

Key Points
Quizartinib at 60 mg/day (vs 30 mg/day) was associated with higher overall response, survival, and bridge to transplant.
The benefit-risk profile of quizartinib in relapsed or refractory FLT3-ITD–mutated AML warrants further evaluation of 60-mg once-daily dose.
Abstract
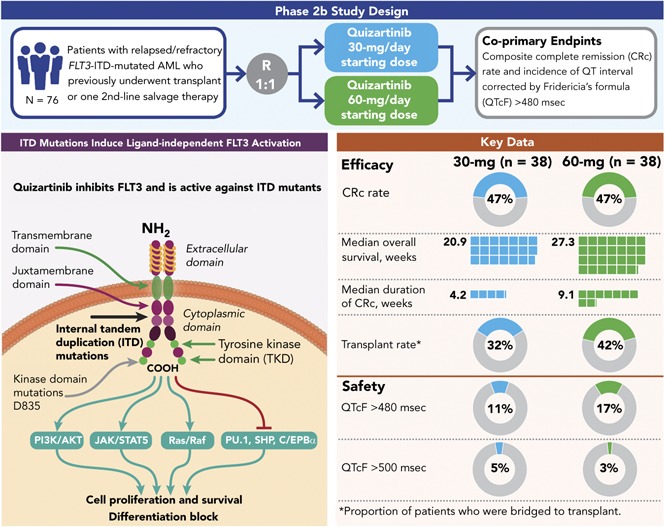
This randomized, open-label, phase 2b study (NCT01565668) evaluated the efficacy and safety of 2 dosing regimens of quizartinib monotherapy in patients with relapsed/refractory (R/R) FLT3-internal tandem duplication (ITD)–mutated acute myeloid leukemia (AML) who previously underwent transplant or 1 second-line salvage therapy. Patients (N = 76) were randomly assigned to 30- or 60-mg/day doses (escalations to 60 or 90 mg/day, respectively, permitted for lack/loss of response) of single-agent oral quizartinib dihydrochloride. Allelic frequency of at least 10% was defined as FLT3-ITD–mutated disease. Coprimary endpoints were composite complete remission (CRc) rates and incidence of QT interval corrected by Fridericia’s formula (QTcF) of more than 480 ms (grade 2 or greater). Secondary endpoints included overall survival (OS), duration of CRc, bridge to transplant, and safety. CRc rates were 47% in both groups, similar to earlier reports with higher quizartinib doses. Incidence of QTcF above 480 ms was 11% and 17%, and QTcF above 500 ms was 5% and 3% in the 30- and 60-mg groups, respectively, which is less than earlier reports with higher doses of quizartinib. Median OS (20.9 and 27.3 weeks), duration of CRc (4.2 and 9.1 weeks), and bridge to transplant rates (32% and 42%) were higher in the 60-mg groups than in the 30-mg group. Dose escalation occurred in 61% and 14% of patients in the 30- and 60-mg groups, respectively. This high clinical activity of quizartinib at the evaluated doses is consistent with previous reports with an improved safety profile. Need to dose-escalate more than half of patients who received quizartinib 30 mg also supports further investigation of treatment with quizartinib 60 mg/day.
Visual Abstract
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease with multiple factors influencing long-term outcome.1,2 FLT3 mutations (predominantly internal tandem duplication [ITD], reported in ∼25% of patients with AML)3-5 are common molecular abnormalities in AML. FLT3-ITD mutations are a key oncogenic driver,6-9 and patients with FLT3-mutated AML have poorer outcomes, lower response rates to chemotherapy, increased risk for relapse, and shorter survival compared with patients without FLT3 mutation.4,10-13 As a consequence, the therapeutic potential of kinase inhibitors targeting FLT3 has been investigated. The recent approval of midostaurin, a first-generation multikinase (including FLT3) inhibitor, in combination with chemotherapy in newly diagnosed FLT3-mutated AML, based on outcomes from the RATIFY trial,14 provides support for FLT3 as a viable target in AML. However, there remains a large unmet need for effective treatment options in patients with relapsed or refractory (R/R) FLT3-ITD–mutated AML.
Quizartinib is an orally administered, highly potent and selective next-generation tyrosine kinase inhibitor that inhibits FLT3 and is active against ITD mutants.15-17 Accumulated clinical experience in phase 1 and 2 clinical trials has shown quizartinib to be highly active in R/R FLT3-ITD–mutated AML.18,19 In a first-in-human phase 1 study, the maximum tolerated dose of quizartinib was 200 mgorally daily in patients with R/R AML with QT interval corrected using Fridericia’s formula (QTcF) prolongation as the dose-limiting toxicity.18 Quizartinib demonstrated encouraging clinical activity and was associated with a manageable safety profile.
Results also suggested the potential for complete and sustained inhibition of FLT3 phosphorylation.18 In a subsequent phase 2 study (NCT00989261), efficacy and safety of quizartinib monotherapy was evaluated in 2 independent cohorts: patients at least 60 years of age with R/R AML within 1 year after first-line therapy (cohort 1) and those at least 18 years of age with R/R disease after salvage chemotherapy or allogeneic hematopoietic stem cell transplant (HSCT; cohort 2).19 Initial treatment with 200 mg/day yielded a higher rate of QTcF prolongation than expected; therefore, lower doses (90 and 135 mg/day) were explored. QTcF prolongation was reversible and successfully managed by treatment interruption and/or dose reductions. QTcF above 500 ms was reported in 17% and 15% of patients treated with 90 and 135 mg/day, respectively.19 These results demonstrated that single-agent quizartinib was highly active (composite complete remission [CRc] rate 46% in FLT3-ITD-positive patients, with 35% of patients bridging to HSCT [cohort 2]) and generally well-tolerated in patients with R/R AML (particularly those with FLT3-ITD mutations).
The study reported here evaluated 2 different dosing regimens of single-agent quizartinib to determine whether the same clinical activity could be achieved while improving the safety profile in patients with R/R FLT3-ITD-mutated AML who had previously received HSCT or 1 second-line salvage therapy.
Methods
Study design
NCT01565668 was a phase 2b open-label, randomized study in adult (age, ≥18 years) patients with morphologically documented primary AML or AML secondary to myelodysplastic syndrome, as defined by the World Health Organization criteria and confirmed by pathology review at the treating institution. Patients were randomly assigned to 1 of 2 dosing regimens with quizartinib dihydrochloride monotherapy: a 30-mg once-daily starting-dose group (equivalent to 26.5 mg free base) or a 60-mg once-daily starting-dose group (equivalent to 53 mg free base). Each group allowed protocol-specified dose escalation for lack/loss of response and dose reduction/interruption for adverse events (AEs). Patients received quizartinib oral solution daily in 28-day cycles until disease progression, intolerance, or HSCT. Quizartinib dihydrochloride powder was reconstituted in sterile water at a concentration of 5 mg/mL with a final volume of 6 or 12 mL (for 30- and 60-mg doses, respectively).
The study was conducted per the Declarations of Helsinki and Good Clinical Practice guidelines. All patients provided written informed consent.
Eligibility criteria.
Patients were required to have been refractory to or relapsed after HSCT or 1 second-line salvage regimen and were required to have FLT3-ITD-activating mutation in bone marrow or peripheral blood. Patients were deemed eligible on the basis of local laboratory results, with all samples subsequently tested at a central laboratory to confirm FLT3 mutation status using a previously published method.20 The FLT3-ITD allelic burden was calculated as the percentage of FLT3-ITD-mutated (dominant allele) to total FLT3 (wild type + FLT3-ITD-mutated) in samples sent to the central laboratory. Patients with an allelic frequency of at least 10% were considered to have FLT3-ITD-mutated disease. Allelic frequencies were not corrected for blast percentages. Patients with FLT3-ITD-mutated allelic ratio lower than 10% identified in the confirmatory FLT3 testing at the central laboratory were allowed to remain in the study and were included in the intent-to-treat (ITT) population. The decision to keep these patients on study was at the discretion of the investigators if they felt the treatment could offer benefit. Patients who had received prior FLT3 inhibitor therapy were allowed. Additional inclusion criteria were Eastern Cooperative Oncology Group performance status of 0 to 2 and adequate renal, hepatic, and coagulation parameters, as indicated by the following laboratory values: aspartate aminotransferase and alanine aminotransferase ≤2.5 × institutional upper limit of normal (ULN), total bilirubin ≤1.5 × institutional ULN, serum creatinine ≤1.5 × institutional ULN, and glomerular filtration rate >30 mL/min (calculated by Cockcroft and Gault formula). Patients with acute promyelocytic leukemia, clinically active central nervous system leukemia, or treatment-related myeloid neoplasm were excluded. Patients with QTcF of at least 450 ms were excluded. Concomitant treatment with drugs that prolonged QT/QTc interval or strong inhibitors/inducers of cytochrome P450-isozyme 3A (CYP3A) were prohibited unless these agents were deemed essential to patient care by the investigator. These agents included, but were not limited to, antibiotics, antifungals, and other antimicrobials that were used as standard of care for the prevention or treatment of infections. (supplemental Appendix 1, supplemental Table 1, available on the Blood Web site). No study drug dose modifications were required in patients receiving such concomitant therapies, but patients underwent additional electrocardiogram monitoring.
Criteria for dose reductions and increases.
Dose escalation from 30 to 60 mg or from 60 to 90 mg was permitted in patients who did not achieve CR, or CR with incomplete platelet recovery (CRp), or CR with incomplete hematologic recovery (CRi) by the end of cycle 1 (ie, day 28), or in those who achieved a response (CR/CRp/CRi/partial remission [PR]) and later relapsed. Dose reduction (from 60 to 30 mg and subsequently to 20 mg, or from 30 to 20 mg) with or without dose interruption was required for grade 2 or higher QTcF prolongation, persistent grade 3 or higher nonhematologic AEs, or myelosuppression in patients achieving CRp/CRi who had received at least 2 cycles of treatment. No dose modifications were required for patients receiving concomitant drugs that prolonged QTcF interval or strong inhibitors/inducers of CYP3A.
Primary and secondary endpoints.
The coprimary objectives were to evaluate the CRc rate (defined as the rate of CR+CRp+CRi) and the rate of grade ≥2 QTcF (>480 ms). Responses to quizartinib were based on the Cheson criteria but were modified for CRi and PR (supplemental Appendix 1).21
Secondary endpoints reported here include the CR rate, overall survival (OS), event-free survival, leukemia-free survival, time to CRc, duration of CRc, rate of patients bridged to HSCT, and overall safety. Other secondary endpoints included pharmacokinetic and pharmacodynamic analyses and will be reported separately. Definitions of efficacy endpoints are presented in supplemental Appendix 1, supplemental Methods.
Study assessments.
Electrocardiograms were obtained at screening, predose, and 2, 4, and 6 hours postdose on days 1 and 15 of cycle 1; predose and 2 hours postdose on day 8 of cycle 1; predose on day 22 of cycle 1; and predose on day 1 of all subsequent cycles and were centrally reviewed. Patients receiving a CYP3A inhibitor or a drug known to cause QT/QTc prolongation during the study were required to undergo additional electrocardiogram monitoring predose and 2 hours postdose on days 1 and days 8 of subsequent cycles. Bone marrow biopsies were obtained at screening and on day 1 of each cycle, starting with cycle 2.
Statistical analysis.
The sample size was based on precision consideration for the rate estimate of CRc and QTcF prolongation (grade ≥2). A sample size of 32 patients per group was estimated to result in a ∼25% width of the 2-sided 90% confidence interval (CI) for both QTcF prolongation (grade ≥2) and CRc rates.
Primary efficacy analyses were conducted in the ITT population, defined as all randomized patients, according to their randomized treatment groups. Safety analyses were conducted in the safety population, defined as all patients who received at least 1 dose of study drug, according to their randomized treatment groups. Efficacy measurements were summarized using descriptive statistics for the initial dose level. Survival curves and medians for the time-to-event analyses were estimated using the Kaplan-Meier method and reported along with the corresponding 95% CIs. In patients achieving a CRc who subsequently relapsed, the duration of CRc was measured from the start of the first observed response to the date of documented relapse; in those patients who did not relapse, the duration of CRc was censored at the last evaluation visit at which the patient was known to be relapse-free, or at the end of treatment of those patients bridging to HSCT. For OS analysis, OS was censored at the date of last contact. Date of last contact was defined as the latest of the following dates: treatment discontinuation date, last dosing administration date, last disease assessment date, or the last follow-up date on which the patient was known to be alive. Long-term survival was defined as an OS ≥ 1 year.
Results
Patient characteristics
Between May 2012 and March 2015, 76 patients were enrolled (ITT population; Figure 1). Baseline characteristics were generally well balanced between the 2 treatment groups (Table 1). Median age was 55 years (range, 19-77 years). The majority of patients (67%) had intermediate cytogenetic risk at baseline. Cytogenetic risk classifications were comparable in the 30-mg group (favorable, 0; intermediate, 68%; unfavorable, 11%; unknown 21%) and 60-mg group (favorable, 5%; intermediate, 66%; unfavorable, 8%; unknown 18%). Comutations (NPM1 and CEBPA) known to influence AML prognosis were also similar across groups (Table 1). The distribution of FLT3-ITD–mutated allelic ratio in the 30- and 60-mg groups, respectively, were ≥10% to ≤ 25% (21% and 11%), ≥25% to ≤50% (53% and 34%), and >50% (18% and 45%). There were 3 patients in each group with FLT3 allele burden < 10% at central review confirmation, 2 of whom had undetectable levels (Table 1). Patients received a median 3 prior chemotherapy regimens for AML (range, 1-9), and 28% of patients had prior HSCT, 92% prior anthracycline, and 15% prior FLT3 inhibitors. Overall, 70% of patients were refractory to their last AML therapy and 30% had documented response (CR or PR), with median duration of response being 6.5 months (range, 0.4-18.0 months).
Figure 1.

CONSORT flowchart.
Table 1.
Patient characteristics and treatment history (ITT population)
| Quizartinib 30-mg group* (n = 38) | Quizartinib 60-mg group† (n = 38) | Total (N = 76) | |
|---|---|---|---|
| Secondary AML, n (%) | 3 (8) | 7 (18) | 10 (13) |
| Median age (range), y | 57 (19-77) | 53 (20-74) | 55 (19-77) |
| Male, n (%) | 22 (58) | 22 (58) | 44 (58) |
| Race, n (%) | |||
| White | 29 (76) | 30 (79) | 59 (78) |
| Black or African American | 1 (3) | 2 (5) | 3 (4) |
| Other or missing‡ | 8 (21) | 6 (16) | 14 (18) |
| Median weight (range), kg | 76.8 (40-116) | 75.1 (47-101) | 75.9 (40-116) |
| ECOG PS, n (%)§ | |||
| Grade 0 | 8 (21.1) | 7 (18.4) | 15 (19.7) |
| Grade 1 | 23 (60.5) | 24 (63.2) | 47 (61.8) |
| Grade 2 | 7 (18.4) | 4 (10.5) | 11 (14.5) |
| FLT3-ITD–mutated allelic ratio, n (%)|| | 37 (97) | 36 (95) | 73 (96) |
| >0 to < 10% | 2 (5) | 2 (5) | 4 (5) |
| ≥10% and ≤ 25% | 8 (21) | 4 (11) | 12 (16) |
| ≥25% and ≤ 50% | 20 (53) | 13 (34) | 33 (43) |
| >50% | 7 (18) | 17 (45) | 24 (32) |
| FLT3-ITD size, median (range) base pairs¶ | 51.0 (21-201) | 54.2 (18-114) | 54.0 (18-201) |
| Risk status with specific cytogenetic patterns, n (%)# | |||
| Favorable | 0 | 2 (5) | 2 (3) |
| Intermediate | 26 (68) | 25 (66) | 51 (67) |
| Unfavorable | 4 (11) | 3 (8) | 7 (9) |
| Unknown | 8 (21) | 7 (18) | 15 (20) |
| AML with recurrent genetic abnormalities | |||
| AML with mutated NPM1 | 8 (21) | 11 (29) | 19 (25) |
| AML with mutated CEBPA | 0 | 0 | 0 |
| Previous HSCT, n (%) | 9 (24) | 12 (32) | 21 (28) |
| Prior AML chemotherapy regimens, median (range)** | 3 (1-6) | 3 (1-9) | 3 (1-9) |
| Prior anthracycline treatment, n (%)** | 35 (92) | 33 (92) | 68 (92) |
| Refractory, n (%)** | 26 (68) | 26 (72) | 52 (70) |
| Relapsed, n (%)** | 12 (32) | 10 (28) | 22 (30) |
| Duration of best response (CR or PR) to last AML therapy, months, median (range)** | 5 (0.4-12) | 8 (1-18) | 6.5 (0.4-18) |
| Prior FLT3 therapy, n (%)**†† | 5 (13) | 6 (17) | 11 (15) |
Quizartinib 30 mg and 60 mg are equivalent to 26.5 mg and 53 mg free base, respectively.
ECOG PS, Eastern Cooperative Oncology Group performance status.
30-mg starting dose with permitted escalation to 60 mg for lack of or loss of initial response.
60-mg starting dose with permitted escalation to 90 mg for lack of or loss of initial response.
Ethnicity was not collected from patients in France per local regulations.
At screening, all patients had ECOG performance status ≤2 per eligibility criteria. The baseline ECOG scores reflects data collected immediately before first on-study treatment dose, and could have changed from the screening ECOG scores.
FLT3-ITD-mutated allele was not detectable (below the assay’s limit of detection) in 1 patient each in the 30-mg group and the 60-mg group. In addition, value is missing for 1 patient in the 60-mg group, who was randomized, specimen was not received at central laboratory, and patient did not receive quizartinib.
Total number of patients, N (%) = 30-mg group, 37 (97); 60-mg group, 36 (95); total, 73 (96).
Cytogenetic information based on available data.
Total number of patients (Safety analysis set), N = 30-mg group, 38; 60-mg group, 36; total, 74.
10 patients received sorafenib and 1 patient received both sorafenib and midostaurin.
Seventy-four patients received at least 1 dose of quizartinib (30-mg group, n = 38; 60-mg group, n = 36; Figure 1; Table 2). Overall, 18 (47%) patients in the 30-mg group and 23 (64%) patients in the 60-mg group had dose reductions/interruptions for management of AEs. As allowed per protocol, 23 (61%) of 38 patients in the 30-mg group were escalated to 60-mg/day quizartinib and 5 (14%) of 36 patients in the 60-mg group were escalated to 90-mg/day quizartinib (Table 2).
Table 2.
Quizartinib treatment exposure and dose modifications (safety population)
| Quizartinib 30-mg group* (n = 38) | Quizartinib 60-mg group† (n = 36)‡ | Total (N = 74) | |
|---|---|---|---|
| Median duration of treatment (range), wk | 9.4 (2.1-32.7) | 10.1 (1.7-109) | 10.0 (1.7-109) |
| Dose interrupted, n (%) | 8 (21) | 13 (36) | 21 (28) |
| Dose reduced, n (%) | 10 (26) | 10 (28) | 20 (27) |
| Dose escalated, n (%) | 23 (61) | 5 (14) | 28 (38) |
Quizartinib 30 mg and 60 mg are equivalent to 26.5 mg and 53 mg free base, respectively.
30-mg starting dose with permitted escalation to 60 mg for lack of or loss of initial response.
60-mg starting dose with permitted escalation to 90 mg for lack of or loss of initial response.
Two patients were randomized but did not receive drug because of ineligibility.
Overall, 24% of patients received concomitant medications with a potential for QTcF prolongation (9 patients in each group) and 53% received a strong CYP3A inhibitor (18 and 21 patients in the 30- and 60-mg groups, respectively; supplemental Appendix 1, supplemental Table 1).
Efficacy results
Of the 76 patients in the ITT analysis set, 47.4% achieved a best response of CRc (18 patients in each group; Table 3). Of the patients achieving CRc, 12 of 18 in the 30-mg group and 11 of 18 in the 60-mg group had CRc at the end of cycle 1. In addition, 5 of 38 patients in the 30-mg group and 9 of 38 patients in the 60-mg group achieved PR. Thus, the overall response rate (ORR; defined as CRc+PR) was 61% in 30-mg group and 71% in the 60-mg group (Table 3). The median durations of CRc and OS were longer in the 60-mg group (4.2 [95% CI, 2.1-9.7] and 9.1 [95% CI, 4.1-22.3] weeks, and 20.9 [95% CI, 17.7-25.3] and 27.3 [95% CI, 17.3-34.9] weeks in the 30- and 60-mg groups, respectively; Table 3; Figure 2). Six patients were considered long-term survivors (total OS duration ≥ 1 year): 1 in the 30-mg group and 5 in the 60-mg group.
Table 3.
Key efficacy outcomes (ITT population)
| Quizartinib 30-mg group* (n = 38) | Quizartinib 60-mg group† (n = 38) | Total (N = 76) | |
|---|---|---|---|
| Overall response (CRc+PR), n (%) | 23 (60.5) | 27 (71.1) | 50 (65.8) |
| CRc (CR+CRp+CRi), n (%) | 18 (47.4) | 18 (47.4) | 36 (47.4) |
| CR, n (%; 95% CI) | 2 (5.3; 0.6-17.7) | 1 (2.6; 0.1-13.8) | 3 (3.9; 0.8-11.1) |
| CRp, n (%; 95% CI) | 0 | 2 (5.3; 0.6-17.7) | 2 (2.6; 0.3-9.2) |
| CRi, n (%; 95% CI) | 16 (42.1; 26.3-59.2) | 15 (39.5; 24.0-56.6) | 31 (40.8; 29.6-52.7) |
| PR, n (%; 95% CI) | 5 (13.2; 4.4-28.1) | 9 (23.7; 11.4-40.2) | 14 (18.4; 10.5-29.0) |
| Median duration of CRc (95% CI), wk‡ | 4.2 (2.1-9.7) | 9.1 (4.1-22.3) | 5.4 (4.1-11.9) |
| Median time to CRc (95% CI), wk | 4.4 (4.1-7.7) | 4.6 (4.1-8.0) | 4.5 (4.3-6.6) |
| Bridge to HSCT transplant rate, n (%) | 12 (31.6) | 16 (42.1) | 28 (36.8) |
| Median OS (95% CI), wk‡,§ | 20.9 (17.7-25.3) | 27.3 (17.3-34.9) | 22.6 (19.9-28.3) |
| Deaths, n (%) | 36 (94.7) | 30 (78.9) | 66 (86.8) |
| Censored, n (%) | 2 (5.3) | 8 (21.1) | 10 (13.2) |
| Median EFS (95% CI), wk | 12.0 (8.3-16.1) | 13.7 (9.7-26.1) | 12.3 (9.7-16.1) |
| Median LFS (95% CI), wk | 4.1 (2.1-9.7) | 9.1 (4.0-22.3) | 5.3 (4.1-11.9) |
Quizartinib 30 mg and 60 mg are equivalent to 26.5 mg and 53 mg free base, respectively. Responses were assessed using modified Cheson criteria as described in supplemental Appendix 1.
EFS, event-free survival; and LFS, leukemia-free survival.
30-mg starting dose with permitted escalation to 60 mg for lack of or loss of initial response.
60-mg starting dose with permitted escalation to 90 mg for lack of or loss of initial response.
From Kaplan-Meier analysis.
Reflects median OS until the time of database lock (study termination).
Figure 2.

Kaplan-Meier plots. (A) Duration of CRc and (B) OS (ITT analysis set). *30-mg starting dose with permitted escalation to 60 mg for lack of or loss of initial response. †60-mg starting dose with permitted escalation to 90 mg for lack of or loss of initial response. QD, once daily.
Twelve (32%) patients in the 30-mg group and 16 (42%) in the 60-mg group bridged to HSCT (Table 3). The last recorded response before discontinuing quizartinib for HSCT was CR in 3 patients, CRi in 12 patients, PR in 6 patients, and no response in 6 patients. One patient did not have a response evaluation before HSCT. Patients who bridged to HSCT were not permitted to restart quizartinib after transplant. Four of the 6 long-term survivors were patients who bridged to HSCT: 1 in the 30-mg group and 3 in the 60-mg group.
Exploratory ad hoc analyses
In an exploratory ad hoc analysis, the CRc rate without dose escalation was 37% (14/38 patients) in patients randomly assigned to the 30-mg group. Of 23 patients with dose escalations to 60 mg, 4 achieved CRc after dose escalation (3 escalated for lack of initial response and 1 for loss of initial response). In patients randomly assigned to the 60-mg group, the CRc rate without dose escalation was 47% (18/38 patients). Dose escalation to 90 mg occurred in 5 patients (14%), and no patient achieved CRc after dose escalation.
Six patients with FLT3-ITD-mutated allelic ratios that were found to be below the protocol-defined cutoff during central laboratory confirmation benefited from quizartinib treatment. Among the 4 patients with FLT3-ITD-mutated allelic ratio lower than 10% in central FLT3 testing, 3 patients achieved a response (1 CR; 2 CRi). Both the patients in whom FLT3-ITD-mutated allele was not detectable achieved a response (1 CR; 1 CRp).
Safety and toxicity results
Assessment of QTcF prolongation showed that 11% of patients in the 30-mg group and 17% of patients in the 60-mg group had QTcF higher than 480 ms (≥ grade 2; Table 4). Grade 3 QTcF prolongation (>500 ms) was reported in 5% and 3% of patients in the 30- and 60-mg groups, respectively; there were no instances of torsades de pointes or other grade 4 ventricular arrhythmias or sudden death. Nine patients had an increase in QTcF of more than 60 ms from baseline; 3 with QTcF values more than 500 ms and 6 with less than 500 ms. Of these, 1 patient with QTcF more than 500 ms experienced ventricular tachycardia 22 days after discontinuing quizartinib and while receiving multiple other QT-prolonging agents for treatment of pneumonia. Of the remaining 8 patients, 2 were receiving 30 mg, 5 were receiving 60 mg, and 1 was receiving 90 mg quizartinib after having been dose escalated from 60 mg. In these 8 patients, median time to QTcF prolongation was 19 days (range, 8-113 days); median age at onset was 59 years, and 5 were women. QTcF prolongation resolved or improved within 7 days in all patients. Resolution/improvement in QTcF occurred in 5 patients with no adjustments in quizartinib dosing, and 3 patients recovered after dose interruption. Two of these 3 patients were successfully re-challenged with no QTcF elevation exceeding Common Terminology Criteria for Adverse Events grade 1. All patients with QTcF higher than 500 ms and/or more than a 60-ms change from baseline had risk factors for QTcF prolongation, including electrolyte abnormalities and concomitant use of medications associated with QTcF prolongation and/or strong CYP3A inhibitors.
Table 4.
QTc prolongations with quizartinib (safety population)
| Quizartinib 30-mg group* (n = 38) | Quizartinib 60-mg group† (n = 36) | |
|---|---|---|
| Maximum value of QTcF, n (%), ms‡ | ||
| ≥450 to ≤480 | 16 (42.1) | 17 (47.2) |
| >480 to ≤500 | 2 (5.3) | 5 (13.9) |
| >500 | 2 (5.3) | 1 (2.8) |
| Maximum change from baseline in QTcF, n (%), ms‡ | ||
| >30 to ≤60 | 18 (47.4) | 15 (41.7) |
| >60 | 2 (5.3) | 7 (19.4) |
Quizartinib 30 mg and 60 mg are equivalent to 26.5 mg and 53 mg free base, respectively.
30-mg starting dose with permitted escalation to 60 mg for lack of or loss of initial response.
60-mg starting dose with permitted escalation to 90 mg for lack of or loss of initial response.
Criteria for maximum QTcF duration and maximum QTcF change from baseline are not mutually exclusive (ie, the same patient could have met either or both criteria).
Grade 3 or higher treatment-emergent adverse events (TEAEs), regardless of relationship to study treatment reported in at least 10% of patients by initial dose group (supplemental Appendix 2, supplemental Table 2) were most frequently hematologic; gastrointestinal TEAEs were typically grade 2 or below. Adverse events were considered treatment-related in 59 (80%) of 74 patients in the Safety Population. Treatment-related TEAEs (TR-TEAEs; includes both “probably” and “possibly” related AEs) were reported at similar rates across both groups (Table 5). The most common TR-TEAEs were hematologic events (anemia [20%], febrile neutropenia [11%]), gastrointestinal events (nausea [16%], diarrhea [11%]), and fatigue (12%). Four (5.4%) patients discontinued treatment because of TR-TEAEs (1 patient with pericardial effusion and pericarditis; 1 patient each with diarrhea, neutropenic sepsis, and pleural effusion), all of whom had been randomly assigned to the 30-mg group. However, in 2 patients, onset of the event leading to discontinuation occurred after dose escalation to 60 mg.
Table 5.
Treatment-related TEAEs (all grades) reported in ≥10% of patients per dose group (safety population)
| Treatment-related TEAE by preferred term* | Quizartinib 30-mg group, n (%)† (n = 38) | Quizartinib 60-mg group, n (%)‡ (n = 36) | Total, n (%) (n = 74) |
|---|---|---|---|
| Overall | 30 (78.9) | 29 (80.6) | 59 (79.7) |
| Anemia | 8 (21.1) | 7 (19.4) | 15 (20.3) |
| Nausea | 4 (10.5) | 8 (22.2) | 12 (16.2) |
| Fatigue | 5 (13.2) | 4 (11.1) | 9 (12.2) |
| Febrile neutropenia | 4 (10.5) | 4 (11.1) | 8 (10.8) |
| Diarrhea | 4 (10.5) | 4 (11.1) | 8 (10.8) |
| Electrocardiogram QT prolonged | 2 (5.3) | 5 (13.9) | 7 (9.5) |
| Thrombocytopenia | 4 (10.5) | 2 (5.6) | 6 (8.1) |
| Abdominal pain | 2 (5.3) | 4 (11.1) | 6 (8.1) |
| Neutropenia | 1 (2.6) | 4 (11.1) | 5 (6.8) |
| Vomiting | 1 (2.6) | 4 (11.1) | 5 (6.8) |
| Dysgeusia | 4 (10.5) | 1 (2.8) | 5 (6.8) |
| Dyspepsia | 4 (10.5) | 0 | 4 (5.4) |
Quizartinib 30 mg and 60 mg are equivalent to 26.5 mg and 53 mg free base, respectively.
Patients may have more than 1 treatment-related TEAE per preferred term. Patients are counted once per preferred term.
30-mg starting dose with permitted escalation to 60 mg for lack of or loss of initial response.
60-mg starting dose with permitted escalation to 90 mg for lack of or loss of initial response.
Serious adverse events (SAEs) were considered by the investigator to be treatment-related in 10 (26%) patients in the 30-mg group and 8 (22%) patients in the 60-mg group. The most common treatment-related SAEs in the 30-mg group were febrile neutropenia (3 events) and thrombocytopenia, pericardial effusion, and gastrointestinal hemorrhage (2 events each). The most common treatment-related SAEs in the 60-mg group included febrile neutropenia and QT prolongation (2 events each). Among deaths not attributed to AML, the leading causes regardless of relationship to treatment were infections and respiratory/thoracic disorders. Two events occurred in the same patient in the 30-mg group who was dose escalated to 60 mg (fatal pericardial effusion and pleural effusion) that were considered possibly related to study drug.
The majority of patients with elevated liver enzymes had values within 3 to 5 times the ULN, and no patient met Hy’s criteria (bilirubin >2 × ULN concurrently with alanine aminotransferase and/or aspartate aminotransferase >3 × ULN with no increase in alkaline phosphatase; supplemental Appendix 3, supplemental Table 3).
Discussion
Relapsed/refractory FLT3-ITD-positive AML has a poor prognosis and a low response rate to salvage therapy.22,23 At present, there are no approved therapies specifically targeting FLT3-ITD mutations in the R/R setting, thereby representing a major unmet need for patients who require more effective and tolerable therapies that offer the possibility to bridge to transplant. The results of this phase 2b study evaluating 2 dosing regimens (30 and 60 mg) of single-agent quizartinib, an oral, highly potent, and selective next-generation FLT3 inhibitor, demonstrated promising antileukemic activity and an improved safety profile, particularly in terms of QTcF prolongation. Quizartinib was generally well tolerated with a manageable safety profile and an observed incidence of grade 3 QTcF prolongation of 3% to 5%.
The overall CRc rate (47%) and ORR (66%) reported here were consistent with those observed in a previous study of higher doses of quizartinib in R/R FLT3-ITD-mutated AML.19 The ORR, duration of CRc, and median OS were numerically higher in the 60-mg group. Responses observed with quizartinib were rapid, and 37% and 47% of patients who achieved CRc did so by the end of cycle 1 in the 30- and 60-mg groups, respectively. Dose escalations to 60 mg were more frequent in the 30-mg group, and more patients in the 60-mg group achieved a CRc without a dose escalation compared with the 30-mg group.
The patients in this study had very poor prognosis as a result of having FLT3-ITD–mutated AML and having heavily pretreated R/R disease (median 3 prior chemotherapy regimens, prior anthracyclines in 92%, and history of HSCT in 28%). Eligible patients were enrolled regardless of the duration of response to prior therapy, with most (70%) patients refractory to their last treatment. The median OS of 21 to 27 weeks observed in this study is clinically relevant as, historically, patients with FLT3-ITD–mutated AML in first relapse were reported to have a median survival of ∼13 weeks after standard chemotherapy.22
As HSCT is an important goal for patients with R/R AML, the ability to provide HSCT to more patients is of benefit to patients with an otherwise low probability of long-term survival.24 In our study, the bridge to HSCT rate in the 60-mg group (42%) was higher than in the 30-mg group (32%) and was substantially higher than historical data (8% in the UK NCRI database).25 This analysis demonstrated that patients who were transplanted had a longer OS and that quizartinib may offer the opportunity of bridging patients to transplant.25 Quizartinib was not restarted after transplant in this study; however, it may provide benefit in the posttransplant setting. On this basis, ongoing studies have been designed to allow for the reinitiation of quizartinib therapy after transplant.26,27
Consistent with prior phase 1 and 2 studies,18,19 responses were documented in 2 patients with undetectable FLT3-ITD mutation. The mechanism by which quizartinib induced remission in patients with undetectable FLT3-ITD mutations is unknown and requires further investigation.
Quizartinib was specifically developed to target FLT3 and demonstrates a high specificity for this kinase in vitro.17 Higher specificity could potentially result in improved therapeutic activity in patients with FLT3-ITD-mutated AML. The promising clinical activity (46%-56% CRc rate) of quizartinib monotherapy (90 or 135 mg) in an earlier phase 2 study19 is reinforced by this study, in which quizartinib dosing regimens of 30 and 60 mg/day (with escalation to 60 and 90 mg/day, as necessary) demonstrated comparable CRc rates (47%). The high CRc rate, ORR (71%), and bridge to HSCT rate (42%) in the 60-mg quizartinib group offer a promising treatment option with this highly potent and selective FLT3 inhibitor, given the limited single-agent efficacy with less-selective FLT3 inhibitors.28-31 The response rates observed with quizartinib in the current study are comparable to those with gilteritinib in a study with a similar patient population and using similar response criteria.31 Taken together, these findings demonstrate clinically meaningful antitumor activity with quizartinib monotherapy in patients with R/R FLT3-ITD–mutated AML.
Quizartinib was generally well tolerated with similar TR-TEAE profiles in both dose groups, possibly because a substantial proportion of patients in the 30-mg group were dose escalated to 60 mg during the study. The grade 2 or greater incidence of QTcF prolongation of 11% in the 30-mg group and 17% in the 60-mg group are relevant given the higher rates in previous reports with higher doses.18,19 Moreover, the incidence of grade 3 QTcF prolongation (>500 ms) was also substantially lower in this study (5% and 3% in 30- and 60-mg groups, respectively, with no grade 4 or higher events) than in the earlier phase 2 trial using higher quizartinib doses (15% and 17% in 135- and 90-mg groups, respectively, with 1 grade 4 event).19 None of the patients in this study experienced arrhythmias associated with QTcF prolongation while receiving quizartinib treatment, supporting an acceptable benefit-risk profile for quizartinib in this difficult-to-treat patient population. Additional analyses of data from this study regarding pharmacokinetics/pharmacodynamics modeling aimed at examining the relationship between quizartinib dose and QTcF prolongation will be reported in a separate publication.
The phase 2 trial design and sample size may limit the generalizability of the results from this study. In addition, the study was not powered to allow for statistical comparative analyses between groups. The numeric difference in efficacy outcomes between the 30- and 60-mg/day dose groups may have been influenced by differences in baseline prognostic factors, lack of standardization in making dose-escalation decisions, the rate of dose escalation for lack/loss of response, and/or the censoring in CRc duration calculations at the end of treatment of patients bridged to HSCT.
In summary, the totality of evidence from the 2 phase 2 studies of quizartinib monotherapy in R/R FLT3-mutated AML suggests quizartinib may be a valuable treatment option for patients with FLT3-ITD–mutated R/R AML. Further investigation of the quizartinib 60-mg once-daily dose is warranted and this dose is currently under evaluation in an ongoing phase 3 clinical trial (QuANTUM-R) to assess the efficacy of quizartinib as monotherapy vs salvage chemotherapy in patients with R/R FLT3-ITD–mutated AML. The dosing regimen in QuANTUM-R incorporates the 60-mg/day dose with a 30-mg/day lead-in to assess QTcF prolongation before dose escalation. Outcomes from QuANTUM-R are awaited.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Vinay Pasupuleti, Accuverus Inc., for his medical editorial assistance with this manuscript.
Financial support for medical editorial assistance was provided by Daiichi Sankyo. This work was supported in part by the National Institutes of Health, National Cancer Institute Leukemia SPORE P50 CA100632 (J.E.C., H.M.K., and M.J.L.) and Cancer Center Support grant P30 CA16672 (J.E.C. and H.M.K.). This study was sponsored by Daiichi Sankyo, Inc., a member of the Daiichi Sankyo Group.
Footnotes
Presented in poster form at the 2014 American Society of Clinical Oncology 50th Annual Meeting, Chicago, IL, 2 June 2014, and the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3 December 2016.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.E.C. and M.J.L. designed the study; J.E.C., M.S.T., D.T., S.L.G., and M.J.L. collected and analyzed data and wrote the paper; J.E.C., M.S.T., D.T., G.G., S.L.G., J.-P.M., H.M.K., and M.J.L. performed the statistical analysis and wrote the paper; J.E.C., M.S.T., G.J.S., D.T., S.L.G., A.E.P., G.M., and M.J.L. collected data and wrote the paper; and all authors critically revised the manuscript for important intellectual content and approved the manuscript for publication.
Conflict-of-interest disclosure: J.E.C. is in a consulting or advisory role for Ambit, Daiichi Sankyo, Astellas, and Novartis and receives research funding (to his institution) from Ambit, Daiichi Sankyo, Astellas, AROG, Flexus, and Novartis. M.S.T. is in a consulting or advisory role for Daiichi Sankyo and receives research funding from AROG, Cellerant, ADC Therapeutics, and Celgene. G.J.S. receives research funding from Ambit. D.T. was employed by Daiichi Sankyo at the time of the conduct of this trial and is employed by Ambit Biosciences at this time. G.G. was employed by Daiichi Sankyo at the time of the conduct of this trial, is currently in a consulting or advisory role for Daiichi Sankyo (as chief executive officer of Guy Gammon Consulting), owns stock or other ownership in Daiichi Sankyo, and accepted travel provided with accommodations and reimbursement for expenses from Daiichi Sankyo. Current conflict-of-interest information for S.L.G. and A.E.P. may be found on the ASCO Web site. J.-P.M. receives honoraria from ICON Clinical. G.M. is in a consulting or advisory role with Celgene, Pfizer, Amgen, and Ariad and is on the speakers’ bureau for Incyte-Teva. H.M.K. receives honoraria from AbbVie, Actinium, Amgen, Ariad, BMS, Immunogen, Orsuex, and Pfizer and research funding (to his institution) from Amgen, Pfizer, Novartis, BMS, Astex, and Ariad. M.J.L. receives honoraria from Daiichi Sankyo, Novartis, and Agios, is in a consulting or advisory role for Daiichi Sankyo, Novartis, and Agios, and receives research funding from Astellas and Novartis.
The current affiliation for G.G. is Guy Gammon Consulting, San Diego, CA.
The current affiliation for G.M. is Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, Meldola, Italy.
Correspondence: Jorge E. Cortes, Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 0428, Houston, TX 77030; e-mail: jcortes@mdanderson.org.
References
- 1.Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5(3):65-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallogly MM, Lazarus HM. Midostaurin: an emerging treatment for acute myeloid leukemia patients. J Blood Med. 2016;7:73-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kainz B, Heintel D, Marculescu R, et al. Variable prognostic value of FLT3 internal tandem duplications in patients with de novo AML and a normal karyotype, t(15;17), t(8;21) or inv(16). Hematol J. 2002;3(6):283-289. [DOI] [PubMed] [Google Scholar]
- 4.Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752-1759. [DOI] [PubMed] [Google Scholar]
- 5.Santos FP, Jones D, Qiao W, et al. Prognostic value of FLT3 mutations among different cytogenetic subgroups in acute myeloid leukemia. Cancer. 2011;117(10):2145-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532-1542. [DOI] [PubMed] [Google Scholar]
- 7.Lazenby M, Gilkes AF, Marrin C, Evans A, Hills RK, Burnett AK. The prognostic relevance of FLT3 and NPM1 mutations on older patients treated intensively or non-intensively: a study of 1312 patients in the UK NCRI AML16 trial. Leukemia. 2014;28(10):1953-1959. [DOI] [PubMed] [Google Scholar]
- 8.Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003;17(9):1738-1752. [DOI] [PubMed] [Google Scholar]
- 9.Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlenk RF, Döhner K, Krauter J, et al. ; German-Austrian Acute Myeloid Leukemia Study Group. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909-1918. [DOI] [PubMed] [Google Scholar]
- 11.Schnittger S, Schoch C, Dugas M, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100(1):59-66. [DOI] [PubMed] [Google Scholar]
- 12.Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a Cancer and Leukemia Group B study. Cancer Res. 2001;61(19):7233-7239. [PubMed] [Google Scholar]
- 13.Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. 2007;110(4):1262-1270. [DOI] [PubMed] [Google Scholar]
- 14.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galanis A, Levis M. Inhibition of c-Kit by tyrosine kinase inhibitors. Haematologica. 2015;100(3):e77-e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee LY, Hernandez D, Rajkhowa T, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129(2):257-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984-2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cortes J, Perl AE, Döhner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial [published online ahead of print 30 May 2018]. Lancet Oncol. doi:10.1016/S1470-2045(18)30240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy KM, Levis M, Hafez MJ, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. 2003;5(2):96-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheson BD, Bennett JM, Kopecky KJ, et al. ; International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia [published correction appears in J Clin Oncol. 2004;22(3):576]. J Clin Oncol. 2003;21(24):4642-4649. [DOI] [PubMed] [Google Scholar]
- 22.Ravandi F, Kantarjian H, Faderl S, et al. Outcome of patients with FLT3-mutated acute myeloid leukemia in first relapse. Leuk Res. 2010;34(6):752-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pratz KW, Levis M. How I treat FLT3-mutated AML. Blood. 2017;129(5):565-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurosawa S, Yamaguchi T, Miyawaki S, et al. Prognostic factors and outcomes of adult patients with acute myeloid leukemia after first relapse. Haematologica. 2010;95(11):1857-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hills R, Gammon G, Trone D, Burnett A. Quizartinib and bridge to transplant in FLT3-ITD AML patients after failure of salvage chemotherapy: a historical comparison with UK National Cancer Research Institute (NCRI) data. Presented at the 22nd Congress of the European Hematology Association; 22-25 June 2017; Madrid, Spain. Abstract S475. [Google Scholar]
- 26.ClinicalTrials.gov. Quizartinib With Standard of Care Chemotherapy and as Maintenance Therapy in Patients With Newly Diagnosed FLT3-ITD (+) Acute Myeloid Leukemia (AML) (QuANTUM-First). https://clinicaltrials.gov/ct2/show/NCT02668653. Accessed 17 April 2018.
- 27.ClinicalTrials.gov. (QuANTUM-R): An Open-label Study of Quizartinib Monotherapy vs. Salvage Chemotherapy in Acute Myeloid Leukemia (AML) Subjects Who Are FLT3-ITD Positive. https://clinicaltrials.gov/ct2/show/NCT02039726. Accessed 17 April 2018.
- 28.Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339-4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith BD, Levis M, Beran M, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(10):3669-3676. [DOI] [PubMed] [Google Scholar]
- 30.Metzelder SK, Schroeder T, Finck A, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia. 2012;26(11):2353-2359. [DOI] [PubMed] [Google Scholar]
- 31.Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18(8):1061-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.