

Atherosclerosis is recognized as a chronic inflammatory disease of the vascular wall, resulting in the narrowing of blood vessels caused by the growth of lipid-rich lesions. Immune cells of all sorts have been identified in atherosclerotic lesions and their individual contributions to the pathogenesis of atherosclerosis continue to be an active topic of cardiovascular disease research. Hyperlipidemia has long been recognized as a source of inflammation that drives atherosclerosis (1). Hyperlipidemia increases cytokine levels in the circulation that promote endothelial cell dysfunction and an upregulation of adhesion molecules that facilitate immune cell recruitment into the vasculature (2). More recently, hyperlipidemia has been recognized as a cause for accelerated hematopoiesis and increased leukocyte burden in the circulation that further drives cytokine production and the recruitment of immune cells into the vessel wall (3). Hyperlipidemia also results in lipid deposits within the vessel wall that become oxidized and avidly taken up by resident and newly recruited myeloid cells. The process of foam cell formation is itself accompanied by cellular activation including via the NF-κB pathway resulting in the secretion of cytokines that further promote lesion inflammation and plaque progression (1).
Non-coding RNA (ncRNA), including microRNA have recently emerged as central participants in the process of vascular inflammation in atherosclerosis (4). Prime examples include miR-33, miR-181b, miR-155 and miR-146a. Through pioneering studies of microRNA in atherosclerosis pathogenesis, Moore and colleagues elucidated contributions of miR-33 in the regulation of hepatic lipid synthesis and plasma lipid metabolism (5), as well as cellular cholesterol homeostasis, energy metabolism and polarity in macrophages (6). These investigators also demonstrated benefits of miR-33 antagonism in suppressing and even reversing atherosclerosis in rodents and primates (7). Furthermore, studies by Nazari-Jahantigh and colleagues revealed detrimental properties of miR-155 in driving macrophage NF-κB activation and atherosclerosis in hyperlipidemic rodents (8). Antagonism of miR-155 was shown to reheve macrophage-derived vascular inflammation and thereby suppress atherosclerosis in hyperlipidemic Apoe−/− mice. Collectively, results of these findings support the value of inflammation control via microRNA as treatments for atherosclerosis (9).
Augmentation of microRNA levels was soon thereafter reported as alternative avenues for atherosclerosis control. Studies by Feinberg and colleagues reported benefits of enhancing miR-181b levels in endothelial cells to control NF-kB-driven inflammation and adhesion molecule expression (10). Furthermore, systemic infusions of miR- 181b mimics emulsified in phospholipid liposomes were shown to enrich miR-181b levels in the endothelium of mice with dyslipidemia resulting in reduced vascular inflammation and atherosclerosis (11). The value of targeting NF-κB-driven inflammation via microRNA was reinforced through studies by Fish and colleagues who demonstrated benefits of miR-146a expression in suppressing inflammatory pathways in the murine endothelium (12).
Building on these observations, our laboratory explored benefits of miR-146a augmentation in controlling NF- κB-driven atherosclerosis in hyperlipidemic mice (13). An impetus for such studies derived from our observations of reduced miR-146a levels and increased NF-κΒ activity in myeloid cells isolated from mice deficient in apolipoprotein (apo) E expression (14). ApoE expression in the macrophage had long since been recognized in protecting against atherosclerosis by preventing cellular activation caused by lipid over-accumulation (14). Our findings of enhanced miR-146a levels in monocytes and macrophages expressing apoE revealed a link between cellular lipid metabolism and microRNA biogenesis in the hematopoietic system (Figure 1). Furthermore, our findings demonstrated that systemic infusions of phospholipid-emulsified miR-146a mimics into hyperlipidemic Apoe−/− mice increased miR- 146a levels and reduced NF-κΒ signaling in peritoneal macrophages. In fact, a three-week course of miR-146a mimic infusion into hyperlipidemi Apoe−/− or Ldlr−/− mice fed a high fat western diet led to a profound suppression of hyperlipidemia-driven hematopoiesis as revealed by reduced levels of circulating Ly6Chi monocytes. This was accompanied by a corresponding suppression of vascular inflammation and atherosclerosis independent of a change in levels of hyperlipidemia (13). However, the importance of the cell specificity in endogenous miR-146a augmentation was not revealed in these studies as systemic miR-146a mimic infusions could have enriched cells of the endothelium as well as those of the hematopoietic system. A study recently published by Cheng et al. (15) addressed this timely and important question that revealed several unexpected outcomes.
Figure 1.
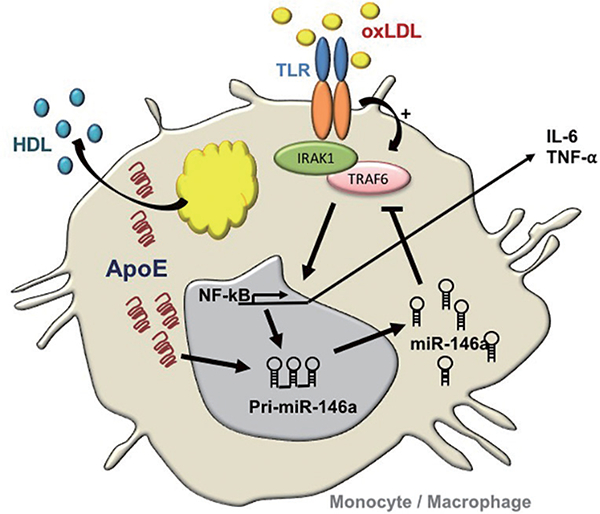
ApoE regulation of myeloid cell activation via microRNA. Environmental ligands of Toll like receptors including oxLDL caused by hyperlipidemia provoke inflammatory signaling in monocytes and macrophages resulting in NF-κΒ activation. Gene transcription from NF-κΒ activity results in the production of inflammatory mediators including pro-atherogenic cytokines. It also results in the production of pri-miR-146a that is subsequently processed to mature miR-146a that silences the expression of key TLR-adaptor molecules IRAK1 and TRAFF6. The production of miR-146a thereby serves as a regulatory feedback loop to suppress NF-κΒ activity and resolve inflammation. Our recent findings have revealed that beyond serving to eliminate pools of cellular lipid levels via plasma HDL, apoE amplifies this regulatory feedback loop by enhancing the biogenesis of miR-146a (13).
The recognition of miR-146a as a guardian of the hematopoietic system emanates from numerous studies by Baltimore and colleagues reporting its ability to control exaggerated NF-κB signaling and premature bone marrow (BM) failure in rodents (16). Guided by this body of literature, Cheng et al. (15) tested consequence of miR-146a deficiency in the BM relative to other cellular compartments in atherosclerosis susceptibility. At the outset, the investigators validated the presence of miR-146a within the vascular wall of Ldlr−/− mice fed a high cholesterol diet (HCD) for 18 weeks, which they found to be associated with both endothelial cells and macrophages within the vascular intima. Interestingly, repeating the assessment at an earlier stage of lesion development revealed increased miR-146a levels at anatomical locations prone to atheroma development including the inner curvature of the aorta. Conceivably, such miR146a enrichment could have derived from a preferential accumulation of myeloid cells in this vascular location, or from increased cellular NF-κB signaling in endothelial cells due to sheer stress forces caused by altered blood flow.
Next, Cheng et al. (15) examined the impact of miR- 146a loss on atherosclerosis susceptibility by comparing lesion sizes in hyperlipidemic Ldlr−/− mice to those in miR- 146-/−.Ldlr−/− double deficient mice henceforth termed DKO mice. Comparisons were made at two-time points; an early 12-week time point and a late 18-week time point following initiation of a HCD. Although no changes in atherosclerosis were noted between the two models at the early time point, the absence of miR-146a protected both male and female DKO mice from atherosclerosis development in the aortic arch 18 weeks after HFD initiation. An explanation for the unexpected phenotype could have rested with an observed reduction in levels of atherogenic very-low density (VLDL) and low density (LDL) lipoprotein-associated cholesterol in the circulation of DKO mice. But despite a protective drop in plasma lipid levels, a global loss of miR-146a expression also led to a substantial increase in systemic inflammatory cytokine levels including IL-6 that should have driven the process of atherosclerosis in these mice. Interestingly, protective effects of global miR146a deficiency were manifest mainly within the aortic arch, a recognized site of atherosclerosis predilection, while other sites within the vascular tree seemed less sensitive to the protective effects.
Such observations could at first glance be construed as heresy to dogma linking miR-146a to the control of NF- κΒ-driven inflammation (17), including in the pathogenesis of atherosclerosis (13). To address this paradox, the investigators resorted to further experimentation including through BM transplant experiments in which wildtype (WT) or miR-146a−/− BM was transplanted into lethally irradiated Ldlr−/− mice, henceforth termed BMT mice that were subsequently fed the HCD and assessed for atherosclerosis burden. Recipient BMT mice displayed similar reconstitution of hematopoiesis 8 weeks after transplantation with either type of BM prior to initiation of a HCD. However, following a 12-week course of HCD, BMT mice reconstituted with miR-146−/− BM displayed fewer lipid-rich plaques in the aortic arch despite increased levels of circulating cytokines including IL-6 and TNF-α. These observations, combined with those of the DKO mice revealed an unexpected benefit of BM miR-146a deficiency in protecting against diet-induced atherosclerosis (Figure 2).
Figure 2.
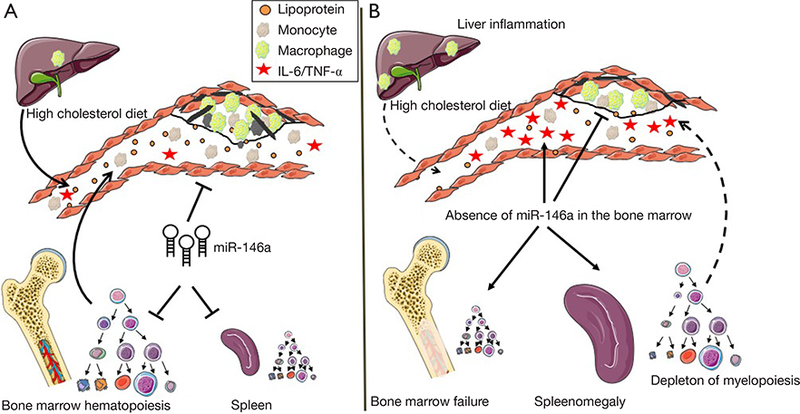
Paradoxical reduction of atherosclerosis in the absence of miR-146a. (A) Recent studies have reported a benefit in enhancing miR146a in myeloid cells (13) and the endothelium (18) to suppress NF-κΒ activity and thereby the progression of atherosclerosis in hyperlipidemic mice. (B) A recent study by Cheng et al. (15) uncovered that an elimination of miR146a in the BM does not enhance diet- induced atherosclerosis but rather results in a reduction of lipid rich lesions in the aortic arch of hyperlipidemic mice. The paradoxical effect appears to stem from diet-induced BM failure and aberrant extramedullary hematopoiesis in the spleen that result in a loss of myeloid cell production. A lack of hematopoietic miR-146a production also resulted in increased cytokine levels in the circulation but reduced plasma lipid levels due to liver inflammation and reduced lipoprotein production. Together, such metabolic and inflammatory dysregulation combined to suppress the growth of atheroma in hyperlipidemic mice.
An explanation to the paradoxical findings emerged at first from the reduced levels of cholesterol and other lipids carried by plasma lipoproteins including VLDL, LDL and HDL in both DKO and miR-146−/− BMT mice. Because lipid levels were not significantly altered in the liver or in feces, Cheng et al. (15) tested gene expression changes in the liver to gain insight into the observed reduced plasma lipids in miR-146−/− BMT mice. Their data showed a marked increase in the expression of macrophage markers as well as inflammatory cytokines that included IL-1β, IL-6 and IL-10 which had previously been associated with reduced hepatic lipid synthesis. Further scrutiny revealed mildly reduced expression of the ApoB gene that is important in the production of VLDL, and robustly increased expression of sortilin-1 (Sort-1) that had previously been identified as a source of macrophage inflammation and as a regulator of plasma LDL levels. Through further experimentation, the investigators confirmed Sort-1 as a new bona-fide target of miR-146a and that an exposure of cultured hepatocytes to IL-6 results in reduced lipoprotein secretion. Collectively, the findings gathered so far suggested that a loss of miR-146a expression in the BM contributed to liver inflammation and thereby to reduced lipoprotein production and lower plasma lipid levels while at the same time resulted in increased systemic inflammation that together protected against atherosclerosis.
In working to resolve a conundrum of inflammatory and metabolic dysregulation that together led to atherosclerosis protection in both DKO and miR-146−/− BMT mice fed a HCD, the investigators paid closer attention to diet- and age-dependent defects in hematopoiesis among miR- 146a deficient mice. Although earlier studies had noted hematopoietic stem cell (HSC) dysfunction and BM failure in LPS-challenged and older miR-146a−/− mice due to unrestrained NF-κΒ signaling (16), the impact that a HCD could exert on precipitating aberrant hematopoiesis had not been previously examined in these mice. In pursuing this mode of reasoning, they observed that extramedullary hematopoiesis and BM failure could be accelerated in young DKO and miR-146−/− BMT mice fed a HCD as evidenced by large spleens and pale femurs. The investigators took a step further to examine the effects that ageing could impart on splenomegaly and its consequences on atherosclerosis. In contrast to young 10-week-old DKO mice that had unaltered plaque burden after a 12-week course of HCD, a similar course of HCD fed to older 20-week-old DKO mice led to a pronounced protection against atherosclerosis in the aortic arch. This protection was accompanied by paling of the femurs and splenomegaly due to an expansion of leukocytes. A similar phenotype of splenomegaly and paling of the femurs was noted in miR-146−/− BMT mice fed a HCD for 12 weeks, which again coincided with a reduction in plasma LDL and cholesterol levels.
A closer inspection of HSC together with multipotent and committed progenitor cells in the BM and spleen revealed a profound dysfunction of hematopoiesis and loss of myelopoiesis in both DKO and miR-146−/− BMT mice fed a HCD. Interestingly, such dysfunction of hematopoiesis occurred more rapidly among miR-146−/− BMT mice than in DKO mice which led to an accumulation of lymphocytes and CD45+ leukocytes in the spleen. Collectively, these observations established the value of miR-146a in guarding against BM failure and extramedullary hematopoiesis caused by HCD feeding.
In seeking to reconcile how diet-induced BM failure combined with accelerated extramedullary hematopoiesis and enhanced systemic inflammation could paradoxically convey protection against atherosclerosis, Cheng et al. (15) monitored levels of circulating immune cells in cohorts of DKO and miR-146−/− BMT mice fed a HCD. By including an earlier time point, the investigators observed a period of robust hematopoiesis 4 weeks after HCD initiation that led to pronounced monocytosis similar to what they had reported in prior models of diet-induced hematopoiesis (19). However, as soon as 12 and 18 weeks of HCD, leukocytosis had taken a downturn in both miR-146−/− BMT and DKO mice respectively indicative of a biphasic response of hematopoiesis in mice deficient in miR-146a in the hematopoietic compartment. A closer look at BM function through competitive BM transplant experiments revealed that miR-146−/− BM outperformed WT BM in producing neutrophils and Ly6Chi monocytes in recipient Ldir−/− BMT mice fed a chow diet or after a short duration of HCD consumption. However, an extended period of HCD consumption resulted in a progressive decline in miR-146a−/− BM derived-leukocytes while leukocytes derived from WT BM persisted. Furthermore, an assessment of leukocytes enzymatically released from advanced atheroma in the aorta of recipient Ldir−/− BMT mice revealed myeloid cells to be predominately of WT BM origin, while lymphocytes of both hematopoietic origins were present at similar levels. This finding was in sharp contrast to what was observed in cells detected in the aorta of recipient Ldlr−/− BMT mice fed a chow diet where the majority of leukocytes were derived from miR-146−/− BM. Consistent with observations in the circulation and atheroma, an examination of the BM revealed that hematopoietic cells were largely of WT origin in mice fed a HCD, but the opposite was seen in mice fed a normal diet.
In a final series of experiments, the investigators sought to look beyond the BM as a site sensitive to miR-146a loss in the pathogenesis of atherosclerosis. This was achieved through BMT experiments performed by transplanting WT BM into lethally irradiated Ldlr−/− mice and DKO mice that were subsequently maintained on a HCD for 12 weeks. Interestingly, there were no noticeable differences in circulating pro-inflammatory cytokines or cholesterol- containing lipoprotein particles between the two models demonstrating that the dysregulation in systemic inflammation and plasma lipoproteins levels are dependent on deletion of miR-146a from the BM rather than other tissue such as hepatocytes. Moreover, levels of leukocytes in the spleen and the circulation were also normalized. Overall, a miR-146a rescue of the hematopoietic system in the WT→DKO BMT mice resulted in increased lipid-rich plaque burden compared to WT→Ldlr−/− mice. Interestingly, the source of this added atherosclerosis likely derived from the inflamed endothelium of WT→DKO BMT mice that displayed increased expression levels of adhesion and chemokines genes consistent with the investigators prior reports demonstrating the value of miR-146a in suppressing endothelial cell activation (12).
In the end, how has this meticulous and exhaustive study succeeded in uncloaking the behavior of miR-146a in the pathogenesis of atherosclerosis? While some data cast miR- 146a as a foe through its ability to sustain myeloid cell output from the BM and spleen that populate the vascular wall of hyperlipidemic mice, other data find it to be a friend by protecting the vascular endothelium from NF- κB inflammatory responses (Figure 2). The conundrum of miR-146a action in atherosclerosis predisposition revealed through studies of these mouse models will thus require further scrutiny to resolve the paradoxical observation. One avenue to explore could include an avoidance of HCD feeding to prevent rapid BM failure and an elimination of myeloid cells in the circulation. In this respect, it would be interesting to explore why a loss of miR-146 in the BM led to myeloid cell depletion but did not exert a similar destructive impact on lymphopoiesis. Also, exploring why the protective properties of miR-146a deficiency were limited to the aortic arch could reveal a flow sensitive nature of miR-146a in the vessel wall as recently reported (20). Furthermore, it would be of interest to test whether systemic delivery of miR-146a mimics in DKO and miR- 146−/− BMT mice fed a HCD would enhance atherosclerosis by preventing BM failure and by enhancing lipoprotein secretion in the liver. How such findings would relate to prior atheroprotective properties ascribed to miR-146a in mature myeloid cells (13) and the endothelium (18) would certainly sustain the mystery surrounding this microRNA. Clearly much more work will be required to fully define the role of miR-146a in atherosclerosis in order to determine whether it may serve as a useful target for human cardiovascular disease.
Acknowledgements
Funding·. This work was supported by grants from the National Institutes of Health, including from the Extracellular RNA Communication Consortium Common Fund grant 5U19CA179512, and grant HL133575 to RLR; the UCSF Diabetes Center grant P30 DK087945; the American Heart Association Grant-in-Aid 16GRNT27640007, which were administered by the Northern California Institute for Research and Education. The work was performed at the Veterans Affairs Medical Center, San Francisco, California.
Footnote
Conflicts of Interest·. The author has no conflicts of interest to declare.
Comment on: Cheng HS, Besla R, Li A, et al. Paradoxical Suppression of Atherosclerosis in the Absence of microRNA-146a. Circ Res 2017;121:354–67.
Provenance: This is an invited Editorial commissioned by the Section Editor Dr. Shengguang Ding (The Second Affiliated Hospital of Nantong University, Nantong, China).
References
- 1.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell 2001;104:503–16. [DOI] [PubMed] [Google Scholar]
- 2.Lichtman AH, Clinton SK, Iiyama K, et al. Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Arterioscler Thromb Vasc Biol 1999;19:1938–44. [DOI] [PubMed] [Google Scholar]
- 3.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rayner KJ, Moore KJ. The plaque “micro” environment: microRNAs control the risk and the development of atherosclerosis. Curr Atheroscler Rep 2012;14:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rayner KJ, Suarez Y, Davalos A, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010;328:1570–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouimet M, Ediriweera HN, Gundra UM, et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest 2015;125:4334–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rayner KJ, Sheedy FJ, Esau CC, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest 2011;121:2921–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nazari-Jahantigh M, Wei Y, Noels H, et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J Clin Invest 2012;122:4190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feinberg MW, Moore KJ. MicroRNA Regulation of Atherosclerosis. Circ Res 2016;118:703–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun X, Icli B, Wara AK, et al. MicroRNA-181b regulates NF-kappaB-mediated vascular inflammation. J Clin Invest 2012;122:1973–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun X, He S, Wara AKM, et al. Systemic delivery of microRNA-181lb inhibits nuclear factor-kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ Res 2014;114:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng HS, Sivachandran N, Lau A, et al. MicroRNA-146 represses endothelial activation by inhibiting pro- inflammatory pathways. EMBO Mol Med 2013;5:1017–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li K, Ching D, Luk FS, et al. Apolipoprotein E Enhances MicroRNA-146a in Monocytes and Macrophages to Suppress Nuclear Factor-kappaB-Driven Inflammation and Atherosclerosis. Circ Res 2015;117:el-ell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raffai RL. Apolipoprotein E regulation of myeloid cell plasticity in atherosclerosis. Curr Opin Lipidol 2012;23:471–8. [DOI] [PubMed] [Google Scholar]
- 15.Cheng HS, Besla R, Li A, et al. Paradoxical Suppression of Atherosclerosis in the Absence of microRNA-146a. Circ Res 2017;121:354–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao JL, Rao DS, O’Connell RM, et al. MicroRNA- 146a acts as a guardian of the quality and longevity of hematopoietic stem cells in mice. Elife 2013;2:e00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boldin MP, Taganov KD, Rao DS, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med 2011;208:1189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma S, Tian XY, Zhang Y, et al. E-selectin-targeting delivery of microRNAs by microparticles ameliorates endothelial inflammation and atherosclerosis. Sci Rep 2016;6:22910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbins CS, Chudnovskiy A, Rauch PJ, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 2012;125:364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong WT, Ma S, Tian XY, et al. Targeted Delivery of Shear Stress-Inducible Micrornas by Nanoparticles to Prevent Vulnerable Atherosclerotic Lesions. Methodist Debakey Cardiovasc J 2016;12:152–6. [DOI] [PMC free article] [PubMed] [Google Scholar]