

The effects of sequence variation on function are elucidated by a study of protein evolution.
If we fully understood how protein function depends on amino acid sequence, we could predict the consequences of genetic variation, design new proteins with desired properties, and forecast a protein’s response to natural selection. A particular challenge is understanding the marginal effects of combinations of mutations, which geneticists call epistasis. New experimental methods like deep mutational scanning can functionally characterize huge protein libraries1, but they chart only relatively tiny regions of the vast landscape of possible sequences. An alternative approach is to computationally analyze the output of evolution, which can be viewed as a massive natural experiment, conducted over eons, in maintaining, optimizing, and diversifying proteins. In this issue, Hopf et al.2 show how a model that statistically identifies epistasis in alignments of present-day protein sequences can illuminate the sequence–function landscape and predict the functional effects of new mutations.
It has long been recognized that the site-specific frequency of each possible amino acid state in an alignment of homologous proteins can provide some insight into the functional effects of mutations, because states that are rarely or never seen have presumably been selected against during evolution. Statistical methods based on this principle have achieved moderate success in predicting pathogenic variants3. A vast literature, however, demonstrates that protein functions emerge from the cooperative interactions of amino acids at different sites, such that the effect of a mutation at one site depends on the amino acids that are present at other sites4. For example, individual residues that cause disease in humans are often the most common state in other species’ orthologous proteins5. Methods that ignore this within-protein epistasis are therefore limited in their explanatory and predictive power.
Experimental studies using deep mutational scanning can address epistasis for low-order combinations at relatively small numbers of sequence sites. They are labor intensive and can be applied only to protein functions that can be rapidly sorted or selected for using bulk laboratory assays. Computational analysis of sequence alignments can help to overcome these limitations. Epistasis produces covariation among sequence sites, because the states found at one site influence the states that are tolerated at another. But reliably identifying signatures of epistasis is a statistical challenge. Covariation can occur between sites that do not interact epistatically, for example, if they each depend on the state at a third site (Fig. 1). Another problem is that the number of epistatic interactions to be evaluated from a limited amount of data is immense. A protein of just 100 residues has almost 2 million potential pairwise interactions between possible amino acid states, and the number of potential third-order interactions is more than a billion.
Figure 1.
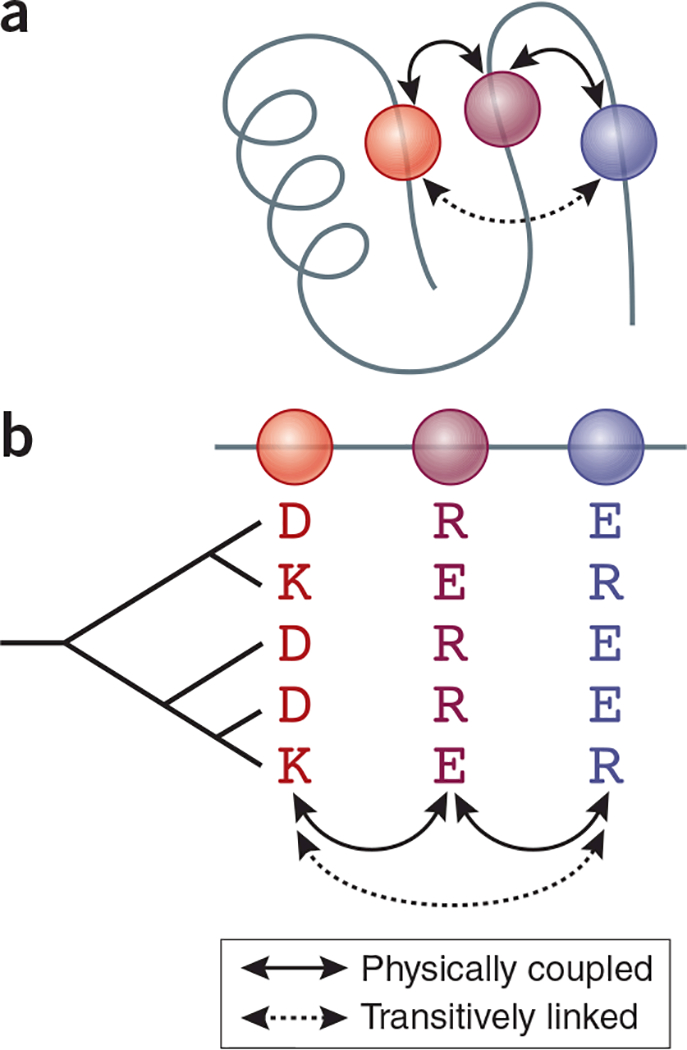
Origins of covariation in a multiple sequence alignment. (a) Sequence sites that are physically adjacent in a three-dimensional protein structure (red and purple, and purple and blue) mutually constrain each other’s set of tolerable amino acid states. (b) These interactions leave signatures of covariation in a multiple sequence alignment. But covariation can also occur between sites that do not interact (red and blue) because they are transitively coupled with a third site (purple). Rows represent protein sequences, whose evolutionary relationships are represented in the cladogram at left.
Recent work has addressed these challenges by fitting maximum-entropy probability models to large sequence alignments. These models, by analogy to statistical physics, express the probability of observing any protein sequence as an exponential function of the contribution of all its individual sequence states (the site-specific parameters) and the marginal contributions of all pairs of states (the epistatic parameters), summed across the entire protein6,7. The model is fit by finding the set of parameter values that maximize the probability of observing all the sequences in an alignment, with regularization to avoid over-fitting. Because the parameters are estimated globally using the entire set of sequence sites, they decouple causal epistatic interactions from the transitive correlations that plague other approaches, like mutual information and statistical coupling analysis, which focus on specific pairs or ‘sectors’ of many sites linked by covariation (Table 1)8.
Table 1.
Methods for covariation analysis
| Methoda | Premise | Description | Applications |
|---|---|---|---|
| Maximum entropy models | Global model of site- specific and interaction effects constructed by analogy with statistical physics | Parameters quantify site-specific residue preferences and epistatic couplings for all possible sequence states and pairs; best estimate maximizes the probability of all data observed in a multiple sequence alignment through the simplest set of couplings | De novo prediction of protein and RNA structure; inference of residues involved in intermolecular interactions; biophysical description of epistasis; prediction of mutations, effects on function |
| Mutual information | Local calculation of covariation score for any pair of sites in an alignment | Identifies pairs of covarying sites in an alignment, whether caused by epistasis or other factors (transitive coupling, or phylogenetic non-independence) | Prediction of physical contact between residues; identification of specificity- determining residues |
| Statistical coupling analysis | Global model of covarying sites interpreted by analogy with economics | Identifies groups of covarying residues by decomposing a pairwise site covariation matrix; covariation may be caused by epistasis or other factors | Identification of protein ‘sectors’ (independent sets of physically interacting residues predicted to cause some functional property); design of artificial proteins with native-like function |
For references and a more exhaustive list of methods, see ref. 8.
The first applications of maximum-entropy models to protein sequence alignments identified epistatically interacting sites, but they did not quantify the interactions between specific amino acid states at these sites. Epistatically linked sites were shown to correspond to residues in close contact in three-dimensional protein structures6, enabling de novo predictions of structure from sequence alignments7. The method has recently been elaborated to make finer-grained inferences of epistatic interactions among specific pairs of states2,9; when applied to TEM-1 β-lactamase, this model predicted with reasonable accuracy the effects of mutations observed in a deep mutational scan9.
Hopf et al.2 have now extended the effort to validate this fine-grained maximum-entropy method. They developed a maximum entropy model called EVmutation, which incorporates epistasis in the analysis of a protein family’s sequence alignment to make predictions about the effects of specific mutations. For each of several dozen protein families, they fit the model to a multiple sequence alignment, predicted the functional properties of new genotypes, and compared these predictions with experimental characterizations, including deep mutational scans, low-throughput assays of mutant function, and databases of human disease-causing mutations. The method predicted the observed effects of mutations reasonably well, with correlation coefficients ranging from 0.4 to 0.7, depending on the data set. The method performed significantly better than methods that ignore epistasis, including several popular approaches that use site-specific amino acid usage profiles and even a version of the authors’ maximum-entropy model that contained no interaction terms.
Although EVmutation is clearly an improved method compared with previous epistasis-free approaches, it still explains less than half the observed variance in protein function among genotypes. Several factors might explain this apparent limitation. First, alignment-based inferences and mutational scans measure different phenotypes. Natural sequence diversity reflects the total set of selective pressures that have affected all of the analyzed sequences over the course of their evolution; in contrast, deep mutational scanning measures growth under a specific set of conditions (like antibiotic challenge) or some specific functional or physical property, like enzyme activity, ligand affinity, or protein stability. Second, the experimental data might be noisy, or the amount of data in the alignment insufficient to precisely parameterize the model. Third, the functional form of the model may not sufficiently represent the relationships between sequence and function. For example, higher-order epistatic interactions may also be important10, and selective pressures on amino acid states may vary among evolutionary lineages.
These possibilities point to ways in which the performance of maximum-entropy models might be improved. First, higher-order interactions could be incorporated. Most of the experimental data sets that Hopf et al.2 used for validation included only single- and double-mutant variants of wild-type proteins; when data on higher-order combinations were available, Hopf et al.2 found that their model predicted the experimental functions of quadruple mutants less accurately than it did of triple mutants. Just as incorporating pairwise epistasis improves accuracy compared to the single-site-based model, incorporating third-order epistasis—or even higher-order interactions—might substantially improve the model’s performance, particularly when predicting the functions of more distant sequences. If such models can be parameterized reliably, they could be particularly useful for engineering new proteins or predicting evolutionary trajectories toward sequences that are multiple mutations away from naturally occurring proteins.
Covariation because of phylogenetic non-independence is another potential source of error in the current method. If two sites that do not functionally interact happen to change along the same evolutionary lineage, the derived states will co-occur in all descendent sequences unless subsequent substitutions break their correlation. If the sites are slowly evolving, then the probability of subsequent evolutionary change might be low, and false epistatic couplings would be inferred. To reduce phylogenetic non-independence, Hopf et al.2 weight sequences inversely to their sequence identity. An approach that explicitly incorporates phylogeny might produce more reliable estimates of epistasis and better predict the functions of new protein sequences.
Deep mutational scanning and maximum entropy models have emerged almost simultaneously; together, they represent a real breakthrough for efforts to explore sequence–function landscapes at unprecedented scales. The capacity to characterize and predict single- and double-mutant phenotypes has immediate implications for genetics and medicine. With further development, we may soon begin to answer deep and unresolved questions at the intersection of protein biochemistry and evolution. How does a protein’s architecture determine the strength and extent of epistasis? Can the evolutionary record embedded in sequence alignments improve efforts to engineer new proteins? How wide are the connected networks of sequences with similar functions, which evolution can explore under neutral drift, point mutation, and purifying selection alone? How distant from each other are networks of sequences with different folds and functions, and what are the properties of proteins along the paths that connect them? Does the set of epistatic interactions in a protein family vary between extant and ancestral proteins, suggesting that a protein’s sequence–function landscape—and therefore its evolutionary potential—changes as it evolves?
To date, only sequence neighborhoods very close to extant proteins have been explored. But progress in these studies allows us to imagine how we might chart more distant regions of sequence-function landscapes as methods continue to improve. Such work may help us to understand how and why evolution has explored these landscapes in the past and to envision proteins of the future.
ACKNOWLEDGMENTS
Supported by NIH R01GM104397 (J.W.T.), NIH training grant T32-GM007183 (T.N.S.), and a Graduate Research Fellowship from the National Science Foundation (T.N.S.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Fowler DM & Fields S Nat. Methods 11, 801–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopf TA et al. Nat. Biotechnol. 35, 128–135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng PC & Henikoff S Annu. Rev. Genomics Hum. Genet. 7, 61–80 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Starr TN & Thornton JW Protein Sci. 25, 1204–1218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kondrashov AS, Sunyaev S & Kondrashov FA Proc. Natl. Acad. Sci. USA 99, 14878–14883 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morcos F et al. Proc. Natl. Acad. Sci. USA 108, E1293–E1301 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marks DS et al. PLoS One 6, e28766 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Juan D, Pazos F & Valencia A Nat. Rev. Genet. 14, 249–261 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Figliuzzi M, Jacquier H, Schug A, Tenaillon O & Weigt M Mol. Biol. Evol. 33, 268–280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinreich DM, Lan Y, Wylie CS & Heckendorn RB Curr. Opin. Genet. Dev. 23, 700–707 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]