

Abstract
DNA acquisition via genetic recombination is considered advantageous as it has the potential to bring together beneficial mutations that emerge independently within a population. Furthermore, recombination is considered to contribute to the maintenance of genome stability by purging slightly deleterious mutations. The prevalence of recombination differs among prokaryotic species and depends on the accessibility of DNA transfer mechanisms. An exceptional example is the human pathogen Mycobacterium tuberculosis (MTB) where no clear transfer mechanisms have been so far characterized and the presence of recombination is questioned. Here, we analyze completely assembled MTB genomes in search for evidence of recombination. We find that putative recombination events are enriched in strains reconstructed by reference-guided assembly and in regions with unreliable alignments. In addition, assembly and alignment artefacts introduce phylogenetic signals that are conflicting the established MTB phylogeny. Our results reveal that the so far reported recombination events in MTB are likely to stem from methodological artefacts. We conclude that no reliable signal of recombination is observed in the currently available MTB genomes. Moreover, our study demonstrates the limitations of reference-guided genome assembly for phylogenetic reconstructions. Rigorously de novo assembled genomes of high quality are mandatory in order to distinguish true evolutionary signal from noise, in particular for low diversity species such as MTB.
Keywords: phylogeny, recombination, Mycobacterium tuberculosis, comparative genomics, assembly, alignment
Introduction
The acquisition of foreign DNA by lateral transfer is widespread among prokaryotes (Gogarten and Townsend 2005; Vos and Didelot 2009). Genetic recombination within a species allows for the combination of beneficial mutations that arose independently and can thereby accelerate the process of adaptation (Fisher 1930; Muller 1932). Furthermore, recombination allows species to escape Muller's ratchet (Muller 1964) by decreasing the accumulation of slightly deleterious mutations in the population (Takeuchi et al. 2014). In the absence of recombination, species adaptation is considered slower due to clonal interference, that is, competition between independently emerging genotypes (Hill and Robertson 1966). For example, experimental evolution studies of prokaryotic organisms showed that adaptation is slower when recombination is genetically hindered (Baltrus et al. 2008; Perron et al. 2012).
The human pathogen Mycobacterium tuberculosis (MTB), the causative agent of tuberculosis, is devoid of known lateral transfer mechanisms. Lateral transfer mechanisms were only observed in close relatives of MTB, for example, in the sister group Mycobacterium canetti (Boritsch et al. 2016) and in other mycobacteria (Mortimer and Pepperell 2014). It is also thought that lateral gene transfer (LGT) events occurred in the branch leading to the ancestor of MTB, for example, in a region with high similarity to a plasmid from distant Mycobacteria (Supply et al. 2013). Hence, this property might have been lost in MTB. MTB genomes are extremely conserved, which results in a low intraspecies genetic diversity. Clonal evolution of MTB is an established fact in the community (Gagneux 2013; Coscolla and Gagneux 2014; Niemann et al. 2016), nonetheless, whole-genome evolutionary reconstruction indicated the presence of recombination footprints in MTB (Namouchi et al. 2012). The results of the latter study were suggested to stem from genome assembly artefacts (Bryant 2014). Other attempts to detect recombination (Liu et al. 2006; Karboul et al. 2008; Phelan et al. 2016) identified highly repetitive regions as recombination targets. However, those events can be traced back to intragenome recombination (Boritsch 2014) or they lack an experimental validation. A systematic study for intergenome recombination is still lacking.
The first complete genome of MTB was determined 20 years ago (Cole et al. 1998). These days, there are two major approaches to genome assembly using high-throughput sequencing; raw reads are either assembled de novo, or by reference-guided assembly using a closely related reference (Tatusova et al. 2014). Most analyses in MTB were conducted without assembly of novel isolates; instead, variants of sequenced isolates have been inferred by aligning the raw reads against a reference. Those analyses contributed to the understanding of drug-resistance mutations (Zhang et al. 2013; Cohen et al. 2015; Gygli et al. 2017) and transmission networks during epidemics (Merker et al. 2015; Manson et al. 2017). The availability of sequenced isolates for numerous MTB strains provides an unprecedented resolution for evolutionary analyses of this species. Here, we exploit completely sequenced MTB genomes to systematically test the presence of recombination in MTB.
Materials and Methods
Data Set
We retrieved 41 closed and complete genomes of M. tuberculosis sensu stricto from the RefSeq database (October 27, 2016, supplementary table S1, Supplementary Material online).
Protein Families
Homologous proteins were identified by using blastp v2.5.0+ (Altschul et al. 1990) all-against-all with an e-value threshold of 1e-10. Proteins sharing at least 60% global amino acid identity as computed by needle from the EMBOSS package (Needleman and Wunsch 1970; Rice et al. 2000) were clustered into homologous families using MCL v14-137 (Enright et al. 2002) with inflation factor 2. To infer our reference phylogeny, we first computed the multiple amino acid alignment of each of the 2,650 universal single copy families using MAFFT v7.31 and the auto option (Katoh et al. 2002). Phyml v20131022 (Guindon et al. 2010) was then used to infer the tree with 100 bootstrap replicates.
Extended Set of Protein Families
The presence of protein families in unannotated regions was identified using tblastn of the amino acid sequences against the annotated pseudogenes. The criteria for retaining a significant hit are 1) e-value < 1e-5, 2) length of the hit >80% length of queried protein, and 3) alignment identity >85%.
Universal Genomic Regions
ProgressiveMauve v2.4.0 (Darling et al. 2010) identifies locally collinear blocks that are conserved and free of rearrangements. Aligning the 41 genomes using progressiveMauve identifies 18 universal blocks, that is, blocks present in all 41 genomes (termed universal genomic regions, supplementary table S2, Supplementary Material online). Universal genomic regions are realigned with MAFFT, resulting in a total alignment length of 4,951,692 nt.
Recombination Detection
Recombination is inferred for the universal genomic regions using ClonalFrameML v1.0-20 and the reference phylogeny. Recombined segments are characterized by the start and end position in the alignment and the branch in the phylogeny where the segment is introduced. We only kept recombined segments of length at least 13 nt.
Block Phylogenies
Phylogenies were estimated based on the MAFFT alignments of the universal genomic regions (supplementary table S2, Supplementary Material online). Phylogenies were inferred using phyml, the substitution model GTR+G and 100 bootstrap replicates. A region is incongruent with the reference phylogeny if its phylogeny displays a conflicting branch with bootstrap support of at least 75. We applied the HoT (Landan and Graur 2007) procedure to identify alignment positions where the alignment is unreliable. For a given multiple sequence alignment, HoT computes the forward and the reverse alignment; thereby reliable columns can be found in both alignments. The HoT score is the percentage of reliable columns and is a representation of alignment quality. The package splitstree4 (Huson and Bryant 2006) was used to infer the splits network, using the neighbor net implementation with the uncorrectedP distances.
Results
We analyzed 41 available completely sequenced MTB genomes having an average length of 4.407 Mb and an average number of 3,997 open reading frames (ORFs; supplementary table S1, Supplementary Material online). To establish a reference phylogeny as the basis for subsequent recombination inference, protein sequences were clustered by similarity into 4,272 homologous protein families. This yielded 2,650 (62%) complete single-copy families (i.e., families present in all genomes compared) and 1,539 (36%) partial protein families (i.e., families that are absent in at least one genome). The low proportion of partial protein families in MTB supports the view of a highly conserved protein content. For comparison, the proportion of partial proteins in Escherichia coli is 88% (panX database, accessed January 10, 2018 [Ding et al. 2017]).
Including 1,399 pseudogenes having sequence similarity to protein family members increased the number of complete single-copy families to 3,293 (77%) and reduced the number of partial protein families to 890 (21%) (supplementary fig. S1B, Supplementary Material online). Notably, many of the partial protein families are characterized by the presence–absence pattern in the reference strain H37Rv (NC_000962.3); 38 partial families are H37Rv-specific, whereas 112 partial families are present in all strains but H37Rv (supplementary fig. S1C, Supplementary Material online). The high proportion of variability of the reference strain H37Rv demonstrates that the level of annotation in this strain is out of the ordinary in comparison to other sequenced isolates.
For the inference of recombination, we reconstructed a reference phylogeny from the complete single-copy families (excluding families with pseudogenes; fig. 1). The resulting topology is concordant with the known evolutionary relationships of MTB lineages (Comas et al. 2013). Recombination was inferred from a concatenation of 18 universal genomic regions in the MTB whole-genome alignment, that is, regions that are common to all compared genomes (supplementary table S2, Supplementary Material online). Inferred recombined segments are stretches in the alignment that do not evolve in the clonal frame (Didelot and Wilson 2015). ClonalFrameML identified a total of 1,297 recombined segments. We find an unexpected high proportion of putative recombined segments that are inferred to occur in terminal branches (i.e., occurring in a single genome) (77%; 993 of 1,297). In addition, the segments are unevenly represented among the strains, where 505 (51%) of the putative recombined segments in terminal branches (993) are inferred in nine genomes that stand out due to their assembly procedure. Seven of these genomes were assembled based on a reference genome. Additionally, two of the strains were removed from the RefSeq database during the time of our study due to a high proportion of frame-shifted ORFs indicating a low-quality assembly (personal communication with NCBI; fig. 2A, supplementary table S1, Supplementary Material online). These results indicate that reference-guided assemblies are a source for bias in the inference of recombination events. We consider the nine genome assemblies noted above as problematic for phylogenetic reconstruction.
Fig. 1.
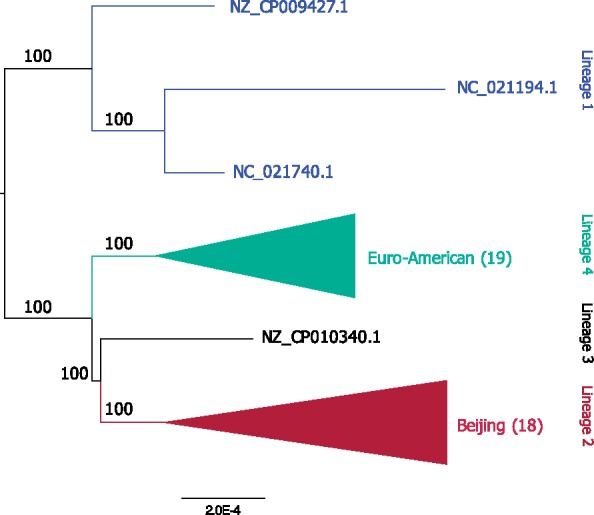
—Phylogenetic tree inferred from the concatenated amino acid alignments of the 2,650 complete single-copy protein families. The total alignment length is 786,198 amino acids, 4,305 (0.55%) of the sites are variable and 1,714 (0.22%) are parsimony informative. The number of strains grouped in the collapsed clade is shown in brackets. See supplementary figure S2, Supplementary Material online for the complete phylogeny. The root position is estimated using midpoint rooting, the MAD method (Tria et al. 2017), and the outgroup method with Mycobacterium canetti based on 2,557 complete single-copy protein families. All approaches infer the root position on the branch splitting lineage 1 from the others; this result is in agreement with previous analyses (Comas et al. 2013).
Fig. 2.

—Properties of recombined segments. Out of the 1,297 recombined segments, 993 (76.56%) segments are inferred to terminal branches and 304 (23.44%) segments are inferred to internal branches. 505 (50.85%) segments from the terminal branches are found on problematic assemblies and 488 (49.15%) segments are found in other strains. (A) Distribution of 993 recombined segments in terminal branches. Colors denote lineages as follows: Blue for lineage 1, red for lineage 2, black for lineage 3, and green for lineage 4 (see also fig. 1). Problematic assemblies are marked with yellow star. (B) Distribution of gap contribution to recombined segments. The proportion of gapped positions in a recombined segment is calculated as the number of alignment positions where at least two strains have no gaps and at least one strain has a gap, divided by the length of the segment (excluding positions with only one strain having no gap).
The universal genomic region alignments are highly conserved with a low proportion of gapped positions (18.55%). Notably, the recombined segments on internal branches exhibit a remarkably high proportion of gapped positions with a mean of 84% gapped positions per segment (median 100%). The high proportion of gapped positions is also observed in recombined segments reconstructed to terminal branches (mean 63%, median 100%; fig. 2B). Because of the high proportion of gaps in recombined segments, we suspect alignment errors have had an influence on the reconstruction of recombined segments. To test for this possibility, we identify alignment positions from the universal region whose alignment is unreliable (Landan and Graur 2007). In total, 682,588 of the 4,951,692 (13.79%) alignment positions are considered unreliable according to this measure. Notably, unreliably aligned positions are overrepresented in putative recombined segments in comparison to the remaining genome alignment (P < 10−6, using χ2 test). Additionally, the proportion of unreliably aligned positions is higher for segments inferred on internal branches in comparison to the putative recombined segments inferred on external branches (P < 10−6, using χ2 test). Our results thus demonstrate that putatively recombined segments largely overlap with genomic regions whose alignment is unreliable. This suggests that the recombination events detected in MTB genomes stem from alignment artefacts.
In summary, we identify two main sources of bias in the inference of recombination in MTB genomes. Recombination events reconstructed in terminal branches are either a sign of a recent recombination (i.e., in the isolate) or an indication for an artefact due to the genome assembly method. The enrichment of recombination events on terminal branches in reference-guided assemblies in comparison to de novo assemblies suggests that reference-guided assemblies are a cause for bias in the recombination detection. We suspect that this is caused by the assembly method that introduces reference variants into the reconstructed sequence. Consequently, all recombination events inferred to terminal branches should be considered with utmost caution. A second source of bias is alignment quality shown by the enrichment of unreliable alignment positions in regions identified as recombination. Thus, the bias introduced at the sequence and alignment level is impeding a reliable inference of recombination in the current MTB data set.
An additional approach to detect lateral transfer is the comparison of gene phylogenies where incongruent topologies constitute a signal of lateral transfer (Ravenhall et al. 2015). The low number of variable sites in the complete single copy amino acid alignments (fig. 1) precludes the reconstruction of reliable single gene phylogenies. Instead, we inferred phylogenies from the aligned universal genomic regions (supplementary table S2, Supplementary Material online). We find six regions with conflicting phylogenetic signals, that is, the phylogenies contain internal branches that are incongruent with the monophyly of one or more of the established MTB lineages (fig. 1). Such conflicting phylogenies suggest an evolutionary history that differs from the established MTB phylogeny. Alternatively, technical, and not biological, reasons can lead to incongruent phylogenies. Alignment errors are one well-known limiting factor in reconstructing reliable phylogenies (Landan and Graur 2007); thus, we excluded unreliable alignment positions from the universal regions and re-estimated the phylogenies. This eliminated the initial conflicting signal in three regions. In region no. 35, the monophyly of the established MTB lineages is recovered (fig. 3A and B, supplementary fig. S3, Supplementary Material online), whereas in region no. 32 and in region no. 45, the bootstrap support of conflicting branches is decreased below our threshold of 75% (supplementary figs. S4 and S5, Supplementary Material online).
Fig. 3.

—Examples of incongruent phylogenies. (A) Phylogeny of universal genomic region 35 inferred from the complete alignment (1,164,225 nt, 1.2% variable sites, 0.92% parsimony informative sites, HoT score: 50.41%). (B) Phylogeny of universal genomic region 35 estimated from the alignment with unreliable positions removed (586,857 nt, 0.71% variable sites, 0.43% parsimony informative sites). See also supplementary figure S3, Supplementary Material online. (C) Phylogeny of universal genomic region 40 inferred from the complete alignment (611,340 nt, 0.68% variable sites, 0.46% parsimony informative sites, HoT score: 98.28%). (D) Splits network of universal genomic region 40 with unreliable positions removed (600,805 nt, 0.62% variable sites, 0.41% parsimony informative sites). See also supplementary figure S5, Supplementary Material online. Five problematic strains are identified: NZ_CP010340.1 and NZ_CP010338.1 were removed from RefSeq, and NZ_CP009100.1, NZ_CP009101.1, and NC_021054.1 from lineage 2 are H37Rv-guided assemblies.
The phylogeny of region no. 40 contains a conflicting branch where the Beijing lineage is not monophyletic (fig. 3C) and this conflict remains after the exclusion of unreliable alignment positions (supplementary fig. S6, Supplementary Material online). In the splits network, the conflicting splits can be traced back to five problematic genomes (supplementary table S1, Supplementary Material online); two genomes that were removed from RefSeq and three genomes of Beijing isolates that were assembled using the H37Rv genome as a reference. These latter genome sequences share many splits with the European lineage (fig. 3D), which is most likely due to the inclusion of H37Rv variants in the reference-guided assembly in this region. We observe a similar phenomenon in region no. 44 (supplementary fig. S7, Supplementary Material online) and in region no. 92 (supplementary fig. S8, Supplementary Material online). Phylogenies reconstructed without the five genomes with problematic assemblies do not contain conflicting branches in region no. 40 (excluding unreliable alignment positions) as well as in region no. 44 and no. 92 (supplementary fig. S9, Supplementary Material online).
Discussion
Here, we systematically exploit completely assembled MTB genomes to test for the presence of DNA acquisition by lateral transfer. We find that the recombination signal appears to be affected by two kinds of artefacts: Unreliable alignments and data quality. Bias in the alignment reconstruction has the potential to introduce a spurious phylogenetic signal, in particular, that of recombination. Additionally, the inclusion of problematic genome assemblies in the analysis leads to the inference of variation that cannot be attributed to biological processes rather it is likely the result of methodological artefacts. In the field of MTB research, the reference strain H37Rv is used as the model organism in most experimental studies. The genome sequence of H37Rv is also commonly used as the reference in epidemiological studies and it has been considered to have a negligible impact on the phylogenetic signal (Lee and Behr 2016). However, the limitations of using H37Rv as a reference are described in the literature. H37Rv is known to evolve in the laboratory and to accumulate genetic variants over time (Ioerger et al. 2010). Consequently, the usage of the reference H37Rv may constrain the detection of virulence-related loci in strains cultivated in the laboratory (O’Toole and Gautam 2017). We demonstrate that the use of H37Rv in reference-guided assemblies biases the phylogenetic reconstruction towards the European lineage and leads to the erroneous inference of DNA acquisition.
The incorrect identification of lateral transfer due to errors in the assembly and the alignment has also been observed for viruses and eukaryotes. For example, the contribution of intrasegmental recombination to rotavirus evolution has been rejected due to the observation that recombined segments occur only once and, thus, might have originated as errors during sequencing (Woods 2015). Similarly, technical chimeras originating from in vitro recombination impede the detection of biological recombination between dengue virus and its host (Peccoud 2018). Assembly errors in the analysis of mixed species samples can result in overestimating the abundance of LGT in the focal genome (Koutsovoulos et al. 2016). Those studies exemplify that quality control is an essential prerequisite for comparative genomics as evolutionary inference is sensitive to the underlying data and analysis pipelines. Aligning reads to a single reference sequence can introduce artefactual variants (Li 2014). Phylogenetic reconstruction from aligned reads to a reference can be improved by the use of multiple reference genomes (Bertels et al. 2014). Nonetheless, phylogenetic artefacts can already emerge at the sequencing stage. The outcome of an evolutionary analysis is greatly influenced by the usage of adequate methodology to deal with sequencing errors. Sequencing errors occur in a nonrandom manner during Illumina sequencing (Nakamura et al. 2011) and accounting for that bias improves subsequent analyses (Benjamini and Speed 2012). Sequencing errors can have a strong impact on population genetic inferences, thus, parameter estimation directly from aligned reads is more accurate than estimation from called polymorphisms (Han et al. 2014). Artefacts are especially severe for data sets of low diversity that show a small ratio of signal to noise (Johnson and Slatkin 2007).
Many pathogens exhibit little sequence diversity which complicates their evolutionary reconstruction (Achtman 2008). In MTB, we expect little amount of recombination and its detection is further impeded by the low genome diversity. Consequently, the signal of recombination in the MTB genomes is expected to be low. However, the distinction between low recombination signal and noise is challenging. Here, we present an approach to detect noise when inferring recombination in completed MTB genomes. We find a high amount of noise that exceeds the signal by far. This data set thus does not allow for an accurate inference of recombination. In order to delineate if recombination signal is low or absent, data sets of high resolution and of high quality are essential.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Bernhard Haubold, Giddy Landan, Thomas A. Kohl, Matthias Merker, and Stefan Niemann for fruitful discussions and Tanita Wein and Christian Woehle for insightful comments on the manuscript. The study was supported by the Leibniz ScienceCampus EvoLUNG, the European Research Council (Grant No. 281357 to T.D.), and the Bioinformatics Network.
Literature Cited
- Achtman M. 2008. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol. 62:53–70. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Baltrus DA, Guillemin K, Phillips PC.. 2008. Natural transformation increases the rate of adaptation in the human pathogen Helicobacter pylori. Evolution 62:39–49. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Speed TP.. 2012. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 4010:e72.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertels F, Silander OK, Pachkov M, Rainey PB, van Nimwegen E.. 2014. Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol Biol Evol. 315:1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boritsch EC. 2014. A glimpse into the past and predictions for the future: the molecular evolution of the tuberculosis agent. Mol Microbiol. 935:835–852. [DOI] [PubMed] [Google Scholar]
- Boritsch EC, et al. 2016. Key experimental evidence of chromosomal DNA transfer among selected tuberculosis-causing mycobacteria. Proc Natl Acad Sci U S A. 11335:9876–9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant J. 2014. Evolutionary genomics of pathogenic mycobacteria. Cambridge: PhD thesis, University of Cambridge. [Google Scholar]
- Cohen KA, et al. 2015. Evolution of extensively drug-resistant tuberculosis over four decades: whole genome sequencing and dating analysis of Mycobacterium tuberculosis isolates from KwaZulu-Natal. PLoS Med. 129:e1001880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 3936685:537–544. [DOI] [PubMed] [Google Scholar]
- Comas I, et al. 2013. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 4510:1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscolla M, Gagneux S.. 2014. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 266:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT.. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 56:e11147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Wilson DJ.. 2015. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol. 112:e1004041.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Baumdicker F, Neher RA.. 2017. panX: pan-genome analysis and exploration. Nucleic Acids Res. 461:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Dongen SV, Ouzounis CA.. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 307:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R. 1930. The genetical theory of natural selection. Oxford: Clarendon Press, p. 143. [Google Scholar]
- Gagneux S. 2013. Genetic diversity in Mycobacterium tuberculosis. Curr Top Microbiol Immunol. 374:1–25. [DOI] [PubMed] [Google Scholar]
- Gogarten JP, Townsend JP.. 2005. Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol. 39:679–687. [DOI] [PubMed] [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 593:307–321. [DOI] [PubMed] [Google Scholar]
- Gygli SM, Borrell S, Trauner A, Gagneux S.. 2017. Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol Rev. 413:354–373. [DOI] [PubMed] [Google Scholar]
- Han E, Sinsheimer JS, Novembre J.. 2014. Characterizing bias in population genetic inferences from low-coverage sequencing data. Mol Biol Evol. 313:723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A.. 1966. The effect of linkage on limits to artificial selection. Genet Res. 83:269–294. [PubMed] [Google Scholar]
- Huson DH, Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 232:254–267. [DOI] [PubMed] [Google Scholar]
- Ioerger TR, et al. 2010. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J Bacteriol. 19214:3645–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PLF, Slatkin M.. 2007. Accounting for bias from sequencing error in population genetic estimates. Mol Biol Evol. 251:199–206. [DOI] [PubMed] [Google Scholar]
- Karboul A, et al. 2008. Frequent homologous recombination events in Mycobacterium tuberculosis PE/PPE multigene families: potential role in antigenic variability. J Bacteriol. 19023:7838–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T.. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 3014:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsovoulos G, et al. 2016. evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini. Proc Natl Acad Sci USA. 11318:5053–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landan G, Graur D.. 2007. Heads or tails: a simple reliability check for multiple sequence alignments. Mol Biol Evol. 246:1380–1383. [DOI] [PubMed] [Google Scholar]
- Lee RS, Behr MA.. 2016. Does choice matter? Reference-based alignment for molecular epidemiology of tuberculosis. J Clin Microbiol. 547:1891–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2014. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 3020:2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gutacker MM, Musser JM, Fu Y-X.. 2006. Evidence for recombination in Mycobacterium tuberculosis. J Bacteriol. 18823:8169–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson AL, et al. 2017. Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into the emergence and spread of multidrug resistance. Nat Genet. 493:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merker M, et al. 2015. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. 473:242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer TD, Pepperell CS.. 2014. Genomic signatures of distributive conjugal transfer among mycobacteria. Genome Biol Evol. 69:2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1932. Some genetic aspects of sex. Am Nat. 66703:118–138. [Google Scholar]
- Muller HJ. 1964. The relation of recombination to mutational advance. Mutat Res Mol Mech Mutagen. 11:2–9. [DOI] [PubMed] [Google Scholar]
- Nakamura K, et al. 2011. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 3913:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namouchi A, Didelot X, Schock U, Gicquel B, Rocha EPC.. 2012. After the bottleneck: genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Res. 224:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD.. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 483:443–453. [DOI] [PubMed] [Google Scholar]
- Niemann S, Merker M, Kohl T, Supply P.. 2016. Impact of genetic diversity on the biology of Mycobacterium tuberculosis complex strains. Microbiol Spectr. 46: doi: 10.1128/microbiolspec.TBTB2-0022-2016. [DOI] [PubMed] [Google Scholar]
- O’Toole RF, Gautam SS.. 2017. Limitations of the Mycobacterium tuberculosis reference genome H37Rv in the detection of virulence-related loci. Genomics 109:471–474. [DOI] [PubMed] [Google Scholar]
- Peccoud J. 2018. A survey of virus recombination uncovers canonical features of artificial chimeras generated during deep sequencing library preparation. G3 Genes Genomes Genet. 8:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron GG, Lee AEG, Wang Y, Huang WE, Barraclough TG.. 2012. Bacterial recombination promotes the evolution of multi-drug-resistance in functionally diverse populations. Proc R Soc B. 2791733:1477–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan JE, et al. 2016. Recombination in pe/ppe genes contributes to genetic variation in Mycobacterium tuberculosis lineages. BMC Genomics. 171:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenhall M, Škunca N, Lassalle F, Dessimoz C.. 2015. Inferring horizontal gene transfer. PLoS Comput Biol. 115:e1004095.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 166:276–277. [DOI] [PubMed] [Google Scholar]
- Supply P, et al. 2013. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet. 452:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi N, Kaneko K, Koonin EV.. 2014. Horizontal gene transfer can rescue prokaryotes from Muller’s Ratchet: benefit of DNA from dead cells and population subdivision. G3 Genes Genomes Genet. 4:325–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova T, Ciufo S, Fedorov B, O’Neill K, Tolstoy I.. 2014. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 42(D1):D553–D559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tria FDK, Landan G, Dagan T.. 2017. Phylogenetic rooting using minimal ancestor deviation. Nat Ecol Evol. 1:s41559-017-0193–017. [DOI] [PubMed] [Google Scholar]
- Vos M, Didelot X.. 2009. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 32:199–208. [DOI] [PubMed] [Google Scholar]
- Woods RJ. 2015. Intrasegmental recombination does not contribute to the long-term evolution of group A rotavirus. Infect Genet Evol. 32:354–360. [DOI] [PubMed] [Google Scholar]
- Zhang H, et al. 2013. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat Genet. 4510:1255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.