

Abstract
Objective
Defects in the apoptotic machinery and augmented survival signals contribute to drug resistance in glioblastoma (GBM). Moreover, another complexity related to GBM treatment is the concept that GBM development and recurrence may arise from the expression of GBM stem cells (GSCs). Therefore, the use of a multifaceted approach or multitargeted agents that affect specific tumor cell characteristics will likely be necessary to successfully eradicate GBM. The objective of this study was to investigate the usefulness of sulforaphane (SFN)—a constituent of cruciferous vegetables with a multitargeted effect—as a therapeutic agent for GBM.
Methods
The inhibitory effects of SFN on established cell lines, early primary cultures, CD133-positive GSCs, GSC-derived spheroids, and GBM xenografts were evaluated using various methods, including GSC isolation and the sphere-forming assay, analysis of reactive oxygen species (ROS) and apoptosis, cell growth inhibition assay, comet assays for assessing SFN-triggered DNA damage, confocal microscopy, Western blot analysis, and the determination of in vivo efficacy as assessed in human GBM xenograft models.
Results
SFN triggered the significant inhibition of cell survival and induced apoptotic cell death, which was associated with caspase 3 and caspase 7 activation. Moreover, SFN triggered the formation of mitochondrial ROS, and SFN-triggered cell death was ROS dependent. Comet assays revealed that SFN increased single- and double-strand DNA breaks in GBM. Compared with the vehicle control cells, a significantly higher amount of γ-H2AX foci correlated with an increase in DNA double-strand breaks in the SFN-treated samples. Furthermore, SFN robustly inhibited the growth of GBM cell–induced cell death in established cell cultures and early-passage primary cultures and, most importantly, was effective in eliminating GSCs, which play a major role in drug resistance and disease recurrence. In vivo studies revealed that SFN administration at 100 mg/kg for 5-day cycles repeated for 3 weeks significantly decreased the growth of ectopic xenografts that were established from the early passage of primary cultures of GBM10.
Conclusions
These results suggest that SFN is a potent anti-GBM agent that targets several apoptosis and cell survival pathways and further preclinical and clinical studies may prove that SFN alone or in combination with other therapies may be potentially useful for GBM therapy.
Keywords: glioblastoma, cancer stem cells, apoptosis, sulforaphane, oncology
Approximately 13,000 people die annually of primary brain tumors in the United States,42 and glioblastoma (GBM) (designated as WHO Grade IV astrocytoma) accounts for the largest group of brain tumors that respond very poorly to current therapies.44 The combination of radiotherapy and adjunct drug temozolomide has increased the survival of patients with GBM, but the median survival of these patients is still only about 14.6 months.8,9,45 Because of this dismal prognosis, there is an urgent need to develop new therapies for this devastating disease. The highly aggressive nature of GBM, which considerably reduces the survival rate, is caused by multiple genetic alterations that result in augmented survival pathways and defects in the apoptosis signaling machinery.17,26,49 Another major problem in treating GBM is the existence of GBM stem cells (GSCs) that may represent subpopulations of tumor cells that are resistant to therapy and crucial for invasive tumor growth.11,33
In this work, we determined the utility of sulforaphane (SFN) to affect the growth of GBM cells in vitro and in vivo. SFN is an isothiocyanate isolated from broccoli and other cruciferous vegetables7 and a multitargeting agent with promising chemopreventive and antitumor activities.14,22,39,47,50 SFN inhibits cancer cell proliferation, retards the growth of tumor xenografts in vivo, and increases apoptosis in a variety of cancer cell lines from different tumor types.5,12,25,31,38,40 Emerging evidence suggests that SFN may target the epigenetic alterations seen in several cancers by reversing aberrant changes in gene transcription through the mechanisms of histone deacetylase inhibition, global demethylation, and the modulation of microRNA.24,25,31,47 SFN induces alterations in the level and activity of chromatin-modifying enzymes, which play a role in cell cycle arrest and apoptosis in cancer cells.47 SFN reduces DNA methyltransferase levels,47 as well as suppresses levels of the polycomb group proteins.24 SFN treatment also results in the generation of reactive oxygen species.1,37 SFN also inhibits the invasion and migration of C6 glioma cells by blocking FAK/JNK-mediated MMP-9 expression.21
While SFN induces apoptosis by activating mitochondrial apoptosis pathways in cancer cells,28 it also plays a significant neuroprotective role.30 Therefore, it is fascinating that while SFN activates mitochondrial and death receptor–mediated apoptotic pathways in cancer cells, it can protect normal brain cells. In this work, we used in vitro and in vivo xenograft studies and assessed the therapeutic potential of SFN for GBM treatment. Our study provides detailed evidence that SFN induces cell death and inhibits the growth of GBM cells and xenografts through the modulation of multiple cell-signaling pathways1,21,24,28,37 and may be potentially useful for the further development for GBM treatment.
Methods
Cells, Cell Culture Conditions, and Drugs
Authenticated human GBM cell lines U87, U373, U118, and SF767 were obtained from American Type Culture Collection. Early-passage GBM10 and GBM43 lines34 were propagated and passaged through NOD.Cg-PrkdcscidIL2rgtm1Wjl/Sz (NSG) mice, and short-term cultures of these cells were used for cell survival and biochemical assays. Cells were maintained in DMEM supplemented with 10% fetal bovine serum and 100 ng/ml each of penicillin and streptomycin (Invitrogen, Inc.) at 37°C in 5% CO2. The stocks were cryopreserved after fewer than 3 passages. The primary patient cell lines GBM10 and GBM43 have been described previously.34 The cell lines identity was confirmed by DNA fingerprint analysis (IDEXX BioResearch) for species and baseline short tandem repeat analysis testing. Both cell lines were 100% human, and a 9-marker short tandem repeat analysis is on file. A panel consisting of the primary GBM cell lines MHBT32 and MHBT161 was established by author A.A.C-G using fresh tumor specimens from patients with pathologically diagnosed GBM.
To establish new primary GBM cultures, clinical samples from 2 patients with WHO Grade IV GBM were collected from the Department of Neurosurgery, Indiana University School of Medicine and Goodman Campbell Brain and Spine. Prior informed consent was obtained in writing from the patients, and all procedures were approved by the institutional review boards in accordance with the general accepted guidelines for the use of human materials. For tissue culture and cell line establishment, fresh tumor tissue was minced using scalpels in DMEM/Ham’s F12 cell culture media that was supplemented with 2.5% fetal bovine serum, 2 mM L-glutamine, and 1% penicillin-streptomycin and passed through a cell strainer to obtain a single cell suspension. The cells were washed with phosphate-buffered saline and seeded in T25 cell culture flasks. The outgrowing cells were detached with trypsin and either frozen or subcultured. Cells were passaged 2 to 3 times and frozen, and experiments were performed with these cells. The MHBT32 and MHBT161 early primary cultures were derived from human primary GBM tissues and maintained in DMEM/Ham’s F12 media. The primary cultures of these cells were propagated through xenograft generation in the NSG mice to establish short-term cultures of GBM cells. Primary cultures of nontumor or normal brain samples (from a patient with epilepsy) were also established as described above to prepare primary cultures of GBM tumors and used as a control to determine the effect of SFN on nontumor brain cells.
SFN was purchased from LKT Laboratories and also synthesized by Chemical Synthesis & Organic Drug Lead Development Core (Indiana University Simon Cancer Center). Rotenone and carbonyl cyanide m-chlorophenylhydrazone (CCCP) were purchased from Sigma Chemical Co.
GBM Stem Cells and Sphere-Forming Assay
GBM spheroids were obtained by plating U87, MHBT161, and early-passage GBM43 primary cultures in 6-well ultralow attachment plates (Corning Inc.) at a density of 1 to 2 × 103 cells/ml in DMEM supplemented with 1% N2 supplement (Invitrogen), 2% B27 supplement (Invitrogen), 25 ng/ml epidermal growth factor (EGF), 25 ng/ml fibroblast growth factor (FGF) (R&D Systems, Inc.), 2 ng/ml platelet growth factor, and 100 ng/ml each of penicillin and streptomycin (Invitrogen, Inc.). The tumor spheres were dispersed as single cells by treating them with Accutase solution (Sigma-Aldrich) for 10 to 15 minutes. The cells were spun down by centrifugation at 1000g for 4 minutes and seeded in fresh sphere-forming media in 96-well plates in a range of 50 to 100 cells per well. After 2 to 3 days, neurospheres containing 6 to 8 cells were formed, which were treated with 5 to 50 μM SFN for 8 to 10 days. Colonies were counted under a Zeiss Axiovert 25 inverted microscope after 5 days of incubation.
Cell Survival Assay
To determine the cytotoxic effect of SFN on the GBM cells, the methylene blue cell survival assay was performed as previously described.2 For each treatment, 1 × 104 cells were seeded in a 96-well plate, and the cells were then treated with or without 5 to 50 μM SFN for 48 hours.
Detection of Apoptosis by DAPI Staining
DAPI staining was performed on untreated and SFN-treated GBM cells as we previously described.2 Apoptotic cells were identified by condensation and fragmentation of nuclei. A minimum of 300 cells were counted for each treatment, and the percentage of apoptotic cells was calculated as the ratio of apoptotic cells to the total cells counted multiplied by 100. The DAPI staining experiments were performed in triplicate.
Isolation of CD133-Positive GBM Cells by Fluorescence-Activated Cell Sorting Analysis
GBM cell lines U87, U373, U118, and SF767 cells were collected using trypsin and analyzed using a standard fluorescence-activated cell sorting (FACS) protocol. The antibody used for the FACS analyses was anti-CD133/1 (AC133) conjugated to phycoerythrin (PE) (Miltenyi Biotech). Normal mouse IgG antibody labeled with PE was used as the isotype control.
Western Blot Analysis
The cells were harvested, rinsed in cold PBS, and lysed in radioimmunoprecipitation assay buffer, and the protein concentrations of the cell lysates were determined with Bradford reagent (Bio-Rad). Western blotting was performed as we previously described.2 The primary antibodies used were as follows: rabbit anti–caspase 3 polyclonal antibody (Cell Signaling Technology) and mouse anti–human caspase 3 and caspase 7 monoclonal antibody (Cell Signaling Technology). Mouse monoclonal anti–γ-H2AX antibody (Ser139; clone JBW301) was obtained from Upstate Biotechnology, anti–β-actin clone AC-74 was obtained from Sigma-Aldrich, and mouse anti–β-actin clone AC-74 monoclonal antibody was obtained from Sigma Chemical Co. The secondary antibodies used were either rabbit anti–mouse or donkey anti–rabbit antibody coupled to horseradish peroxidase (Amersham).
Analysis of Reactive Oxygen Species and Apoptosis
This method was performed as previously described by our laboratory.29 Levels of intracellular reactive oxygen species (ROS) were measured using dichlorodihydrofluorescein diacetate (Molecular Probes, Inc.). To determine if the increase in ROS generated was responsible for apoptosis, the cells were treated with or without 10 mM of the antioxidant N-acetylcysteine (NAC) for 3 hours prior to treatment with these agents, and cell survival was measured as described above. To determine if SFN-mediated ROS is produced through a mitochondrial pathway, the cells were exposed to increasing concentrations of SFN for 3 hours or treated with 10 and 30 μM SFN in the absence or presence of the combination of 1 μM rotenone and 1 μM CCCP (a mitochondrial respiratory chain [MRC] Complex I inhibitor and a mitochondrial respiration uncoupler, respectively) for 3 hours and immediately assayed for ROS generation.
Comet Assays for Assessing SFN-Triggered DNA Damage
To determine the extent of DNA damage, individual U87 cells were treated with a vehicle (0.05% dimethyl sulfoxide [DMSO]) or 10 and 30 μM SFN, respectively, for 24 hours, and the cells were processed for the comet assay under alkaline as well as neutral conditions, as previously described,48 using the single cell gel electrophoresis/comet assay kit (Trevigen). Comets were visualized by epifluorescence microscopy using a fluorescein isothiocyanate filter. The slides were stained with SYBR Green for 5 minutes while protected from light and dried at room temperature before visualization. Fluorescence images were captured using a Nikon Diaphot 200 fluorescence microscope. The comet lengths of the individual cells per group were measured in each treatment group. Two hundred comets were randomly analyzed by measuring the distance of DNA migration in each cell from the nuclear core to the trailing edge of the comet using TriTek CometScore TM Freeware v1.5C software (TriTek Corporation).
Confocal Microscopy
For CD133 detection, U87 spheroids were incubated with mouse anti–human CD133 antibody from Miltenyi Biotec and subsequently Texas Red secondary anti–mouse antibody. For SOX2 detection, the U87 spheroids were incubated with SOX2 (D6D9) XP rabbit monoclonal antibody (Alexa Fluor 488 Conjugate). Confocal images were acquired with the confocal/2-photon FLUOVIEW FV1000MPE system (Olympus America) at the Indiana Center for Biological Microscopy facility using an Olympus XLUMPLFL (×20, numerical aperture 0.95, water immersion objective lens). Images were collected in a sequential illumination mode using 405-, 488-, and 559-nm laser lines. Fluorescence emission was collected in 3 PMTs at a filter range set to 425 to 475 nm for DAPI, 500 to 545 nm for green dye, and 570 to 670 nm for red dye. Optical sections (Z-stacks) were collected using optimal step size settings with images comprised of 512 × 512 pixels (634 × 634 μm2).
In Vivo Studies
Eight-week-old female NSG mice were used for the in vivo studies. Ectopic and xenografts were established by implanting 1 × 106 GBM10 cells into the flank of each NSG mouse. For the in vivo studies, 6 to 13 animals per group were used, and drugs were administered starting on Day 7 after tumor implantation. Group 1 was comprised of vehicle-treated control animals (0.1 ml saline containing 0.5% DMSO). For Group 2, SFN (100 mg/kg in 0.1 ml saline containing 0.5% DMSO) was administered by gavage for 5 days per week throughout the duration of the experiment. At 21 days postinjection, tumor size was determined, the animals were killed, and the tumors were harvested. Toxicity associated with in vivo SFN therapy was analyzed using toxicological variables, including animal weight, tissue analysis, and animal appearance and behavior. Hematoxylin and eosin staining was performed to detect toxicity to the mouse tissues (liver, lung, brain, spleen, and kidney).
Histological Analysis
For light microscopy of the GBM10 xenografts, the tumors were placed in 10% zinc formalin and stored at 4°C overnight prior to paraffin processing. Six-micrometer-thick tissue sections obtained from the control and SFN-treated animals were placed on charged slides and dried, the paraffin was removed, and the tissues were stained with hematoxylin and eosin.
Statistical Analysis
For the in vitro and in vivo studies, the data graphed with error bars represent the mean and standard deviation of the experiments conducted in triplicate. The student t-test was used to determine the significance of the difference between samples.
Results
SFN Causes Apoptosis and Inhibits the Survival of GBM
We first determined if SFN affects the survival of the GBM cell lines. As shown in Fig. 1A, treatment with various concentrations of SFN for 48 hours triggered the significant inhibition of cell survival in the U87MG, SF767, U118, and U373 cell lines. SF767 cells were more sensitive to SFN treatment. We next determined to what extent SFN inhibits the survival of the early-passage primary culture GBM cells. The methylene blue cell survival assay revealed that SFN induced the dose-dependent inhibition of cell survival in the early-passage GBM10, GBM43, MHBT32, and MHBT161 cells (Fig. 1B). The 50% inhibition of cell survival (IC50) due to SFN in the established GBM cell lines and early-passage primary culture GBM cells were determined from the cell survival plots of the cells treated with increasing concentrations of SFN for 48 hours and are shown in Figure 1A and B. The concentrations of SFN that triggered IC50 in the established U87MG, U118, U373, and SF767 GBM cell lines were 35.1, 29.6, 23.7, and 9.6 μM, respectively. The SFN IC50 values for the MHBT161, GBM10, GBM43, MHBT32, and MHBT161 cells were 44, 22.2, 27.9, and 9.3 μM, respectively. Comparing the results shown in Figure 1 with the information provided in the molecular profile of the GBM cells provided in Table 1 suggest that there are no obvious trends that associate certain genotypes or phenotypes of the GBM cell lines with sensitivity to SFN.
Fig. 1.
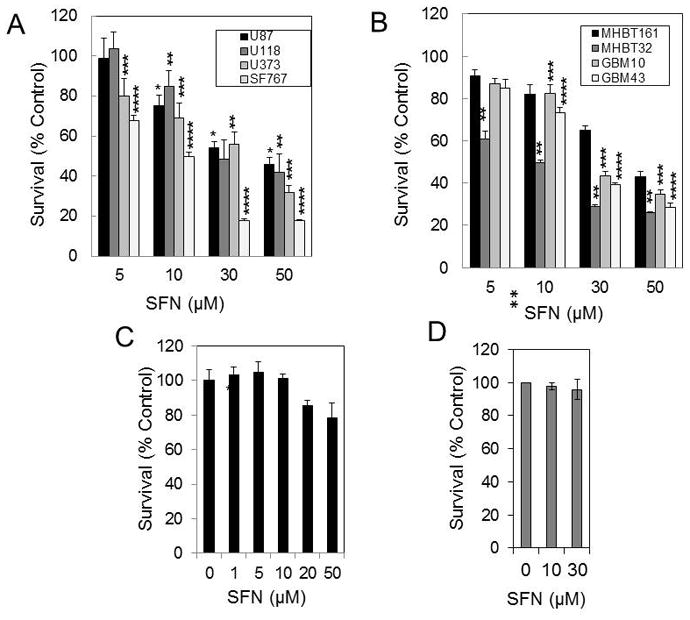
SFN reduces survival in GBM cell lines but does not affect the survival of the normal human brain cells or normal human MSCs. Established U87MG, SF767, U118, U373 cell lines (A), and MHBT32, MHBT161, GBM10, and GBM43 early-passage GBM primary cultures (B) were treated with 5 to 50 μM SFN for 48 hours. Cell survival was determined by the methylene blue cytotoxicity assay (*p < 0.0001; **p < 0.0001; ***p < 0.0001; ****p < 0.0001for SFN vs vehicle [n = 6]). C: Normal brain cells were treated with 1 to 50 μM SFN for 48 hours, and cell survival was measured by the methylene blue cytotoxicity assay (*p < 0.0001 vs vehicle [n = 6]; bars and whiskers indicate median survival ± SD, respectively). D: MSCs were treated with 10 and 30 μM SFN for 24 hours. Cell survival was determined by the methylene blue cytotoxicity assay (p > 0.05 for SFN vs vehicle).
TABLE 1.
Molecular profile of the GBM cell lines
| Cell line | EGFR Amplification | P53 Status | P73 status | HDM2 status | MGMT expression |
|---|---|---|---|---|---|
| U87MG | No | WT | WT | WT | Negative |
| SF767 | No | WT | WT | WT | High |
| U118 | No | Mutant | WT | WT | Low |
| U373 | No | Mutant | WT | WT | Negative |
| GBM10 | No | WT | TBD | WT | Negative |
| GBM43 | No | P270C | TBD | WT | Negative |
| M-HBT32 | TBD | R273H | TBD | TBD | TBD |
| M-HBT161 | TBD | TBD | TBD | TBD | TBD |
TBD (TO Be Determined)
We next determined if SFN affects the viability of normal brain cells. As shown in Fig. 1C, SFN at 20 and 50 μM decreased the survival of the nontumor and normal brain tumor primary cultures by only 18% and 23%, respectively. Similarly, SFN did not affect the survival of normal human mesenchymal stromal cells (MSCs) (Fig. 1D).
To determine if SFN induces apoptosis, the U87MG and MHBT32 cells were treated with 5 to 30 μM SFN for 24 hours, and apoptosis was determined by DAPI staining using U87 and MHBT32 cells. As shown in Fig. 2A and B, SFN induced significant dose-dependent apoptosis in the U87 and MHBT32 cells. To determine if SFN triggered apoptosis in correlation with caspase activation, Western blot analysis was performed which indicated that the SFN-induced apoptosis in the U87MG and MBT-32 cells was associated with caspase 3 and caspase 7 activation (Fig. 2C and D).
Fig. 2.
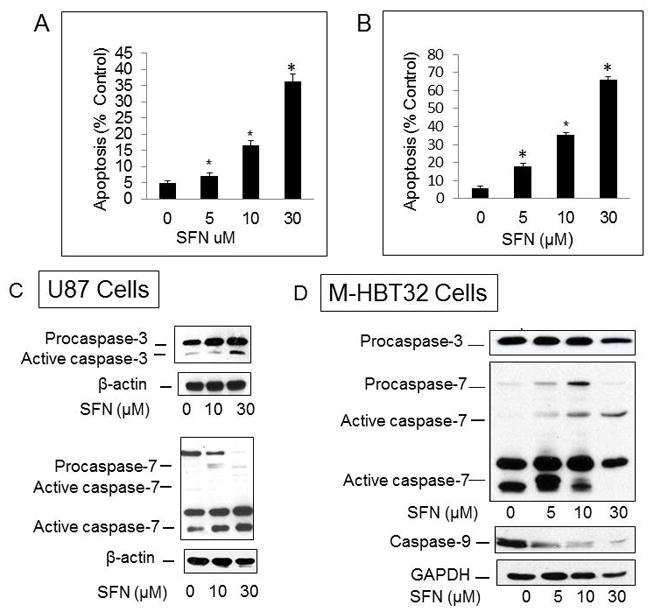
SFN triggered apoptosis as well as caspase 3, caspase 7, and caspase 9 activation in SFN-treated GBM cells. SFN induced apoptosis in U87MG (A) and M-HBT-32 cells (B) treated with 5 to 30 μM for 24 hours. Apoptotic cells with condensed and fragmented nuclei were identified by DAPI staining. A minimum of 300 cells were counted for each treatment, and the percentage of apoptotic cells was calculated (*p < 0.0001 vs vehicle [n = 6]). C and D: U87MG and MHBT32 cells were treated with SFN for 24 hours and processed for Western blotting using antibodies to caspase 3, caspase 7, and caspase 9 as described in the Methods.
SFN Generated ROS in GBM Cells
To determine if the SFN-induced apoptosis is triggered by the increased production of ROS, we first measured the formation of ROS at various concentrations of SFN. The results shown in Fig. 3A revealed that 10 to 30 μM SFN triggered the formation of intracellular ROS in the U87MG cells treated or 4 hours. Moreover, when the cells were pretreated with 10 mM NAC for 2 hours and then treated with 30 μM SFN for 24 hours, ROS production was maintained at levels similar to those observed prior to SFN treatment (Fig. 3A). Moreover, as shown in Fig. 3B, while SFN significantly decreased cell survival, treatment with NAC inhibited the effect of SFN on cell survival (Fig. 3B), revealing that SFN’s inhibitory effect on cell survival is ROS dependent. Furthermore, SFN-triggered ROS production was significantly reduced when the cells were depleted of respiring mitochondria by treating them with the combination of 1 μM rotenone (an MRC Complex I inhibitor) and 1 μM CCCP (a mitochondrial respiration uncoupler), and the combination of these compounds decreased the level of ROS by 82% (Fig. 3C). Therefore, the source of the SFN-generated ROS in the U87MG cells was primarily mitochondrial in origin.
Fig. 3.
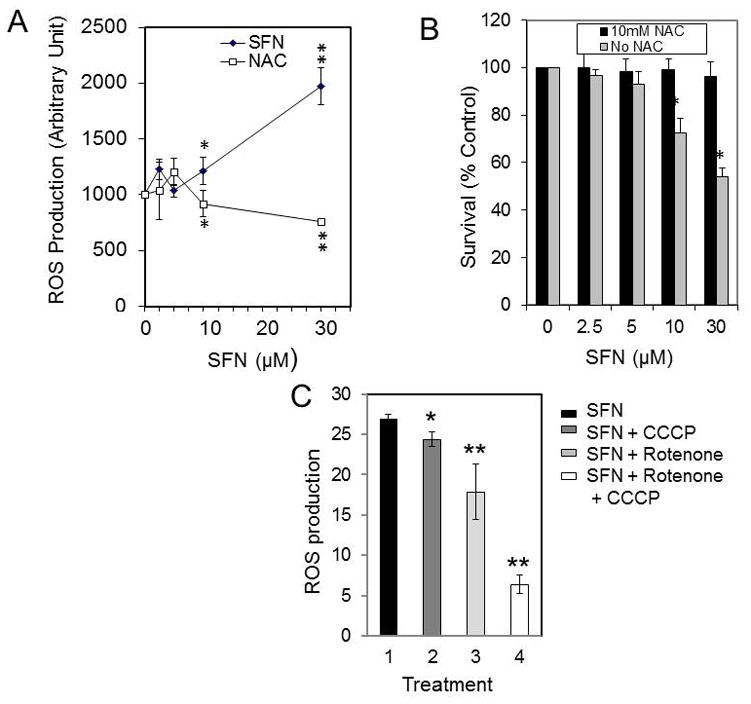
SFN mediated ROS formation and the effect of NAC. A: U87-MG cells were plated at a density of 5 × 105 in 60-mm dishes, allowed to attach overnight, and exposed to either 5 to 30 μM SFN for 24 hours or pretreated with 10 mM NAC for 3 hours and 5 to 30 μM SFN for 24 hours, and ROS was determined as described in the Methods. B: U87 cells were treated with 10 mM NAC, and the survival fractions of the NAC-treated cells compared with the untreated control sample were determined as described in the Methods section (*p < 0.001; **p < 0.0001 for SFN + NAC vs SFN [n = 3]). C: U87 cells were pretreated with 1 mM CCCP, rotenone, or CCCP + rotenone, respectively, then treated with 10 μM SFN, and ROS was determined as described in the Methods (*p < 0.01 for SFN + CCCP vs SFN alone; **p < 0.0001 for SFN + rotenone vs SFN alone; **p < 0.0001 for SFN + rotenone + CCCP vs SFN alone [n = 3]). Bars and whiskers indicate median survival ± SD, respectively.
SFN Triggers DNA Strand Breaks in GBM Cells
To determine if SFN triggers DNA strand breaks, we treated U87MG cells with 10 to 30 μM SFN for 24 hours and then performed the comet assay under alkaline conditions to assess DNA with single-strand breaks (SSBs) and double-strand breaks (DSBs).48 Comet assays under alkaline conditions showed that SFN induces DNA SSBs (Fig. 4A). We also performed comet assays under neutral conditions to exclusively detect DSBs48 and found that 10 to 30 μM SFN induced increases in DNA DSBs compared with control cells (Fig. 4B). To assess if SFN treatment increases DNA tail length, 200 comet cells were counted and compared with vehicle-treated cells. The results shown in Figure 4C clearly show that SFN treatment increased DNA tail length due to DNA DSBs. One of the earliest indicators of DNA DSBs is the phosphorylation of H2AX on serine139 (γ-H2AX).18 The γ-H2AX protein localizes near DNA strand breaks and recruits other proteins to the damaged site. To demonstrate the formation of DSBs by SFN, we detected γ-H2AX in U87MG GBM cells by performing Western blot analysis using the mouse anti–phospho-γ-H2AX antibody (Ser139) (Fig. 4D). As shown in Fig. 4C, the increase in DNA breaks in the U87MG cells treated with 10 μM SFN for 24 hours was detected by Western blot analysis. Compared with the untreated control cells, a significantly higher amount of γ-H2AX and, as consequence, a higher number of breaks were observed in the SFN-treated sample.
Fig. 4.
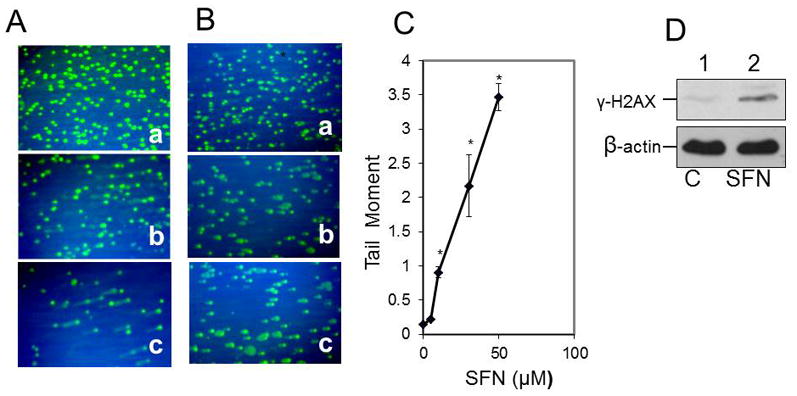
SFN induced DNA SSBs and triggered the induction of γ-H2AX and DSBs in U87 glioblastoma cells. A: Control vehicle-treated U87-MG cells (a) and cells treated with 10 and 30 μM SFN, respectively (b and c), were processed for the comet assay under alkaline conditions as described in the Methods. B: The comet assay was performed under neutral conditions. Control U87 cells (a) and cells treated with 10 and 30 μM SFN, respectively (b and c), were processed for the comet assay under neutral conditions. C: Under neutral conditions, SFN treatment increased the DNA tail length. At least 200 comet cells were counted and compared with vehicle-treated cells (*p < 0.001 for 10, 30, and 50 μM SFN vs vehicle alone [n = 3]). D: U87 cells (3 × 105 cells) were treated with the vehicle (0.05% DMSO) (Lane 1) or 10 μM SFN (Lane 2) for 24 hours. Cell lysates (50 μg protein) were analyzed via Western blot using the antiphospho-H2AX antibody (Ser139) and anti–β-actin.
SFN Induces Apoptosis in GSCs
To determine if the CD133-positive GBM stem cells are sensitive to SFN, we isolated CD133-positive stem cells from the regular cultures of U87MG, SF767, U118, and U373 cell lines by FACS (Fig. 5A), as described in the Methods. CD133-positive GBM cells were cultured in serum-free neural growth media containing basic FGF and EGF. The percentages of the CD133-positive cells isolated from the U373, SF767, U118, and U87MG cells were 0.04%, 0.06%, 0.27%, and 0.6%, respectively. Neurosphere-like brain tumor spheres (i.e., spheroids) obtained from the CD133-positive U87MG cells are shown in Figure 5B. Immunofluorescent staining with the human CD133-PE antibody showed that the cultured cells grown in the neural growth media are CD133-positive cancer stem cells (Fig. 5C). We next confirmed that these CD133-positive stem cells from the brain tumor spheres that were sorted from these cell lines are sensitive to SFN. As illustrated in Fig. 5C SFN at 30 and 70 μM induced substantial levels of apoptosis as detected by DAPI staining in the CD133-positive stem cells isolated from the GBM cell lines. DAPI staining was used to detect apoptosis since it is a sensitive method and can be used on low numbers of cells.
Fig. 5.
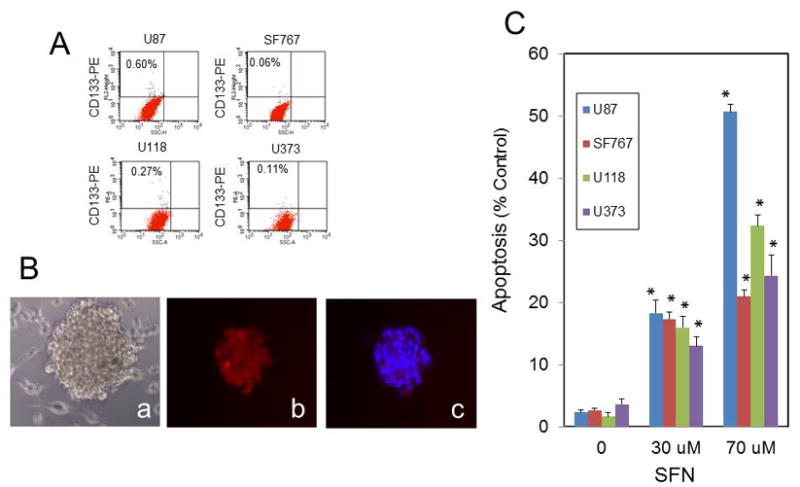
SFN triggers apoptosis in CD133-positive GSCs. A: The frequency of CD133-positive cells in unsorted GBM cell populations. The GBM cells were stained with the CD133 antibody conjugated with phycoerythrin (anti–CD133-PE) and analyzed by flow cytometry. B: The CD133-positive cells were isolated from U87 cells by FACS and cultured in sphere-forming neural growth media, as described in the Methods (a). The spheroids formed after 7 days and were processed for fluorescence microscopy for CD133 detection (b) and CD133 and DAPI staining (c). C: SFN induced apoptosis in GBM CD133-positive cells. The CD133-positive stem cells were treated with 30 and 70 μM SFN for 16 hours; after harvesting the cells, apoptosis was determined by DAPI staining. At least 3 microscopic fields of 300 cells were scored per condition per experiment by counting the total number of CD133-positive cells, and the percentage of those that showed a fragmented or dense DAPI-stained nucleolus representing the apoptotic cells was calculated (*p < 0.0001 vs vehicle [n = 3]).
SFN Induces Apoptosis in GBM Spheroids
To determine the cytotoxic and antiproliferative effects of SFN on GBM spheroids, we grew GBM spheroids from the U87, MHBT161, and GBM43 cells in the spheroid culture media as described in Methods. These spheroids consist of a heterogeneous population of GSCs and nonstem cells, and confocal microscopic examination revealed that these spheroids express CD133 and SOX2, a marker for GSCs. Figure 6 shows the expression of these markers, which overlapped in many cells in the individual U87 and MHB-161 spheroids. As shown in the left side of Fig. 7, treatment with various concentrations of SFN for 5 days triggered the significant inhibition of cell survival in U87 spheroids. Interestingly, consistent with the data shown in Fig. 4B and C, pretreating the spheroids with 1 mM NAC for 3 hours abrogated the inhibitory effect of 5 to 30 μM SFN on GBM spheroid cell survival, indicating that scavenging ROS prevents SFN-mediated apoptosis in the GBM spheroids. However, NAC only partially inhibited the effect of 50 μM SFN treatment, suggesting that at this concentration of SFN other mechanisms are also involved in its cytotoxic effect on GBM spheroids. We next determined to what extent SFN inhibits the survival of GBM43 and MHBT32 spheroids. The data shown in Fig. 6C and D revealed that SFN induced the dose-dependent inhibition of GBM43 and MHBT32 survival and only 5% to 10% of the spheroids survived with 5 μM SFN treatment.
Fig. 6.
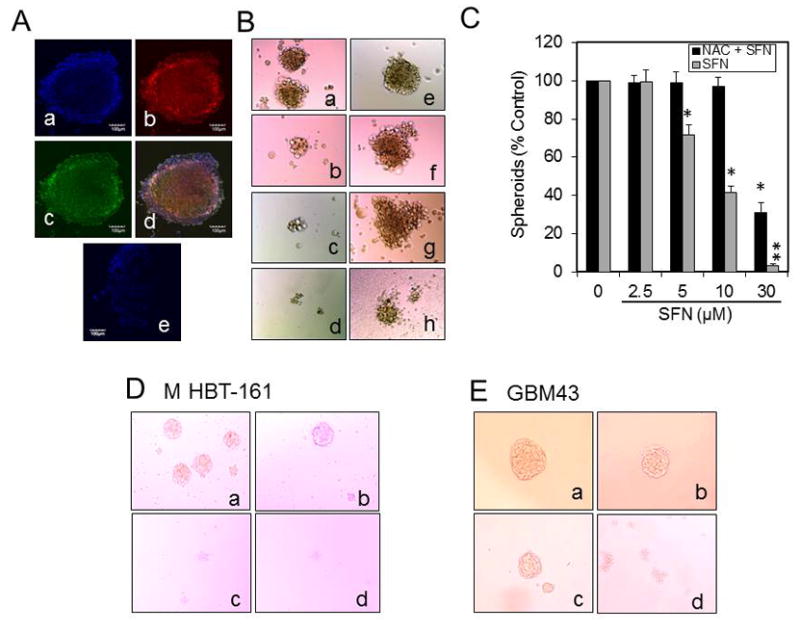
Confocal microscopy of U87 spheroids and the effect of SFN or SFN + NAC on the survival of the spheroids. A: The spheroids were stained with the CD133 or SOX2 antibody, and the confocal images were acquired with a confocal/2-photon Olympus FLOVIEW FV1000 MPE system, as described in the Methods (a, DAPI staining; b, CD133 expression; c, SOX2 expression; d, overlapping CD133 and SOX2 expression; e, isotype control antibody [magnification ×20]; scale bar = 100 μm). B: SFN inhibits the survival of U87 spheroids. The U87 primary spheroids were isolated as described in the Methods and dispersed to single cells by treating them with Accutase for 15 to 20 minutes. The cells were spun down by centrifugation at 1000g for 4 minutes and seeded in fresh sphere-forming media in 96-well plates at a range of 20 to 50 cells per well. After 2 to 3 days, the spheroids containing 6 to 8 cells were formed, which were not treated (a, control), treated with 5 to 30 μM SFN (b–d,) pretreated with 1 mM NAC for 3 hours (e), or pretreated with 1 mM NAC and then treated 5 to 30 μM SFN (f–h) for 8 to 10 days. C: SFN inhibits growth and reduces the survival of the spheroids isolated from U87 cells, and NAC prevents SFN’s effect (*p < 0.001 SFN vs vehicle alone; **p < 0.001 SFN + NAC [n = 3]). Tumor primary neurospheres from M-HBT-161 (D) and GBM43 (E) early-passage primary cultures isolated and treated with SFN as described above. The untreated spheroids (a) or spheroids treated with 10, 30, and 50 μM SFN (b, c, and d, respectively) inhibits growth and reduces the survival of spheroids isolated from early-passage GBM primary cultures.
Fig. 7.
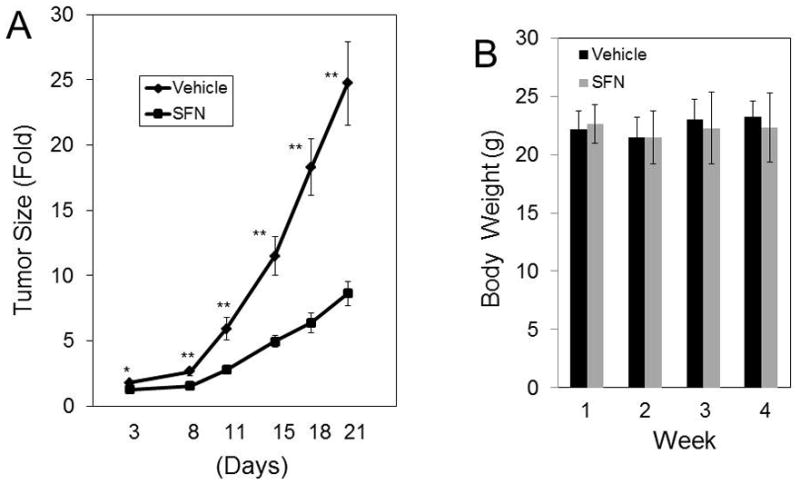
SFN administration significantly decreases the growth of ectopic GBM10 xenografts. Left: NSG mice were implanted with GBM10 cells in the flank. When the tumors reached approximately 100 mm3, the cohorts of mice (n = 13 for SFN treatment and n = 6 for control) were treated for 3 cycles with the vehicle or SFN. Each treatment cycle consisted of 5 days of treatment followed by a 2-day rest period. SFN was delivered via oral gavage at 100 mg/kg for Cycles 1, 2, and 3 (*p < 0.01; **p < 0.0001). Right: The body weights of the mice treated with the vehicle or SFN were measured every week. The sizes of the tumors in the control mice reached 1 to 1.5 cm3 when the experiment was terminated.
SFN Inhibits the In Vivo Growth of GBM Tumor Xenografts
To determine if SFN inhibits the in vivo growth of GBM primary cultures, we established GBM10 xenografts by using early-passage human GBM10 primary cultures that were implanted in the flank of NSG mice. We determined the effect of oral SFN administration in these mice using a 100 mg/kg/day dose of SFN as described in Methods. SFN retarded the growth of ectopic GBM10 xenografts (Fig. 7 right). Our results show that SFN robustly inhibits the in vivo growth of ectopic GBM xenografts. As shown in Fig. 6A, SFN significantly enhanced tumor cell kill, which was clearly observed in the SFN-treated mice versus vehicle-treated mice (p > 0.001). Moreover, the body weights of the control and SFN-treated animals were similar (Fig. 7 right). Histological analysis of the tumor sections from SFN-treated xenografts revealed the absence of blood vessels (data not shown). Histological examinations of the mouse tissues showed no cytotoxicities in the liver, lung, brain, spleen, and kidney.
Discussion
There is accumulating evidence demonstrating that SFN can be considered a hormetic agent,38 a compound that is able to trigger biologically opposite effects depending on its concentration. Thus, at low concentrations it exerts chemopreventive, indirect antioxidant, and cytoprotective effects, while at higher doses it triggers cytotoxic and antitumor properties.4 This paradoxical property has garnered support to exploit SFN as a chemopreventive compound and as an anticancer agent. SFN’s ability to permeate the blood-brain barrier10 makes it an excellent candidate for modulating the antioxidant response during brain inflammation and potentially useful for anti-GBM therapy.
Our results clearly demonstrate that SFN significantly inhibits the survival of GBM cells and induces apoptosis in GBM cells that was associated with the increased activity of caspase 3 and caspase 7. We also found that SFN had a modest effect on the nontumor brain cells and did not affect normal human MSCs. These data corroborate those reported by Sharma et al.,41 which show that the treatment of normal human lymphocytes with up to 100 μM SFN for 24 hours did not result in a cytotoxic effect.
Cancer cells generate increases in ROS; however, these ROS levels are still below that which causes overt cellular damage, and this range of ROS is capable of increasing tumorigenesis by activating signaling pathways that regulate cellular proliferation, metabolic alterations, and angiogenesis.46 Our data show that SFN triggered the increased formation of intracellular ROS, and when the cells were pretreated with NAC ROS production in and cell survival of the GBM cells were similar to the untreated control U87 cells, revealing that SFN-triggered cell death is ROS dependent. Furthermore, our results revealed that combination with rotenone inhibited most of SFN-triggered ROS generation. Therefore, the SFN-generated ROS in GBM cells were formed at the MRC level. Our results corroborate previous results that show that SFN-triggered ROS were formed at the mitochondrial MRC level and, in fact, rotenone or myxothiazol (MRC Complex I and III inhibitors, respectively) abolished ROS formation.37 These data also corroborate a report that rotenone or myxothiazol abrogated SFN-mediated ROS formation in Jurkat cells.38 Moreover, we showed that NAC pretreatment abrogated the survival and proliferation of GSC spheroids. Interestingly, the therapeutic benefit of ROS has been attributed, in part, to the inhibition of GSC’s self-renewal and stemness.43 The ROS threshold theory states that in cancer cells the intensive SFN-triggered ROS production could pass the threshold level to induce cell death. However, in normal cells, due to the lower initial ROS level, the same elevation of the ROS level will evoke a cytoprotective effect.37
As revealed by the comet assay, we demonstrated that SFN increased SSBs and DSBs in GBM cells. Compared with the untreated control cells, a significantly higher level of γ-H2AX expression, as a consequence of a higher number of DSBs, was observed in the SFN-treated sample. These results reveal that one of the mechanisms by which SFN triggers apoptosis in GBM cells is DNA damage. Moreover, the formation of ROS was demonstrated to be linked to the induction of SSBs and that preventing SFN-mediated ROS formation also prevented SSBs.36 SFN treatment also contributed to genomic instability in MG63 osteosarcoma and colon cancer cells, as revealed by the increased number of DNA breaks.6
Growing evidence suggests that GSCs comprise a small subpopulation of highly tumorigenic cells with stem cell properties, which play significant roles in tumor aggressiveness as well as chemoresistance and radiation resistance.20,27,32 A histopathological characteristic feature of high-grade gliomas, including GBM, is intratumoral central necrosis, which is indicative of hypoxia and an extremely poor oxygen supply within the tumor.20,32,35 GSCs are preferentially localized in the perinecrotic hypoxic area (i.e., the hypoxic niche),27,32 and CD133 is largely used as a cancer stem cell(CSC) marker in various tumors, including GBM.3 Furthermore, silencing CD133 in human GBM neurospheres impaired the self-renewal and tumorigenic capacity of neurosphere cells.3 Another marker of GSCs is the key stemness transcription factor, SOX2.32,33 In this study, for the first time, we showed that SFN targets CD133-positive GSCs obtained from GBM cell lines and induced apoptotic cell death in these cells. Moreover, it significantly inhibited the survival of the CD133-positive and SOX2-expressing GBM spheroids obtained from U87MG, GBM43, and MHBT161 cell lines. These results support previous reports demonstrating that SFN triggers cell death in CSCs from other tumors.23 Recent results also demonstrated that SFN significantly reduced miR-21 expression by inhibiting the Wnt/β-catenin/TCF4 pathway in GBM cells.19 Therefore, because Wnt/β-catenin signaling is a key downstream mediator of MET signaling in GSCs,16 it is tempting to speculate that SFN may also target GSCs through the Wnt/β-catenin pathway. Moreover, it is well documented that hypoxia is the driving force behind GBM development and the hypoxia-mediated maintenance of GSCs through the upregulation of HIF-1α.15 Since SFN downregulates the expression of HIF-1α13 and GSCs preferentially reside within the hypoxic niche,32,36 it may play a pivotal role in eliminating GSCs through HIF-1α.
Our results clearly demonstrated that SFN uses various strategies to inhibit the growth of and induces cell death in GBM cells. It is tempting to speculate that SFN-induced mitochondrial ROS is a key instigator of triggering DNA damage and apoptosis in GBM cell lines and GSC-containing spheroids. Further investigation of the role of SFN-induced ROS in eliminating these cells should delineate detailed molecular mechanisms of SFN-triggered cell death and the signaling pathways involved.
Conclusions
Our results show that SFN robustly and selectively inhibits the survival of GBM cells and GSCs in vitro in established cell cultures and early-passage primary cultures through generating ROS and triggering DNA damage. SFN is also highly effective in eliminating GSCs as well as GBM spheroids, and it significantly inhibits the growth of GBM xenografts in vivo. These data show that SFN is a multitargeting compound that shows promise as a therapeutic option for treating GBM. Since other laboratories have demonstrated30 that SFN can cross the blood-brain barrier, future investigations will focus on the efficacy of SFN as a single agent, as well as in combination with frontline temozolomide and radiation therapy, using orthotopic GBM xenograft models.
Acknowledgments
We thank the staff at the Methodist Research Institute Biorepository and the In Vivo Therapeutics (IVT) Core for their expert assistance. The research in this publication was supported by the National Cancer Institute of the National Institutes of Health under award numbers R01CA101743 (awarded to A.R.S.), R01CA138798 (awarded to H.W. and K.P.), and the IUPUI Signature Center Initiative for the Cure of Glioblastoma. In vivo work was performed by the IVT Core, a core laboratory of the Indiana University Melvin and Bren Simon Cancer Center, and supported by the National Cancer Institute (P30 CA082709). We greatly appreciate the generous support of the Mary Ann and Gene Zink Family Glioblastoma Multiforme Research Fund, Team JOEY, Heroes Foundation Program, Riley Children’s Foundation, and Jeff Gordon Foundation (awarded to H.W. and K.P.).
ABBREVIATIONS
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- DMSO
dimethyl sulfoxide
- DSB
double-strand break
- EGF
epidermal growth factor
- FACS
fluorescence-activated cell sorting
- FGF
fibroblast growth factor
- GBM
glioblastoma
- GSC
glioblastoma stem cell
- IC50
50% inhibition of cell survival
- MRC
mitochondrial respiratory chain
- MSC
mesenchymal stromal cell
- NAC
N-acetylcysteine
- NSG
nonobese diabetic scid gamma
- PE
phycoerythrin
- ROS
reactive oxygen species
- SSB
single-strand break
- SFN
sulforaphane
Footnotes
Author Contributions
Conception and design: Safa, Bijangi-Vishehsaraei, Saadatzadeh, Cohen-Gadol, Pollok. Acquisition of data: Safa, Bijangi-Vishehsaraei, Saadatzadeh, Wang, Nguyen, Kamocka, Cai. Analysis and interpretation of data: Safa, Bijangi-Vishehsaraei, Saadatzadeh, Wang, Nguyen, Kamocka, Cai, Pollok. Drafting the article: Safa, Bijangi-Vishehsaraei, Saadatzadeh, Pollok. Critically revising the article: Safa, Cohen-Gadol, Halum, Sarkaria, Pollok. Reviewed submitted version of manuscript: Safa, Bijangi-Vishehsaraei, Saadatzadeh, Wang, Kamocka, Cohen-Gadol, Halum, Sarkaria, Pollok. Approved the final version of the manuscript on behalf of all authors: Safa. Statistical analysis: Safa, Bijangi-Vishehsaraei. Study supervision: Safa, Cohen-Gadol, Pollok. Surgical removal and providing GBM tumors: Cohen-Gadol. Providing the GBM primary cultures: Sarkaria.
Disclosures
The authors report the following. Dr. Cohen-Gadol is a consultant for Zeiss and Meditec. Dr. Sarkaria received clinical or research support for this study from Novartis, Basilea, Genentech, Sanofi, Beigene, Lilly, Glaxo-Smith-Kline, Peloton, Glionova, Bristol-Myers Squibb, Cavion, and Curtana.
References
- 1.Balasubramanian S, Chew YC, Eckert RL. Sulforaphane suppresses polycomb group protein level via a proteasome-dependent mechanism in skin cancer cells. Mol Pharmacol. 2011;80:870–878. doi: 10.1124/mol.111.072363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bijangi-Vishehsaraei K, Saadatzadeh MR, Huang S, Murphy MP, Safa AR. 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) targets mRNA of the c-FLIP variants and induces apoptosis in MCF-7 human breast cancer cells. Mol Cell Biochem. 2010;342:133–142. doi: 10.1007/s11010-010-0477-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brescia P, Ortensi B, Fornasari L, Levi D, Broggi G, Pelicci G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells. 2013;31:857–869. doi: 10.1002/stem.1317. [DOI] [PubMed] [Google Scholar]
- 4.Calabrese V, Cornelius C, Dinkova-Kostova AT, Iavicoli I, Di Paola R, Koverech A, et al. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim Biophys Acta. 2012;1822:753–783. doi: 10.1016/j.bbadis.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Cho NP, Han HS, Leem DH, Choi IS, Jung JY, Kim HJ, et al. Sulforaphane enhances caspase-dependent apoptosis through inhibition of cyclooxygenase-2 expression in human oral squamous carcinoma cells and nude mouse xenograft model. Oral Oncol. 2009;45:654–660. doi: 10.1016/j.oraloncology.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Ferreira de Oliveira JM, Remédios C, Oliveira H, Pinto P, Pinho F, Pinho S, et al. Sulforaphane induces DNA damage and mitotic abnormalities in human osteosarcoma MG-63 cells: correlation with cell cycle arrest and apoptosis. Nutr Cancer. 2014;66:325–334. doi: 10.1080/01635581.2014.864777. [DOI] [PubMed] [Google Scholar]
- 7.Gamet-Payrastre L, Lumeau S, Gasc N, Cassar G, Rollin P, Tulliez J. Selective cytostatic and cytotoxic effects of glucosinolates hydrolysis products on human colon cancer cells in vitro. Anticancer Drugs. 1998;9:141–148. doi: 10.1097/00001813-199802000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Hegi ME, Diserens AC, Godard S, Dietrich PY, Regli L, Ostermann S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–1874. doi: 10.1158/1078-0432.ccr-03-0384. [DOI] [PubMed] [Google Scholar]
- 9.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 10.Innamorato NG, Rojo AI, García-Yagüe AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- 11.Jackson M, Hassiotou F, Nowak A. Glioblastoma stem-like cells: at the root of tumor recurrence and a therapeutic target. Carcinogenesis. 2015;36:177–185. doi: 10.1093/carcin/bgu243. [DOI] [PubMed] [Google Scholar]
- 12.Jackson SJ, Singletary KW. Sulforaphane: a naturally occurring mammary carcinoma mitotic inhibitor, which disrupts tubulin polymerization. Carcinogenesis. 2004;25:219–227. doi: 10.1093/carcin/bgg192. [DOI] [PubMed] [Google Scholar]
- 13.Jeon YK, Yoo DR, Jang YH, Jang SY, Nam MJ. Sulforaphane induces apoptosis in human hepatic cancer cells through inhibition of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase4, mediated by hypoxia inducible factor-1-dependent pathway. Biochim Biophys Acta. 2011;1814:1340–1348. doi: 10.1016/j.bbapap.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 14.Jin CY, Moon DO, Lee JD, Heo MS, Choi YH, Lee CM, et al. Sulforaphane sensitizes tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis through downregulation of ERK and Akt in lung adenocarcinoma A549 cells. Carcinogenesis. 2007;28:1058–1066. doi: 10.1093/carcin/bgl251. [DOI] [PubMed] [Google Scholar]
- 15.Kalkan R. Hypoxia is the driving force behind GBM and could be a new tool in GBM treatment. Crit Rev Eukaryot Gene Expr. 2015;25:363–369. doi: 10.1615/critreveukaryotgeneexpr.2015015601. [DOI] [PubMed] [Google Scholar]
- 16.Kim KH, Seol HJ, Kim EH, Rheey J, Jin HJ, Lee Y, et al. Wnt/β-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro Oncol. 2013;15:161–171. doi: 10.1093/neuonc/nos299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer. 2010;9:135. doi: 10.1186/1476-4598-9-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuo LJ, Yang LX. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo. 2008;22:305–309. [PubMed] [Google Scholar]
- 19.Lan F, Yu H, Hu M, Xia T, Yue X. miR-144-3p exerts anti-tumor effects in glioblastoma by targeting c-Met. J Neurochem. 2015;135:274–286. doi: 10.1111/jnc.13272. [DOI] [PubMed] [Google Scholar]
- 20.Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell. 2011;8:482–485. doi: 10.1016/j.stem.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee CS, Cho HJ, Jeong YJ, Shin JM, Park KK, Park YY, et al. Isothiocyanates inhibit the invasion and migration of C6 glioma cells by blocking FAK/JNK-mediated MMP-9 expression. Oncol Rep. 2015;34:2901–2908. doi: 10.3892/or.2015.4292. [DOI] [PubMed] [Google Scholar]
- 22.Lenzi M, Fimognari C, Hrelia P. Sulforaphane as a promising molecule for fighting cancer. Cancer Treat Res. 2014;159:207–223. doi: 10.1007/978-3-642-38007-5_12. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Zhang T. Targeting cancer stem cells with sulforaphane, a dietary component from broccoli and broccoli sprouts. Future Oncol. 2013;9:1097–1103. doi: 10.2217/fon.13.108. [DOI] [PubMed] [Google Scholar]
- 24.Meeran SM, Patel SN, Li Y, Shukla S, Tollefsbol TO. Bioactive dietary supplements reactivate ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS One. 2012;7:e37748. doi: 10.1371/journal.pone.0037748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood) 2007;232:227–234. [PMC free article] [PubMed] [Google Scholar]
- 26.Nagy A, Eder K, Selak MA, Kalman B. Mitochondrial energy metabolism and apoptosis regulation in glioblastoma. Brain Res. 2015;1595:127–142. doi: 10.1016/j.brainres.2014.10.062. [DOI] [PubMed] [Google Scholar]
- 27.Nakano I. Stem cell signature in glioblastoma: therapeutic development for a moving target. J Neurosurg. 2015;122:324–330. doi: 10.3171/2014.9.JNS132253. [DOI] [PubMed] [Google Scholar]
- 28.Negrette-Guzmán M, Huerta-Yepez S, Tapia E, Pedraza-Chaverri J. Modulation of mitochondrial functions by the indirect antioxidant sulforaphane: a seemingly contradictory dual role and an integrative hypothesis. Free Radic Biol Med. 2013;65:1078–1089. doi: 10.1016/j.freeradbiomed.2013.08.182. [DOI] [PubMed] [Google Scholar]
- 29.Park SJ, Wu CH, Gordon JD, Zhong X, Emami A, Safa AR. Taxol induces caspase-10-dependent apoptosis. J Biol Chem. 2004;279:51057–51067. doi: 10.1074/jbc.M406543200. [DOI] [PubMed] [Google Scholar]
- 30.Ping Z, Liu W, Kang Z, Cai J, Wang Q, Cheng N, et al. Sulforaphane protects brains against hypoxic-ischemic injury through induction of Nrf2-dependent phase 2 enzyme. Brain Res. 2010;1343:178–185. doi: 10.1016/j.brainres.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 31.Rajendran P, Kidane AI, Yu TW, Dashwood WM, Bisson WH, Löhr CV, et al. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics. 2013;8:612–623. doi: 10.4161/epi.24710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Emerging targets for glioblastoma stem cell therapy. J Biomed Res. 2015;30:30. doi: 10.7555/JBR.30.20150100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015;2:152–163. doi: 10.1016/j.gendis.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–2271. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 35.Schiffer D, Mellai M, Annovazzi L, Caldera V, Piazzi A, Denysenko T, et al. Stem cell niches in glioblastoma: a neuropathological view. BioMed Res Int. 2014;2014:725921. doi: 10.1155/2014/725921. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Sekine-Suzuki E, Yu D, Kubota N, Okayasu R, Anzai K. Sulforaphane induces DNA double strand breaks predominantly repaired by homologous recombination pathway in human cancer cells. Biochem Biophys Res Commun. 2008;377:341–345. doi: 10.1016/j.bbrc.2008.09.150. [DOI] [PubMed] [Google Scholar]
- 37.Sestili P, Fimognari C. Cytotoxic and antitumor activity of sulforaphane: the role of reactive oxygen species. BioMed Res Int. 2015;2015:402386. doi: 10.1155/2015/402386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sestili P, Paolillo M, Lenzi M, Colombo E, Vallorani L, Casadei L, et al. Sulforaphane induces DNA single strand breaks in cultured human cells. Mutat Res. 2010;689:65–73. doi: 10.1016/j.mrfmmm.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Shan Y, Wu K, Wang W, Wang S, Lin N, Zhao R, et al. Sulforaphane down-regulates COX-2 expression by activating p38 and inhibiting NF-κB-DNA-binding activity in human bladder T24 cells. Int J Oncol. 2009;34:1129–1134. doi: 10.3892/ijo_00000240. [DOI] [PubMed] [Google Scholar]
- 40.Shankar S, Ganapathy S, Srivastava RK. Sulforaphane enhances the therapeutic potential of TRAIL in prostate cancer orthotopic model through regulation of apoptosis, metastasis, and angiogenesis. Clin Cancer Res. 2008;14:6855–6866. doi: 10.1158/1078-0432.CCR-08-0903. [DOI] [PubMed] [Google Scholar]
- 41.Sharma C, Sadrieh L, Priyani A, Ahmed M, Hassan AH, Hussain A. Anti-carcinogenic effects of sulforaphane in association with its apoptosis-inducing and anti-inflammatory properties in human cervical cancer cells. Cancer Epidemiol. 2011;35:272–278. doi: 10.1016/j.canep.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 43.Singer E, Judkins J, Salomonis N, Matlaf L, Soteropoulos P, McAllister S, et al. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015;6:e1601. doi: 10.1038/cddis.2014.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 45.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 46.Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tortorella SM, Royce SG, Licciardi PV, Karagiannis TC. Dietary sulforaphane in cancer chemoprevention: the role of epigenetic regulation and HDAC inhibition. Antioxid Redox Signal. 2015;22:1382–1424. doi: 10.1089/ars.2014.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitaker SJ. DNA damage by drugs and radiation: what is important and how is it measured? Eur J Cancer. 1992;28:273–276. doi: 10.1016/0959-8049(92)90432-2. [DOI] [PubMed] [Google Scholar]
- 49.Wojton J, Meisen WH, Kaur B. How to train glioma cells to die: molecular challenges in cell death. J Neurooncol. 2016;126:377–384. doi: 10.1007/s11060-015-1980-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu C, Shen G, Chen C, Gélinas C, Kong AN. Suppression of NF-κB and NF-κB-regulated gene expression by sulforaphane and PEITC through IκBα, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24:4486–4495. doi: 10.1038/sj.onc.1208656. [DOI] [PubMed] [Google Scholar]