

Abstract
The mouse has become an indispensable and versatile model organism for the study of development, genetics, behavior, and disease. The application of comprehensive gene expression profiling technologies to compare normal and diseased tissues or to assess molecular alterations resulting from various experimental interventions has the potential to provide highly detailed qualitative and quantitative descriptions of these processes. Ideally, to interpret experimental data, the magnitude and diversity of gene expression for the system under study should be well characterized, yet little is known about the normal variation of mouse gene expression in vivo. To assess natural differences in murine gene expression, we used a 5406-clone spotted cDNA microarray to quantitate transcript levels in the kidney, liver, and testis from each of 6 normal male C57BL6 mice. We used ANOVA to compare the variance across the six mice to the variance among four replicate experiments performed for each mouse tissue. For the 6 kidney samples, 102 of 3,088 genes (3.3%) exhibited a statistically significant mouse variance at a level of 0.05. In the testis, 62 of 3,252 genes (1.9%) showed statistically significant variance, and in the liver, there were 21 of 2,514 (0.8%) genes with significantly variable expression. Immune-modulated, stress-induced, and hormonally regulated genes were highly represented among the transcripts that were most variable. The expression levels of several genes varied significantly in more than one tissue. These studies help to define the baseline level of variability in mouse gene expression and emphasize the importance of replicate microarray experiments.
Keywords: DNA microarray|variation|transcript|ANOVA
The use of DNA microarrays to obtain qualitative and quantitative profiles of gene expression has increased dramatically over the past several years. Microarrays can provide rapid and accurate measurements of thousands of distinct transcripts simultaneously. Most of the microarray expression studies performed to date have used relatively controlled systems that are manipulable in vitro, such as single-cell organisms (e.g., yeast) and clonal cell lines (1–3). The technology has also been applied to more complex in vivo systems involving mammalian tissues and organs. Many of these studies have been performed by using the mouse as a model organism, in part because of the relative ease of genetic manipulation coupled with the extensive genomic, anatomical, and physiological synteny with humans. Microarrays have been used to analyze gene expression in murine liver (4–8), kidney (9), brain (10), adipose tissue (11), pancreas (12), placenta (13), skeletal muscle (14), and heart (15). It is likely that murine applications for microarray analysis will continue to expand as functional genomics efforts increasingly use the mouse for determining genotypic and phenotypic relationships in the context of development and disease.
Although the use of DNA microarray technology for the study of gene expression in complex mouse tissues is certainly informative, several concerns are apparent that do not exist for single-celled organisms or clonal cell populations. Tissues are comprised of several distinct cell types that may be present in different proportions in different mice. Second, the environment of an organ cannot be controlled. Even genetically identical mice housed under the same conditions are likely to have a different hormonal milieu. The state of the immune system and the degree of inflammatory activity in a given tissue are also likely to vary from mouse to mouse. Third, the process of killing the animal may itself cause global changes in gene expression that are inconsistent from one mouse to the next, especially if time-intensive dissection of the organ is necessary. This process is particularly problematic in studies concerned with stress-responsive genes.
These problems with in vivo studies of gene expression are not new but they are of great importance when using DNA microarrays or other comprehensive expression profiling technologies because of the sheer number of genes analyzed. When assaying the expression of thousands of transcripts, there is a high likelihood of finding “differentially expressed” genes that actually vary as a result of technical limitations of the method or that vary normally in the tissues under study. Before microarray data from complex tissues and organisms can be interpreted meaningfully, it is first necessary to define the normal physiological variance in gene expression. Natural variability is also interesting from a biological standpoint. A component of the variability in gene expression for outbred populations such as humans is likely the result of genotypic variation. However, inbred mouse populations are genetically alike, allowing one to study how gene expression varies independently of genetics.
In this report, we describe the results of using cDNA microarrays to ascertain the variance in transcript levels for several thousands of genes expressed in normal mouse tissues. By using ANOVA, we determined that 0.8, 1.9, and 3.3% of all transcripts assayed were normally variable in the liver, testis, and kidney, respectively. The expression levels of several genes varied significantly in more than one tissue. Several of these genes have been reported previously as differentially expressed in microarray studies of murine development or disease states. These results emphasize the requirement for rigorous experimental design when using microarrays to study gene expression in complex tissues.
Materials and Methods
Animal Studies and RNA Preparation.
Six male C57BL6 mice were individually killed in a CO2 chamber at 15 weeks of age. The liver, kidney, and testis were removed, in that order, and in each case the left kidney and left testis were used only. Care was taken to ensure the time between time of death and harvest of each of the organs was as rapid and consistent as possible. All organs were harvested in the same sitting, and only 30 min elapsed between the time of death of the first mouse to the last. Organs were snap-frozen in liquid nitrogen immediately after harvest. Total RNA was extracted from tissue by using the TRIzol reagent (Life Technologies, Grand Island, NY) according to the manufacturer's protocol. For an RNA reference standard, equal quantities of total RNA from the livers, testes, and kidneys of each mouse were combined to produce a composite RNA pool representing 18 organs. The same reference RNA was used for synthesizing cDNA reference probes for each microarray experiment.
Microarray Preparation.
Replicate-spotted cDNA microarrays were prepared on polylysine-coated glass microscope slides by using a robotic spotting tool as described (2). Each array consisted of 5,285 mouse cDNAs chosen from the Research Genetics sequence-verified set of image clones (http://www.resgen.com/products/SVMcDNA.php3). The clone inserts were amplified by PCR, purified, and verified by gel electrophoresis. Additional control and reference cDNAs were included for a total of 5,406 unique genes represented on the array.
Probe Construction, Microarray Hybridization, and Data Acquisition.
The protocol used for indirect labeling of cDNAs was a modification of a protocol described elsewhere (http://cmgm.stanford.edu/pbrown/protocols/aadUTPCouplingProcedure.htm). Briefly, cDNA probes were made from 30 μg of total RNA in a reaction volume of 30 μl containing oligo(dT) (16) primer/0.2 mM 5-(3-aminoallyl)-2′-deoxyuridine-5′-triphosphate (amino acid-dUTP; Sigma-Aldrich)/0.3 mM dTTP/0.5 mM each dATP, dCTP, and dGTP/380 units of Superscript II reverse transcriptase (Life Technologies). Purified cDNA was combined with either Cy3 or Cy5 monoreactive fluors (Amersham Pharmacia) that covalently couple to the cDNA-incorporated aminoallyl linker in the presence of 50 mM NaHCO3 (pH 9.0). Reference and experimental probes were combined and competitively hybridized to microarrays under a coverslip for 16 h at 63°C. Slides were washed in graded SSC (1 × SSC = 0.15 M sodium chloride/0.015 M sodium citrate, pH 7) and spun dry. Fluorescent array images were collected for Cy3 and Cy5 emissions by using a GenePix 4000A fluorescent scanner (Axon Instruments, Foster City, CA). Image intensity data were extracted and analyzed by using GENEPIX 3.0 microarray analysis software. Each experiment was performed in quadruplicate (two experiments with each fluorescent label to account for dye effects).
Quantitative Real-Time PCR.
cDNA was generated from 30 μg of total RNA from each sample by using the same protocol described previously for array probe synthesis except that amino acid-dUTP was not added to the reaction. After removal of primers and salts with a Microcon 30 filter (Amicon), the cDNA was quantitated in duplicate by using 2 μl of undiluted cDNA in a Gene-Spec III spectrophotometer (Hitachi, Tokyo). Real-time PCR reactions were performed in quadruplicate by using 5 ng of cDNA template, 0.3 μM of each primer, and 1× SYBR green PCR master mix (Applied Biosystems) in a volume of 50 μl. Reactions were analyzed on an Applied Biosystems 5700 sequence detector by using a fluorescence threshold corresponding to the middle of the exponential range. For each primer set, a standard curve was generated by using 10, 1, and 0.1 ng of cDNA. For the 10-fold dilutions the difference in threshold cycle number was always between 3.2 and 3.4, indicating high PCR efficiency. Control reactions with RNA as template and with template omitted did not produce significant amplification products. Primers to ribosomal protein S16 were used to normalize cDNA loading as described (16). The sequences of the primers used in this study are CisH forward, 5′-GGTGGGGCACAACATAGAGA-3′; CisH reverse, 5′-GGTGGCCAGACAGACAGGAG-3′ (102-bp amplicon); Bcl-6 forward, 5′-CACACCCGTCCATCATTGAA-3′; Bcl-6 reverse, 5′-TGTCCTCACGGTGCCTTTTT-3′ (50-bp amplicon); complement factor D forward, 5′-CCACGTGAGACCCCTACCCT-3′; complement factor D reverse, 5′-CCGGGTTCCACTTCTTTGTC-3′ (50-bp amplicon); S16 forward, 5′-AGGAGCGATTTGCTGGTGTGGA-3′; and S16 reverse, 5′-GCTACCAGGCCTTTGAGATGGA-3′ (102-bp amplicon).
Data Analysis.
A gene was considered expressed if the spot had at least 6 foreground pixels greater than 4 standard deviations above background on every array. For each spot, the expression levels of Cy5 and Cy3 probes were obtained by subtracting median backgrounds from median foregrounds. The logarithm (log) base 2 ratios of these two channels were taken to quantify the relative expression levels of genes between experimental and control samples. To allow for interarray comparisons, each array was normalized to remove systematic sources of variation. Instead of using global mean or median gene expression values for each array, a print-tip-specific intensity-based normalization method was used (17). A scatter-plot smoother, which uses robust locally linear fits, was applied to capture the dependence of the log ratios on overall log-spot intensities. The log ratios were normalized by subtracting the fitted values based on the print-tip-specific scatter-plot smoother from the log ratios of experimental and control samples. To assess potential systematic experimental variation resulting from different batches of arrays, different RNA preparations, or other unanticipated factors, we examined the scales of the normalized log ratios for each gene from every experiment. A comparison of boxplots of the log ratios across all arrays for each tissue indicated that the spread of log ratios varied somewhat from array to array with some variation attributable to different array printings (data available at www.pedb.org). We did not observe a correlation with any other identified experimental variable. To account for these differences in the overall analysis, each array was scale-adjusted so that the median of the deviation from the median, a robust estimate of scale, was the same for all arrays from each tissue. It is important to note that other systematic sources of variation may still exist which could influence the experimental results obtained by using microarray methods. For example, there may be temporal changes in individual pin-tip performance or in the concentration of spotting material during a microarray printing run.
ANOVA models were used to identify genes whose variability in expression among mice is greater than zero. Dye effect was incorporated as a covariate to account for the possible systematic difference in expression values between the two dyes. “Array variance,” defined as the mean of squared errors that are not explained by either dye or mouse effect, and “mouse variance,” defined as one-fourth the difference between the mean square for mouse and the array variance were estimated for each gene according to the following formulas:
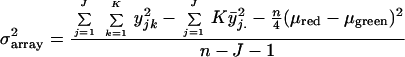 |
1 |
 |
where yjk denotes the log ratio for the jth mouse and kth replicate, and n is the total number of arrays, J⋅K. Subscript j. refers to the mean of all arrays for a given mouse and .. refers to the grand mean of all mice and all replicates. μred and μgreen refer to the mean of all arrays in which the experimental RNA was labeled with Cy5 or Cy3, respectively. F values were obtained by the division of mean square for mouse and the array variance. The degrees of freedom associated with the F values are (no. of mice − 1) and (no. of arrays − no. of mice − 1).
The Westfall and Young step down-adjusted P values were used to adjust for the multiple comparisons. A permutation algorithm proposed by Dudoit et al. (17) was adopted for the calculation of these adjusted P values, where the t test statistics were replaced by the F statistics. A total of 1,000 permutations were conducted. A significance level of 0.05 is used throughout this article unless otherwise stated. For simplicity, the analysis was restricted to the subset of genes that have complete data on all arrays for each tissue. Additional array analyses and statistical data may be viewed in Figs. 4–12, which are published as supporting information on the PNAS web site, www.pnas.org.
Results
Experimental Outline.
To identify genes whose expression levels vary normally in the mouse, we isolated RNA from the kidney, liver, and testis of 6 genetically identical male C57BL6 mice. An experimental reference was created by combining equivalent amounts of RNA from each of the three organs of each mouse. This reference RNA was used as the control for every array experiment to make all experiments comparable. Four separate microarray assays were conducted for each organ from each animal, for a total of 24 arrays per organ. For half of the replicate arrays, the experimental RNA was labeled with the Cy3 dye and the reference RNA with the Cy5 dye; for the other half, the labeling scheme was reversed to control for any dye-based bias.
Determining Variance: Finding Normally Variable Genes.
For each expressed gene, the variance of log ratios among the six mice (mouse variance) was calculated as described in Materials and Methods. Looking across all expressed genes, the testis showed the highest average gene expression variance among mice at 0.054. The average gene expression variance in the kidney was a little lower than the testis at 0.038, whereas the liver showed more stable gene expression with an average variance of 0.018 (Table 1). An F value was calculated for each gene corresponding to the ratio of expression differences among mice divided by the experimental error (array variance). The higher the F value of a gene, the more likely it is that the gene is truly variable among mice. The distribution of F values for all expressed genes shows that the kidney and testis had larger F values overall than the liver, suggesting more gene expression variability in the kidney and testis and less in the liver (Fig. 1). Comparing the observed F distribution to the null distribution demonstrates that all three organs are shifted toward higher F values than would be expected by chance. P values adjusted for multiple comparisons were calculated based on the F values as described in Materials and Methods. The majority of genes did not vary significantly among mice (P > 0.05), but an unexpected number of genes showed considerable variance. The kidney had the greatest number of variable genes with 3.3% of all genes having a P ≤ 0.05. Of testis genes, 1.9% varied significantly among the mice, and 0.8% of the genes expressed in liver were variable (Table 1). The most highly variable genes from each of the three tissues are listed in Fig. 2. Additional significantly variable genes can be viewed at http://www.pedb.org.
Table 1.
Summary of results
| Government parameter | Kidney | Liver | Testis |
|---|---|---|---|
| No. of mice sampled | 6 | 6 | 6 |
| Replicate arrays per sample | 4 | 4 | 4 |
| Total no. of expressed genes on array | 3,088 | 2,514 | 3,252 |
| Variable genes at P < 0.05 | 102 (3.3%) | 21 (0.8%) | 62 (1.9%) |
| Average mouse variance | 0.038 | 0.018 | 0.054 |
Figure 1.

Histograms of F values. The frequency of log2-transformed F values is shown for all of the expressed genes in the kidney, liver, and testis. A higher F value indicates a higher likelihood that a gene is variable among the six mice. For each gene, the F value is calculated as the ratio of the mean square of log ratios among the six mice divided by the experimental error (array variance) among replicate experiments (see Materials and Methods and text). For comparison, an F distribution under the null hypothesis with degrees of freedom 5 and 17 is also plotted. The circle on the null distribution corresponds to a type I error rate of P = 0.05.
Figure 2.

Variable genes in the kidney, liver, and testis. The most statistically significant variable genes in the kidney, liver, and testis are listed with a graphical depiction of the relative expression of each gene in the six mice. Red indicates higher relative expression and green indicates lower expression. Relative intensity refers to the average spot intensity of the gene relative to the mean spot intensity of all expressed genes on the array. ΔFold refers to the difference in gene expression levels among the mice with the highest and lowest measurements. The P values listed are adjusted for multiple comparisons. Relative expression levels of genes in bold face were confirmed by alternative methods. Genes that are variable in both the kidney and the liver are marked with a 1. The gene marked with a 2 is variable in both the kidney and the testis.
One concern is that highly expressed genes will tend to have higher F values as a result of a greater variance. However, plots of F value vs. intensity revealed little correlation between intensity and F value (data available at www.pedb.org). The majority of the statistically significant variable genes had an average intensity value near the mean of all expressed genes on the array (Fig. 2).
Genes with Variable Expression in the Kidney.
Of 3,088 genes with detectable expression in the kidney, 102 (3.3%) were significantly variable among mice. Among the most highly variable were several immune-modulated and stress-responsive genes, including BCL-6, complement factor D, uromodulin, and CisH (Fig. 2). CisH belongs to the SOCS family of proteins that negatively regulate cytokine signaling. The transcription of this gene is known to be induced by a variety of cytokines, suggesting that the variability in gene expression seen among mice may be related to the cytokine milieu of the animal (18). SOCS genes are also known to be regulated by growth hormone (GH) by means of STAT5b activation (19). BCL-6 is a ubiquitously expressed transcriptional repressor that participates in the repression of STAT6-dependent IL4-induced genes (20). Mice deficient in BCL-6 develop an inflammatory disease characterized by abnormal expression of the T helper (Th)-2 cytokines IL-4, -5, and -13 (21).
To confirm the microarray results, the transcript levels of complement factor D, CisH, and BCL-6 were assessed by quantitative real-time reverse transcription (RT)-PCR in each of the kidney samples by using SYBR green as a fluorescent reporter (Fig. 3A). Replicate reactions produced highly reproducible results with standard deviations of 0.2-fold. The assays confirmed the variable expression of these genes, and the direction of the relative fold differences was concordant with the microarray data. In general, the magnitude of relative expression differences was greater in the RT-PCR assays compared with microarray results, a finding we have also observed with Northern analysis (22). The transcript levels for the ribosomal S16 gene, a gene not expected to exhibit tissue expression variation, was also measured in the kidney and liver samples. RT-PCR results demonstrated very consistent levels of S16 expression among all samples assayed (Fig. 3C).
Figure 3.

Confirmation of microarray data by quantitative real-time RT-PCR. Kidney (A) and liver (B) RNAs were reverse transcribed and amplified by the PCR, using real-time quantitative amplicon measurements with primers specific for complement factor D, CisH, BCL-6, and ribosomal protein S16 genes. S16 expression levels were used to normalize real-time PCR data although there was not more than a 1.5-fold difference in S16 expression between any two mice in the kidney or the liver (C). Results are expressed relative to the lowest expressing mouse for each gene (adjusted to a value of 1). Error bars indicate the standard deviation of four microarray or four real-time PCR experiments. Some error bars are not visible because of small standard deviations.
Genes with Variable Expression in the Liver.
The liver had the fewest variable genes of the 3 organs studied with only 21 of 2,514 genes showing statistically significant variability among the 6 mice (Fig. 2). Two of the genes, BCL-6 and CisH, were variable in both the kidney and the liver. As in the kidney, there was a relatively large representation of immune-modulated and stress-responsive genes that showed variability including Gadd45, MKP-1, CisH, BCL-6, and Cyp4a12. Gadd45 is inducible by growth arrest and DNA damage, and there is evidence that it is induced at the transcriptional level by the stress kinase p38 (23). p38 has also been shown to activate the dual specificity mitogen-activated protein (MAP) kinase phosphatase MKP-1, suggesting there may be a correlation in the variability of Gadd45 and MKP-1 (24).
The Sin3-associated protein, Sap30, functions in the Sin3/Rpd3 histone deacetylase complex (25). Histone deacetylases are thought to act as global repressors of transcription by restricting access of transcription factors to their target sites. Interestingly, the transcriptional repressor BCL-6 has also been linked to histone deacetylases through its interaction with the SMRT and N-CoR corepressors (26). That Sap30 and BCL-6 were quite variable in untreated mice suggests that there may be global differences in transcriptional regulation even in the absence of genetic heterogeneity.
The relative expression levels of BCL-6 and CisH among the six mouse livers were confirmed by quantitative real-time RT-PCR (Fig. 3B). As with the analysis of these genes in the kidney, the direction of the relative fold differences were concordant with the microarray data, and the magnitude of relative expression differences was greater in the RT-PCR assays compared with microarray results. The relative transcript levels of the Gadd45 gene were also confirmed by Northern analysis, and the results were in agreement with the microarray data (results not shown).
Genes with Variable Expression in the Testis.
Of 3,252 genes with detectable expression in the testis, 62 genes (1.9%) were significantly variable. Genes exhibiting the greatest variance represent a diverse range of functions including vesicle transport (coatomer, ARF-4), proteasomal (Psma1), immune function (LPAAT-4, β-2 microglobulin), cell stress (DnaJ homolog 2/HSP40, DNA-PK, Pidd) nuclear transport (RAN-binding protein 16), transcription regulation (USF2), and lipid transport [apolipoprotein (apo) C1] (Fig. 2). Two genes varied in both the kidney and the testis including β-2 microglobulin and an uncharacterized expressed sequence tag (GenBank AI450295).
The most variable genes in the testis were LPAAT-4, ApoC1, and calmodulin-3, with LPAAT-4 and ApoCI each varying by more than 50-fold (Fig. 2). LPAAT-4 is an acyltransferase involved in lipid metabolism that has been shown to enhance cytokine-induced signaling responses (27). The apoCI is a component of very low-density lipoprotein (VLDL) and high density lipoprotein (HDL) that is predominantly expressed in the liver, although it has been shown to be expressed in the testis (28). Calmodulin is a ubiquitously expressed and abundant Ca2+-binding protein that functions in many cellular processes, including transcriptional regulation through CaM kinase and the transcription factor CREB (29).
Discussion
If these mice are genetically identical, the same age, and housed under the same conditions, then to what can we attribute the variability in gene expression? Several possibilities exist, all of which are important to consider when analyzing gene expression alterations in response to experimental manipulations. Mouse to mouse differences in immune status may be one of the largest sources of variation. We found that many of the variable genes encode components of the immune system and are mediators of the acute immune response. The inflammatory state of the tissue at the time of death may vary depending on environmental influences like injury or infection. The measured variability may be the result of differential gene expression in the primary cellular components of the tissue or may reflect the presence of different numbers of tissue-infiltrating lymphoid cells.
We hypothesized that many normally variable genes would be regulated by cytokines or hormones. This hypothesis is supported by the differences observed in measurements of cytokine-inducible genes, as well as a cohort of putative hormone-responsive genes. For example, the recently characterized short-chain dehydrogenase/reductase PSDR1 was discovered as an androgen-regulated gene in human prostate (22). Another variable gene, complement factor D, is known to be regulated by insulin (30). The cytokine-inducible genes CisH and BCL-6 are also regulated by GH (31). Some evidence suggests that Gadd45 and the p38 stress kinase are induced by GH in the liver (32). In male mice, GH is produced by the pituitary in a pulsatile fashion, and fluctuations in the amount of GH may have contributed to the variability in expression of these genes.
Another factor that may contribute to expression variability is the process of killing the animal. Although great care was taken to remove the organs and snap-freeze them quickly, it is difficult to control this process perfectly. A host of gene expression changes may occur immediately after death as a result of hypoxia and ischemia. Stress-induced genes that were variable, such as DNA-activated protein kinase, Pidd, and heat shock protein 40 in the testis, and MKP-1, Gadd45, and cytochrome p450 gene cyp4a12 in the liver, may be variable as a result of differences in the response to the process of death. Examining the pattern of variance in the six mice reveals a nonrandom mouse effect for multiple genes. For example, kidney gene expression patterns from the first four mice are fundamentally different from the last two mice (Fig. 2). In the testis, the first two mice are systematically different from the last four mice (Fig. 2). No pattern was observed in the liver. Although all mice were killed within a 30-min time period, it is possible that the time of death had an effect on gene expression levels, perhaps from natural hourly fluctuations in hormone levels or from a response induced by removing (and killing) littermates.
Importantly, many of the genes that we found to vary normally have been reported previously to be differentially expressed because of a pathological process or experimental intervention. One recent study used microarrays to investigate the effect of aging and caloric restriction on gene expression in the skeletal muscle of male C57BL6 mice (14). Heat shock proteins, including DnaJ Homolog 2 (Hsp40) and other stress-responsive genes such as Gadd45, were reported to be differentially expressed as a result of dietary alterations. Calmodulin 3 and 60S ribosomal protein L32 were described as differentially expressed with age, genes we found to vary normally in the testis and kidney, respectively. Although it is likely that many of the genes reported in this study are truly differentially expressed with age and/or caloric restriction, we found the same kinds of genes, and indeed some of the very same genes, among age-matched C57BL6 mice without dietary intervention. Also, the small sample size (n = 3) makes this study particularly vulnerable to misinterpretation due to normal variation in gene expression.
Several studies have used microarrays to profile gene expression in mouse liver. One report described the effects of thyroid hormone treatment on liver gene expression and found that tctex-1, β-globin, and two FK506-binding proteins were regulated by T3 (4). These were all genes we have found to be normally variable in at least one tissue. Another study investigated the effects of acetaminophen on gene expression in the mouse liver (8). Eight of the genes reported to differ in response to acetaminophen, including Gadd45, CisH2, and Hsp40, were genes we found to vary normally. A third study used microarrays to compare apoE-deficient mice to controls and reported that apoCI was differentially expressed as well as several heat shock proteins (6). These expression differences may accurately reflect the influence of apoE, but the high level of normal variance in these genes makes this difficult to determine without rigorous control experiments.
Genetically diverse populations such as humans are likely to show even greater variability in gene expression than what we have observed among inbred mice. In addition, environmental conditions cannot be carefully controlled in humans. These factors present challenges for microarray-based studies of human gene expression in vivo. Meaningful interpretation of global gene expression in humans will require an extensive characterization of normal variability. Our data suggests that both specific genes and functional classes of genes will be consistently variable, even in multiple tissue types. To assist future investigations of gene expression, a comprehensive database of normally variable genes could be created for both mouse and human tissues and organs. This database might be used to caution investigators about highly variable genes, and could also identify and catalog cohorts of genes with relatively stable expression.
Supplementary Material
Acknowledgments
We thank Cassie Neal and Ryan Basom in the DNA microarray facility at the Fred Hutchinson Cancer Research Center and Hugh Arnold for their assistance with cDNA array experiments. We thank Barbara Trask and Stan Nelson for critical reviews of the manuscript. We thank Jerry Radich and Charles Kooperberg for helpful discussions. This work was supported in part by National Institutes of Health Grants DK59125 and CA75173 (to P.S.N.) and a Poncin Foundation Award (to C.C.P.).
Abbreviations
- RT
reverse transcription
- GH
growth hormone
- log
logarithm
- apo
apolipoprotein
References
- 1.Lasharki D A, DeRisi J L, McCusker J H, Namath A F, Gentile C, Hwang S Y, Brown P O, Davis R W. Proc Natl Acad Sci USA. 1997;94:13057–13062. doi: 10.1073/pnas.94.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 3.Ross D T, Scherf U, Eisen M B, Perou C M, Rees C, Spellman P, Iyer V, Jeffrey S S, Van de Rijn M, Waltham M, et al. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 4.Feng X, Jiang Y, Meltzer P, Yen P M. Mol Endocrinol. 2000;14:947–955. doi: 10.1210/mend.14.7.0470. [DOI] [PubMed] [Google Scholar]
- 5.Callow M J, Dudoit S, Gong E L, Speed T P, Rubin E M. Genome Res. 2000;10:2022–2029. doi: 10.1101/gr.10.12.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang G S, Yang S M, Hong M Y, Yang P C, Liu Y C. Life Sci. 2000;68:19–28. doi: 10.1016/s0024-3205(00)00912-7. [DOI] [PubMed] [Google Scholar]
- 7.McNeish J, Aiello R J, Guyot D, Turi T, Gabel C, Aldinger C, Hoppe K L, Roach M L, Royer L J, de Wet J, et al. Proc Natl Acad Sci USA. 2000;97:4245–4250. doi: 10.1073/pnas.97.8.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reilly T P, Bourdi M, Brady J N, Pise-Masison C A, Radonovich M F, George J W, Pohl L R. Biochem Biophys Res Commun. 2001;282:321–328. doi: 10.1006/bbrc.2001.4576. [DOI] [PubMed] [Google Scholar]
- 9.Nagasawa Y, Takenaka M, Kaimori J, Matsuoka Y, Akagi Y, Tsujie M, Imai E, Hori M. Nephrol Dial Transplant. 2001;16:923–931. doi: 10.1093/ndt/16.5.923. [DOI] [PubMed] [Google Scholar]
- 10.Yoshikawa T, Nagasugi Y, Azuma T, Kato M, Sugano S, Hashimoto K, Masuho Y, Muramatsu M, Seki N. Biochem Biophys Res Commun. 2000;275:532–537. doi: 10.1006/bbrc.2000.3330. [DOI] [PubMed] [Google Scholar]
- 11.Soukas A, Cohen P, Socci N D, Friedman J M. Genes Dev. 2000;14:963–980. [PMC free article] [PubMed] [Google Scholar]
- 12.Dusetti N J, Tomasini R, Azizi A, Barthet M, Vaccaro M I, Fiedler F, Dagorn J C, Iovanna J L. Biochem Biophys Res Commun. 2000;277:660–667. doi: 10.1006/bbrc.2000.3734. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka T S, Jaradat S A, Lim M K, Kargul G J, Wang X, Grahovac M J, Pantano S, Sano Y, Piao Y, Nagaraja R, et al. Proc Natl Acad Sci USA. 2000;97:9127–9132. doi: 10.1073/pnas.97.16.9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee C K, Klopp R G, Weindruch R, Prolla T A. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 15.Friddle C J, Koga T, Rubin E M, Bristow J. Proc Natl Acad Sci USA. 2000;97:6745–6750. doi: 10.1073/pnas.100127897. . (First Published May 30, 2000; 10.1073/pnas.100127897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foley K P, Leonard M W, Engel J D. Trends Genet. 1993;9:380–385. doi: 10.1016/0168-9525(93)90137-7. [DOI] [PubMed] [Google Scholar]
- 17.Dudoit, S., Yang, Y. H., Callow, M. J. & Speed, T. P. (2000) Technical report, Department of Statistics, University of California at Berkeley.
- 18.Krebs DL, Hilton D J. J Cell Sci. 2000;113:2813–2819. doi: 10.1242/jcs.113.16.2813. [DOI] [PubMed] [Google Scholar]
- 19.Choi H K, Waxman D J. Growth Horm IGF Res. 2000;10, Suppl. B:S1–S8. doi: 10.1016/s1096-6374(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 20.Harris M B, Chang C C, Berton M T, Danial N N, Zhang J, Kuehner D, Ye B H, Kvatyuk M, Pandolfi P P, Cattoretti G, et al. Mol Cell Biol. 1999;19:7264–7275. doi: 10.1128/mcb.19.10.7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dent A L, Shaffer A L, Yu X, Allman D, Staudt L M. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 22.Lin B, White J T, Ferguson C, Wang S, Vessella R, Bumgarner R, True L D, Hood L, Nelson P S. Cancer Res. 2001;61:1611–1618. [PubMed] [Google Scholar]
- 23.Oh-Hashi K, Maruyama W, Isobe K. Free Radical Biol Med. 2001;30:213–221. doi: 10.1016/s0891-5849(00)00461-5. [DOI] [PubMed] [Google Scholar]
- 24.Hutter D, Chen P, Barnes J, Liu Y. Biochem J. 2000;352:155–163. [PMC free article] [PubMed] [Google Scholar]
- 25.Laherty C D, Billin A N, Lavinsky R M, Yochum G S, Bush A C, Sun J M, Mullen T M, Davie J R, Rose D W, Glass C K, et al. Mol Cell. 1998;2:33–42. doi: 10.1016/s1097-2765(00)80111-2. [DOI] [PubMed] [Google Scholar]
- 26.Huynh K D, Bardwell V J. Oncogene. 1998;17:2473–2484. doi: 10.1038/sj.onc.1202197. [DOI] [PubMed] [Google Scholar]
- 27.West J, Tompkins C K, Balantac N, Nudelman E, Meengs B, White T, Bursten S, Coleman J, Kumar A, Singer J W, Leung D W. DNA Cell Biol. 1997;16:691–701. doi: 10.1089/dna.1997.16.691. [DOI] [PubMed] [Google Scholar]
- 28.Jong M C, Hofker M H, Havekes L M. Arterioscler Thromb Vasc Biol. 1999;19:472–484. doi: 10.1161/01.atv.19.3.472. [DOI] [PubMed] [Google Scholar]
- 29.Toutenhoofd S L, Foletti D, Wicki R, Rhyner J A, Garcia F, Tolon R, Strehler E E. Cell Calcium. 1998;23:323–338. doi: 10.1016/s0143-4160(98)90028-8. [DOI] [PubMed] [Google Scholar]
- 30.Miner J L, Byatt J C, Baile C A, Krivi G G. Physiol Behav. 1993;54:207–212. doi: 10.1016/0031-9384(93)90100-t. [DOI] [PubMed] [Google Scholar]
- 31.Waxman D J. Novartis Found Symp. 2000;227:61–74. doi: 10.1002/0470870796.ch5. and 75–81. [DOI] [PubMed] [Google Scholar]
- 32.Thompson B J, Shang C A, Waters M J. Endocrinology. 2000;141:4321–4324. doi: 10.1210/endo.141.11.7874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}