS differ in pathogenic site and clinical manifestations, they can coexist in the same individual and share the common pathogenesis. Excessive airway Th2-type cytokine production-induced abnormal wound-healing event may be the common pathway underlying the pathogenesis of AADs.
EPITHELIUM-DERIVED INTERLEUKIN-25/INTERLEUKIN-33/THYMIC STROMAL LYMPHOPOIETIN INVOLVE IN THE PATHOGENESIS OF ALLERGIC AIRWAY DISEASES
The key regulatory factors and molecular events underlying abnormal airway wound-healing process in patients with AADs have not been well clarified. However, it is now hypothesized that epithelial-mesenchymal trophic unit-mediated communication between epithelial and mesenchymal cells plays a key role in the pathogenesis of AADs, and the communication media mainly depend on epithelium-derived triple cytokines, namely IL-25, IL-33, and TSLP.
Toll-like receptors (TLRs) expressed on epithelial cells can recognize structurally conservative pathogen-associated molecular patterns (PAMPs). Epithelial cells respond to PAMP–TLR interaction by secreting various immunocytokines, which further bind various potential target cells in the surrounding milieu and form communication links to induce innate or adaptive immune response.[15] Epithelial-derived IL-25 (IL-17E) belongs to the family members of the IL-17 cytokines. IL-25, via binding with its heterodimer receptor (IL-17RA/IL-17RB) and activation of NF-κB signal pathway, plays a critical role in airway chronic inflammation and remodeling process in asthmatic patients by promoting the expression of Th2 cytokines (IL-4, IL-5, and IL-13).[16,17,18] By nasal drip of IL-25, we successfully build chronic asthma mouse model that mimics the main features of classical ovalbumin (OVA)-induced chronic asthma model. We showed that IL-25 can induce airway hyperresponsiveness, promote peripheral blood and airway eosinophil infiltration, and promote the excessive production of IgE, Th2 cytokines (IL-4/5/13), fibrotic cytokines (transforming growth factor beta/connective tissue growth factor/insulin-like growth factor-1), and angiogenesis factors (amphiregulin, angiogenin, endothelin-1, transcription factor ERG, basic fibroblast growth factor, epidermal growth factor, and vascular endothelial growth factor). Furthermore, IL-25-treated mice showed airway mucosal epithelial shedding, goblet cell hyperplasia, and excessive subepithelial deposition of collagen I/III/V and fibronectin.[19,20,21]
IL-33 belongs to the family of IL-1 cytokines, and its receptor complex contains 2 transmembrane proteins, namely, IL-1R1 (also known as ST2L) and il-1RAcP. When IL-33 binds to its receptor, it can activate the MyD88 and NF-κB pathways, thereby promoting the Th2 cytokine response.[22] Previous study showed that the plasma level of IL-33 is elevated in patients with asthma.[23] In accordance with this, we also showed that nasal administration with IL-33 can replicate the typical characteristics of the airway Th2 immune response and abnormal airway remodeling.[24] Furthermore, IL-33 monoclonal antibody can significantly attenuate allergen-induced eosinophil infiltration, mucus oversecretion, and Th2 cytokine response in asthma mouse model.[25]
TSLP is a member of the IL-2 cytokine family, and its high affinity receptor complexes are TSLPR and IL-7R. Previous study found that the expression of TSLP in airway epithelial cells in asthmatic patients was significantly increased compared with that of normal controls.[26] OVA failed to induce airway inflammation and remodeling in TSLPR-defect mice, and reconstruction of CD4+ TSLPR+ cells could restore the OVA's effect on mouse airway inflammation and remodeling, indicating that TSLP plays an important role in abnormal asthmatic airway wound-healing process.[27] Collectively, IL-25/IL-33/TSLP involves in airway inflammation and abnormal airway remodeling by inducing a Th2 immune response in patients with asthma and asthmatic animal models.
In view of the “one airway theory”, many subsequent studies have focused on the role of IL-25/IL-33/TSLP in allergic upper airway disease (AR and CRS with allergic endotype). It was shown that TSLP mRNA level expressed in nasal polyp epithelial cells is remarkably increased in patients with CRSwNP.[28] Furthermore, the expression levels of TSLP receptors in mucosal epithelial cells and subepithelial inflammatory cells were significantly increased in CRSwNP and CRS patients without nasal polyps,[29] indicating a potential role of TSLP in the pathogenesis of CRS. Another study showed that IL-25 in nasal epithelial cells was significantly upregulated in CRSwNP mouse model, and IL-25 neutralizing antibodies can significantly reduce the number of polyps; attenuate mucosal edema, collagen deposition, and inflammatory cell infiltration; and reduce airway expression of CCL11/CXCL2/ICAM (intercellular cell adhesion molecule-1/vascular cell adhesion molecule-1), indicating a potential correlation between IL-25 and upper airway inflammation and remodeling.[30] Recently, one Chinese study showed that compared with the control group and noneosinophilic CRSwNP patients, the expression of TSLP/TSLPR/IL-7Ra and ST2L/sST2 in eosinophilic CRSwNP patients was significantly increased, while the expressions of IL-33 and IL-25/IL-17RB were all upregulated in both noneosinophilic-CRSwNP and eosinophilic-CRSwNP patients.[31] Furthermore, the expression level of TSLP/TSLPR/ST2L was positively correlated with the patients’ symptoms, the sinus CT score, and the expression of Th2 cytokines, suggesting that the triple epithelium-derived cytokines (IL-25/IL-33/TSLP) may play an important role in the upper airway changes in certain subgroups of CRSwNP patients.[31] Another study found that the serum level of IL-33 and IL-33R (ST2L) in nasal epithelial cells in patients with AR was significantly elevated, indicating that IL-33/ST2 pathway may be a novel potential interventional target for AP patients.[32] In addition, a previous study also suggested that IL-33/ST2 and TSLP/TSLPR participate in the acute-phase response of AR by modulating IgE production and nasal Th2 activation.[33] Specifically, IL-33/ST2 can mediate early nasal Th2 immune response after allergen exposure, while TSLP/TSLPR and IL-33/ST2 pathways are both involved in the Th2 immune response of chronic AR.[33] In conclusion, IL-25/IL-33/TSLP may also participate in the upper airway inflammation and remodeling process of patients with AP and CRS.
CIRCULATING FIBROCYTES ACT AS INTERLEUKIN-25-TARGETED CELLS
The underlying mechanisms of IL-25/IL-33/TSLP on chronic airway inflammation and remodeling of AADs still remain largely unknown. This depends on further interpretation of IL-25/IL-33/TSLP-targeted cells. Abnormal epithelial-mesenchymal communication in AADs depends on IL-25/IL-33/TSLP expressed by the damaged and activated epithelial cells, while IL-25/IL-33/TSLP requires targeted cells expressing their corresponding receptors to exert their functions. Generally speaking, cells residing in airway mucosa can be divided into infiltrated inflammatory/immune/remodeling cells and resident structural cells. Previous studies have found that a variety of different cell types can express IL-25 receptor (IL-17RB), including dendritic cells, eosinophils, smooth muscle cells, airway epithelial cells, and fibroblasts.[34] It was also shown that IL-33 receptor (ST2) can be expressed by mast cells, macrophages, hematopoietic stem cells, natural killer T (NKT)-cells, eosinophilic/basophilic cells, innate lymphoid cells, and fibroblasts,[35] while TSLP receptor (TSLP) can be expressed by CD4/CD8+ T-cells, B-cells, mast cells, NKT-cells, monocytes, eosinophils, smooth muscle cells, and airway epithelial cells.[36] Thus, a variety of different structural and inflammatory cells can both express IL-25/IL-33/TSLP receptors, but which targeted cell plays a key role in the abnormal airway chronic inflammation and remodeling process in AADs is still unclear.
In our previous work, we found that bone marrow-derived circulating fibrocytes can also express the receptor of IL-25 (IL-17RB) and can respond to exogenous IL-25 (unpublished data). Fibrocytes are a group of mesenchymal stem cells/progenitor cells that have directional differentiation potential and enormous paracrine function. They can be isolated from peripheral blood mononuclear cells (PBMCs) and express cell surface marker of hematopoietic stem cells/myeloid cells (such as CD34, CD45, CD45RO, CD11b, and CD13) and cytoplasmic marker of type I collagen and alpha-smooth muscle actin.[37] A previous study showed that the number of CD45+ CD34+ collagen1+-circulating fibrocytes was significantly increased in asthmatic patients with acute exacerbation and in patients with chronic obstructive asthma/uncontrolled asthma, indicating that recruitment of fibrocytes correlates with the endotype and severity of asthma.[38] Various chemokines and cytokines (including the CCL2/5/11/19/24, CXCL12, and platelet-derived growth factor) can be secreted by airway epithelial cells, inflammatory cells (e.g., eosinophils, macrophages, and mast cells), and airway smooth muscle cells, which form a chemokine/cytokine concentration gradient and promote the migration process of fibrocytes from blood vessels into the diseased airway.[39] We previously showed that the total number of IL-17RA+/RB+ fibrocytes in patients with asthma (especially GINA-high asthma) was significantly higher than that of healthy controls, and the recruitment of IL-17RA+/RB+ fibrocytes predicted poor lung function of asthmatic patients [Figures 1 and 2]. This indicates that airway epithelial cells and circulating fibrocytes may form a communication pathway by IL-25–IL-17RB interaction and contribute to airway inflammation and abnormal remodeling.
Figure 1.

The number of circulating IL-17RA+/IL-17RB+fibrocytes in asthmatic patients was greater than that of control group. PBMC cells from peripheral blood of 25 asthmatic patients and 10 normal controls were isolated using lymphocyte separation medium. Then, anti-CD3 immunomagnetic beads were used to eliminate lymphocytes. The cells then received adherent culture for 2 h to remove the adherent cells. Nonadherent non-T PBMCs were marked with eFluor™-anti-CD45, FITC-anti-CollgenI, APC-antiCD217 (IL-17RA), PE-antiIL-17RB antibodies for flow cytometry analysis of CD45+CollgenI+IL-17RA+IL-RB+fibrocytes. (a) Gating with isotype control antibodies; (b) Gating with specific antibodies; (c) Comparison of CD45+CollgenI+IL-17RA+IL-17RB+fibrocytes between asthmatic patients and controls. Data were shown as mean ± standard error, n = 10 (controls) versus n = 25 (asthmatic patients), *P < 0.001, unpaired t-test with Welch's correction. PBMCs: Peripheral blood mononuclear cells.
Figure 2.

(a) The number of CD45+CollgenI+ fibrocyte was significantly negatively correlated with FEV1/FVC in asthmatic patients, n=25, Spearman r correlation test, P<0.05; (b) The number of CD45+CollgenI+ fibrocyte was significantly negatively correlated with FEV1/pred in asthmatic patients, n=25, Spearman r correlation test, P<0.05. (c) The number of CD45+CollgenI+ IL-17RA+IL-17RB+fibrocyte was significantly negatively correlated with FEV1/FVC in asthmatic patients, n=25, Spearman r correlation test, P<0.05; (d) The number of CD45+CollgenI+ IL-17RA+IL-17RB+ fibrocyte was significantly negatively correlated with FEV1/pred in asthmatic patients, n=25, Spearman r correlation test, P<0.05. FVC: Forced vital capacity; FEV1: Forced expiratory volume in 1 s; pred: Predicted.
INTERLEUKIN-25/INTERLEUKIN-33/THYMIC STROMAL LYMPHOPOIETIN (IL-17RB+ST2L+TSLPR+) FIBROCYTE AXIS MAY CONTRIBUTE TO THE PATHOGENESIS OF ALLERGIC AIRWAY DISEASES BY MEDIATING ABNORMAL EPITHELIAL-MESENCHYMAL CROSSTALK
Fibrocyte is an important effector cell that can mediate chronic airway inflammation and pathologic remodeling in AADs. On the other hand, IL-25/IL-33/TSLP is the key regulatory factor involved in the abnormal wound-healing process. Therefore, we propose a new idea that abnormal communication may exist between IL-25/IL-33/TSLP and fibrocyte, which forms a functional pathologic pathway (i.e., IL-25/IL-33/TSLP-[IL-17RB+ ST2L+ TSLPR+] fibrocyte axis) to exert the pathogenesis of airway changes in AADs [Figure 3]. To further verify this hypothesis, we suggest that future studies should be focused on the following topics:[1] building the biological sample library of AAD (AA/AR/CRS) patients and detecting the expression level of IL-17RB/ST2L/TSLPR on the circulating and airway fibrocytes;[2] establishing chronic AA/AR/CRS mouse model, detecting the numbers of (IL-17RB/ST2L/TSLPR)+ fibrocytes, assessing their correlation with airway/nasal mucosal inflammation, airway/nasal mucosa remodeling and basement membrane (degree of fibrosis or smooth muscle layer thickness); exploring whether neutralizing antibodies against IL-17RB/ST2L/TSLPR could attenuate airway inflammation and remodeling;[3] and establishing a co-culture system between airway/nasal mucosal epithelial cells and fibrocytes to explore the potential role of IL-25/IL-33/TSLP in the abnormal epithelial–mesenchymal communication in vitro. By emphasizing the above questions, we will further clarify the underlying key molecular events and functional pathways contributing to the abnormal airway wound-healing process in the AADs (AA/AR/CRS). This will provide a potential new interventional target for the common treatment of the heterogeneous diseases sharing similar pathogenesis.
Figure 3.
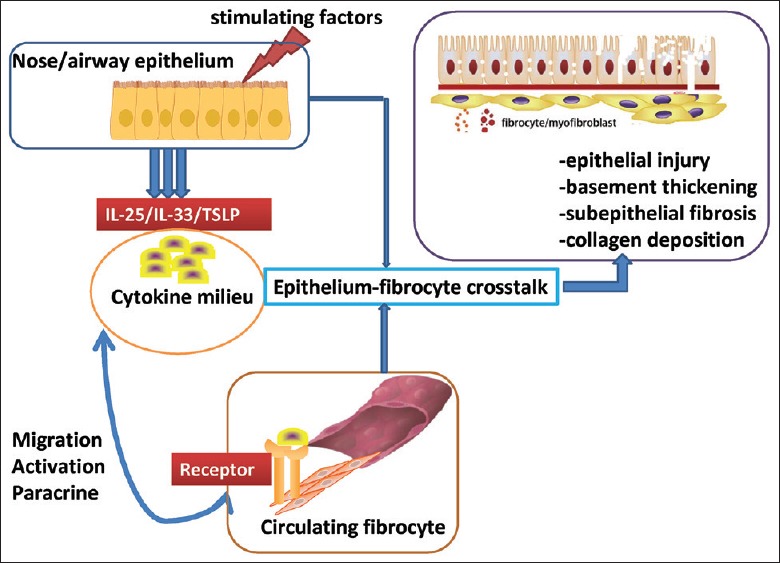
Schema diagram illustrating the hypothesis that IL-25/IL-33/TSLP-(IL-17RB/ST2L/TSLPR)+fibrocyte axis modulates epithelial–mesenchymal crosstalk in AADs. Damaged or activated nasal/airway mucosal epithelial cells can secrete excessive amount of cytokines (including IL-25, IL-33, and TSLP) that can promote type Th2 immune response. These cytokines form a chemotaxis gradient in local airway milieu and promote the recruitment of bone marrow-derived circulating fibrocytes which express IL-25/IL-33/TSLP receptors (IL-17RB/ST2L/TSLPR). Abnormal epithelial–mesenchymal interaction will be mediated by the functional IL-25/IL-33/TSLP-(IL-17RB/ST2L/TSLPR)+fibrocyte axis. Activated fibrocytes can further differentiate into collagen-productive myofibroblasts. In addition, fibrocytes can promote the abnormal airway remodeling by their powerful paracrine functions, which ultimately induce epithelial cell damage, basement membrane thickening, lamina propria collagen deposition/fibrosis, submucosal gland hyperplasia, and hypertrophy and hyperplasia of airway smooth muscle and blood vessels. Thus, targeted therapy against IL-25/IL-33/TSLP-(IL-17RB/ST2/TSLPR)+fibrocyte axis may be the novel strategy for the common treatment of refractory AADs. IL: Interleukin; TSLP: Thymic stromal lymphopoietin; AADs: Allergic airway diseases.
CONCLUSIONS
We suggest that IL-25/IL-33/TSLP-(IL-17RB/ST2/TSLPR)+ fibrocyte axis may exist in the core pathogenesis of AADs and may serve as the master switch for the abnormal airway inflammation and remodeling in AADs. Thus, patients with refractory AADs would benefit from targeted therapy against IL-25/IL-33/TSLP-(IL-17RB/ST2/TSLPR)+ fibrocyte axis in the future.
Financial support and sponsorship
This study was supported by grants from the Natural Science Foundation of China (No. 81641003) and Application of Clinical Features in Capital City by the Beijing Municipal Science and Technology Commission (No. Z131107002213135).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Yi Cui
REFERENCES
- 1.Feng CH, Miller MD, Simon RA. The united allergic airway: Connections between allergic rhinitis, asthma, and chronic sinusitis. Am J Rhinol Allergy. 2012;26:187–90. doi: 10.2500/ajra.2012.26.3762. doi: 10.2500/ajra.2012.26.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kariyawasam HH, Rotiroti G. Allergic rhinitis, chronic rhinosinusitis and asthma: Unravelling a complex relationship. Curr Opin Otolaryngol Head Neck Surg. 2013;21:79–86. doi: 10.1097/MOO.0b013e32835ac640. doi: 10.1097/MOO.0b013e32835ac640. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell PD, O’Byrne PM. Biologics and the lung: TSLP and other epithelial cell-derived cytokines in asthma. Pharmacol Ther. 2017;169:104–12. doi: 10.1016/j.pharmthera.2016.06.009. doi: 10.1016/j.pharmthera.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Leynaert B, Neukirch C, Kony S, Guénégou A, Bousquet J, Aubier M, et al. Association between asthma and rhinitis according to atopic sensitization in a population-based study. J Allergy Clin Immunol. 2004;113:86–93. doi: 10.1016/j.jaci.2003.10.010. doi: 10.1016/j.jaci.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Lin DC, Chandra RK, Tan BK, Zirkle W, Conley DB, Grammer LC, et al. Association between severity of asthma and degree of chronic rhinosinusitis. Am J Rhinol Allergy. 2011;25:205–8. doi: 10.2500/ajra.2011.25.3613. doi: 10.2500/ajra.2011.25.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bresciani M, Paradis L, Des Roches A, Vernhet H, Vachier I, Godard P, et al. Rhinosinusitis in severe asthma. J Allergy Clin Immunol. 2001;107:73–80. doi: 10.1067/mai.2001.111593. doi: 10.1067/mai.2001.111593. [DOI] [PubMed] [Google Scholar]
- 7.Barnes PJ. Pathophysiology of allergic inflammation. Immunol Rev. 2011;242:31–50. doi: 10.1111/j.1600-065X.2011.01020.x. doi: 10.1111/j.1600-065X.2011.01020.x. [DOI] [PubMed] [Google Scholar]
- 8.Scadding G. Cytokine profiles in allergic rhinitis. Curr Allergy Asthma Rep. 2014;14:435. doi: 10.1007/s11882-014-0435-7. doi: 10.1007/s11882-014-0435-7. [DOI] [PubMed] [Google Scholar]
- 9.Van Bruaene N, Pérez-Novo CA, Basinski TM, Van Zele T, Holtappels G, De Ruyck N, et al. T-cell regulation in chronic paranasal sinus disease. J Allergy Clin Immunol. 2008;121:1435–41. doi: 10.1016/j.jaci.2008.02.018. 1441e1-3 doi: 10.1016/j.jaci.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 10.Hansbro PM, Scott GV, Essilfie AT, Kim RY, Starkey MR, Nguyen DH, et al. Th2 cytokine antagonists: Potential treatments for severe asthma. Expert Opin Investig Drugs. 2013;22:49–69. doi: 10.1517/13543784.2013.732997. doi: 10.1517/13543784.2013.732997. [DOI] [PubMed] [Google Scholar]
- 11.Girodet PO, Ozier A, Bara I, Tunon de Lara JM, Marthan R, Berger P, et al. Airway remodeling in asthma: New mechanisms and potential for pharmacological intervention. Pharmacol Ther. 2011;130:325–37. doi: 10.1016/j.pharmthera.2011.02.001. doi: 10.1016/j.pharmthera.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Gras D, Bourdin A, Chanez P, Vachier I. Airway remodeling in asthma: Clinical and functional correlates. Med Sci (Paris) 2011;27:959–65. doi: 10.1051/medsci/20112711011. doi: 10.1051/medsci/20112711011. [DOI] [PubMed] [Google Scholar]
- 13.Hupin C, Gohy S, Bouzin C, Lecocq M, Polette M, Pilette C, et al. Features of mesenchymal transition in the airway epithelium from chronic rhinosinusitis. Allergy. 2014;69:1540–9. doi: 10.1111/all.12503. doi: 10.1111/all.12503. [DOI] [PubMed] [Google Scholar]
- 14.Kim TH, Lee JY, Lee HM, Lee SH, Cho WS, Ju YH, et al. Remodelling of nasal mucosa in mild and severe persistent allergic rhinitis with special reference to the distribution of collagen, proteoglycans, and lymphatic vessels. Clin Exp Allergy. 2010;40:1742–54. doi: 10.1111/j.1365-2222.2010.03612.x. doi: 10.1111/j.1365-2222.2010.03612.x. [DOI] [PubMed] [Google Scholar]
- 15.Hansel TT, Johnston SL, Openshaw PJ. Microbes and mucosal immune responses in asthma. Lancet. 2013;381:861–73. doi: 10.1016/S0140-6736(12)62202-8. doi: 10.1016/S0140-6736(12)62202-8. [DOI] [PubMed] [Google Scholar]
- 16.Su J, Chen T, Ji XY, Liu C, Yadav PK, Wu R, et al. IL-25 downregulates Th1/Th17 immune response in an IL-10-dependent manner in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:720–8. doi: 10.1097/MIB.0b013e3182802a76. doi: 10.1097/MIB.0b013e3182802a76. [DOI] [PubMed] [Google Scholar]
- 17.Tang W, Smith SG, Beaudin S, Dua B, Howie K, Gauvreau G, et al. IL-25 and IL-25 receptor expression on eosinophils from subjects with allergic asthma. Int Arch Allergy Immunol. 2014;163:5–10. doi: 10.1159/000355331. doi: 10.1159/000355331. [DOI] [PubMed] [Google Scholar]
- 18.Cheng D, Xue Z, Yi L, Shi H, Zhang K, Huo X, et al. Epithelial interleukin-25 is a key mediator in Th2-high, corticosteroid-responsive asthma. Am J Respir Crit Care Med. 2014;190:639–48. doi: 10.1164/rccm.201403-0505OC. doi: 10.1164/rccm.201403-0505OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao X, Wang W, Li Y, Huang P, Zhang Q, Wang J, et al. IL-25 induces airways angiogenesis and expression of multiple angiogenic factors in a murine asthma model. Respir Res. 2015;16:39. doi: 10.1186/s12931-015-0197-3. doi: 10.1186/s12931-015-0197-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao X, Wang W, Li Y, Lv Z, Guo R, Corrigan CJ, et al. Characteristics of IL-25 and allergen-induced airway fibrosis in a murine model of asthma. Respirology. 2015;20:730–8. doi: 10.1111/resp.12546. doi: 10.1111/resp.12546. [DOI] [PubMed] [Google Scholar]
- 21.Yao XJ, Huang KW, Li Y, Zhang Q, Wang JJ, Wang W, et al. Direct comparison of the dynamics of IL-25- and ‘allergen’-induced airways inflammation, remodelling and hypersensitivity in a murine asthma model. Clin Exp Allergy. 2014;44:765–77. doi: 10.1111/cea.12298. doi: 10.1111/cea.12298. [DOI] [PubMed] [Google Scholar]
- 22.Yagami A, Orihara K, Morita H, Futamura K, Hashimoto N, Matsumoto K, et al. IL-33 mediates inflammatory responses in human lung tissue cells. J Immunol. 2010;185:5743–50. doi: 10.4049/jimmunol.0903818. doi: 10.4049/jimmunol.0903818. [DOI] [PubMed] [Google Scholar]
- 23.Préfontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased expression of IL-33 in severe asthma: Evidence of expression by airway smooth muscle cells. J Immunol. 2009;183:5094–103. doi: 10.4049/jimmunol.0802387. doi: 10.4049/jimmunol.0802387. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Wang W, Huang P, Zhang Q, Yao X, Wang J, et al. Distinct sustained structural and functional effects of interleukin-33 and interleukin-25 on the airways in a murine asthma surrogate. Immunology. 2015;145:508–18. doi: 10.1111/imm.12465. doi: 10.1111/imm.12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizutani N, Nabe T, Yoshino S. Interleukin-33 and alveolar macrophages contribute to the mechanisms underlying the exacerbation of IgE-mediated airway inflammation and remodelling in mice. Immunology. 2013;139:205–18. doi: 10.1111/imm.12071. doi: 10.1111/imm.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uller L, Leino M, Bedke N, Sammut D, Green B, Lau L, et al. Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-beta in bronchial epithelial cells from donors with asthma. Thorax. 2010;65:626–32. doi: 10.1136/thx.2009.125930. doi: 10.1136/thx.2009.125930. [DOI] [PubMed] [Google Scholar]
- 27.Al-Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. J Exp Med. 2005;202:829–39. doi: 10.1084/jem.20050199. doi: 10.1084/jem.20050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagarkar DR, Poposki JA, Tan BK, Comeau MR, Peters AT, Hulse KE, et al. Thymic stromal lymphopoietin activity is increased in nasal polyps of patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2013;132:593–600e512. doi: 10.1016/j.jaci.2013.04.005. doi: 10.1016/j.jaci.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boita M, Garzaro M, Raimondo L, Riva G, Mazibrada J, Vizio B, et al. The expression of TSLP receptor in chronic rhinosinusitis with and without nasal polyps. Int J Immunopathol Pharmacol. 2011;24:761–8. doi: 10.1177/039463201102400322. doi: 10.1177/039463201102400322. [DOI] [PubMed] [Google Scholar]
- 30.Shin HW, Kim DK, Park MH, Eun KM, Lee M, So D, et al. IL-25 as a novel therapeutic target in nasal polyps of patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2015;135:1476–85.e1477. doi: 10.1016/j.jaci.2015.01.003. doi: 10.1016/j.jaci.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Liao B, Cao PP, Zeng M, Zhen Z, Wang H, Zhang YN, et al. Interaction of thymic stromal lymphopoietin, IL-33, and their receptors in epithelial cells in eosinophilic chronic rhinosinusitis with nasal polyps. Allergy. 2015;70:1169–80. doi: 10.1111/all.12667. doi: 10.1111/all.12667. [DOI] [PubMed] [Google Scholar]
- 32.Kamekura R, Kojima T, Takano K, Go M, Sawada N, Himi T, et al. The role of IL-33 and its receptor ST2 in human nasal epithelium with allergic rhinitis. Clin Exp Allergy. 2012;42:218–28. doi: 10.1111/j.1365-2222.2011.03867.x. doi: 10.1111/j.1365-2222.2011.03867.x. [DOI] [PubMed] [Google Scholar]
- 33.Akasaki S, Matsushita K, Kato Y, Fukuoka A, Iwasaki N, Nakahira M, et al. Murine allergic rhinitis and nasal Th2 activation are mediated via TSLP- and IL-33-signaling pathways. Int Immunol. 2016;28:65–76. doi: 10.1093/intimm/dxv055. doi: 10.1093/intimm/dxv055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao X, Sun Y, Wang W, Sun Y. Interleukin (IL)-25: Pleiotropic roles in asthma. Respirology. 2016;21:638–47. doi: 10.1111/resp.12707. doi: 10.1111/resp.12707. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell PD, O’Byrne PM. Epithelial-derived cytokines in asthma. Chest. 2017;151:1338–44. doi: 10.1016/j.chest.2016.10.042. doi: 10.1016/j.chest.2016.10.042. [DOI] [PubMed] [Google Scholar]
- 36.Watson B, Gauvreau GM. Thymic stromal lymphopoietin: A central regulator of allergic asthma. Expert Opin Ther Targets. 2014;18:771–85. doi: 10.1517/14728222.2014.915314. doi: 10.1517/14728222.2014.915314. [DOI] [PubMed] [Google Scholar]
- 37.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 38.Shipe R, Burdick MD, Strieter BA, Liu L, Shim YM, Sung SS, et al. Number, activation, and differentiation of circulating fibrocytes correlate with asthma severity. J Allergy Clin Immunol. 2016;137:750–57e753. doi: 10.1016/j.jaci.2015.07.037. doi: 10.1016/j.jaci.2015.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang CH, Punde TH, Huang CD, Chou PC, Huang TT, Wu WH, et al. Fibrocyte trafficking in patients with chronic obstructive asthma and during an acute asthma exacerbation. J Allergy Clin Immunol. 2015;135:1154–62e1151-5. doi: 10.1016/j.jaci.2014.09.011. doi: 10.1016/j.jaci.2014.09.011. [DOI] [PubMed] [Google Scholar]