

Abstract
Bone cements for treatment of fractures at weight-bearing sites are subjected to dynamic physiological loading from daily activities. An ideal bone cement rapidly sets after injection, exhibits bone-like strength, stimulates osteogenic differentiation of endogenous cells, and resorbs at a rate aligned with patient biology. However, currently available materials fall short of these targeted properties. Nanocrystalline hydroxyapatite (nHA) enhances osteogenic differentiation, new bone formation, and osteoclast differentiation activity compared to amorphous or micron-scale crystalline hydroxyapatite. However, the brittle mechanical properties of nHA precludes its use in treatment of weight-bearing bone defects. In this study, we report settable nHA-poly(ester urethane) (PEUR) nanocomposites synthesized from nHA, lysine triisocyanate (LTI), and poly(caprolactone) triol via a solvent-free process. The nanocomposites are easily mixed and injected using a double-barrel syringe, exhibit mechanical properties exceeding those of conventional bone cements, enhance mineralization of osteoprogenitor cells in vitro, and undergo osteoclast-mediated degradation in vitro. This combination of properties cannot be achieved using other technologies, which underscores the potential of nHA-PEUR nanocomposites as a new approach for promoting bone healing at weight-bearing sites.
Introduction
Bone substitutes utilized in the treatment of fractures at weight-bearing sites, such as intra-articular joints, are subjected to repetitive, dynamic physiological loading from daily activities.1 An ideal bone cement rapidly sets after injection, provides bone-like strength, stimulates osteogenic differentiation of endogenous cells, and resorbs at a rate aligned with patient biology. Bone substitutes that remodel to form new bone while maintaining load-bearing capacity would significantly improve patient care.2 However, biomaterials that provide both functionality and load-bearing capability are currently not available. Poly(methyl methacrylate) (PMMA) cement is indicated for use in arthroplasty procedures of the hip, knee, and other weight-bearing joints due to its high strength (≥70 MPa).3,4 However, PMMA is non-resorbable and does not integrate with host bone, resulting in device loosening and long-term failure.5–9
Injectable and settable calcium phosphate cements (CPCs) showed promise in early weight-bearing applications due to their bone-like compressive strength.2,10 Calcium phosphate cements (CPCs) are osteoconductive and resorbable, but their low bending strength and fatigue resistance preclude their use at weight-bearing sites.2,11 Biphasic CPCs reinforced with a polymeric or metallic phase exhibit enhanced mechanical properties compared to monophasic cements.12–16 Reinforcement of CPCs with polymer or metal fibers increased bending strength to 139 MPa,16 but there are a limited number of studies evaluating osteogenic differentiation and osteoclast-mediated resorption of these materials. Recently, bioactive glass-polymer hybrids characterized by covalent nanoscale-interactions between the inorganic and organic phases have been reported to exhibit compressive strengths as high as 300 MPa and tunable degradation.17–22 However, bioactive glass-polymer composites undergo uncontrolled degradation by hydrolysis, and the sol-gel approach requires solvents and elevated temperatures that preclude injection. Thus, there is a compelling need for settable bone cements that exhibit bone-like strength, stimulate osteogenic differentiation and mineralization of endogenous cells, and undergo osteoclast-mediated resorption at a rate aligned with patient biology.
Nanocrystalline (grain size < 100 nm) hydroxyapatite (nHA) enhances osteogenic differentiation, new bone formation, and osteoclast differentiation activity compared to micron-scale crystalline HA.23–32 The reactive hydroxyl (OH) group on the surface of HA can be used to graft organic molecules, such as polyesters and polyisocyanates.33–38 Grafting organic molecules to the surface of ceramic nanoparticles enhances their dispersion in organic polymers as well as interfacial binding between the polymer and ceramic phases, resulting in improved mechanical properties and stimulation of cell attachment, proliferation, and differentiation.37,39–45 However, the effects of surface grafting on mechanical and biological properties of reactive and settable nHA/polymer composite cements have not been investigated. Furthermore, studies investigating grafting of polyisocyanates to nHA have utilized solvents such as dimethyl formamide (DMF), which are cytotoxic and not suitable for injectable biomaterials.36,38 We have previously reported that lysine-derived poly(ester urethane)s (PEUR) undergo cell-mediated oxidative degradation.46,47 In this study, we grafted lysine triisocyanate (LTI) to the surface of nHA by a solvent-free process to yield a viscous prepolymer that can be further crosslinked with a polyester triol by injection through a double-syringe static mixer to form a nHA-PEUR nanocomposite cement. We investigated the effects of grafting LTI to nHA on nHA dispersion, mechanical properties, osteoblast mineralization, and osteoclast- and ROS-mediated resorption of the nHA-PEUR cements.
Experimental
Materials
nHA particles were purchased from Sigma-Aldrich and dried at 80 °C under vacuum for 48 hours before use. LTI was supplied by Jinan Haohua Industry Co., Ltd (Jinan, China). Before use, LTI was purified by dissolving in tert-butyl methyl ether (TBME, Across-Organic) and refluxing with decolorizing carbon (Fisher Scientific) at 63°C for 22 hours. α-MEM, FBS, trypsin-EDTA and Antibiotic-Antimycotic were purchased from ThermoFisher Scientific. Mesenchymal stem cell media were purchased from PromoCell, Inc. Iron acetylacetonate (FeAA) catalyst, fibronectin, vitronectin, Alizarin Red S and 1α,25-Dihydroxyvitamin D3 were purchased from Sigma-Aldrich and used as received. The mouse MC3T3 osteoblast cell line was purchased from ATCC. Human bone marrow derived mesenchymal stem cells (hMSCs) were purchased from Extem Bioscience (San Francisco, CA). Dentin was acquired from the donation of a surrendered elephant tusk from the National Eagle and Wildlife Property Repository of the United States Department of the Interior, U.S. Fishes and Wildlife Service.
Synthesis and characterization of nHA-LTI/LTI prepolymer
nHA particles (Sigma) were mixed with LTI and FeAA (0.027 wt%) catalyst using a SpeedMixer (FlackTek, Inc, Landrum, SC) for 10 min and maintained at 50 °C for 3 h. %NCO of the resulting prepolymer was measured by titration.48,49 ATR-FTIR was performed using a Seagull Variable Angle Reflection Accessory (Harrick Scientific) attached to a Tensor 27 FTIR (Bruker) from 750 to 4000 cm−1. The prepolymer was dissolved in dichloromethane and centrifuged to recover grafted nHA-LTI particles. XPS was performed using an Ulvac-PHI Versaprobe 5000. Particle samples were pressed into indium foil and mounted onto sample pucks before introducing them into the instrument. Monochromatic Al Kα x-rays (1486 eV), a 100-γm diameter x-ray spot, and a takeoff angle of 60° off sample normal were used. Pass energies of 187.7 eV and 23.5 eV were utilized for the survey and high-resolution acquisitions, respectively. Charge neutralization was accomplished using 1.1 eV electrons and 10 eV Ar+ ions. Placing the -CH2- type bonding in the carbon 1s spectrum at 284.8 eV corrected any minor energy shifts due to charging. Relative atomic concentrations were calculated using peak areas and PHI handbook sensitivity factors.50 nHA-LTI particles (> 200 per group) were re-dispersed in ethanol (0.1 wt%), sputter-coated, imaged by SEM (Zeiss Merlin), and measured by Image J. XRD was conducted using a Scintag XGEN-4000 X-ray diffractometer (λ = 154 nm). The peak at 2θ = 26.04° was chosen for calculation of crystallite size, which corresponds to (0 0 2) Miller’s plane family and shows the crystal growth along the c-axis of the HA crystalline structure.51 Viscosity samples were loaded between 25-mm plates with 500-μm gap size on a TA AR2000ex rheometer.
Synthesis of nHA/PEUR nanocomposites
To fabricate the nHA/PEUR nanocomposites, nHA-LTI/LTI prepolymer was mixed with poly(ε-caprolactone) triol (PCL triol, 300 g mol−1) using a SpeedMixer (FlackTek, Inc, Landrum, SC) for 1 min. Although the nanocomposites rapidly set after mixing, they were maintained at 50 °C overnight to ensure full conversion. The isocyanate index (ratio of NCO: OH equivalents * 100) was either 115 or 140. Alternatively, ungrafted nHA particles were added to the reactive LTI/poly(ε-caprolactone) mixture without the prepolymer step. These materials are denoted as nHA-LTI and nHA nanocomposites, respectively.
Mechanical testing
nHA-LTI and nHA nanocomposites were cured and soaked in PBS at 37°C for 24 h. Quasi-static compression testing was conducted using an MTS 858 Bionix Servohydraulic test system (Eden Prairie, MN). Specimens were preloaded to 12 N and compressed until failure at 25 mm min−1. Compressive modulus was calculated from the initial slope of the stress-strain curve, and compressive strength was determined from the maximum stress. Flexural strength and moduli were measured by 4-point bending of rectangular (36.9 mm x 7.4 mm x 3.7 mm) specimens. The support and load spans were maintained at 30.8 mm and 10.3 mm (1/3 of the support span), respectively (ASTM D6272-10).52 Bending strength (σB) was calculated from the following equation:
| (1) |
where F = load at fracture (N), L = support span length (mm), w = width (mm), and d = thickness (mm). Bending modulus (EB) was calculated according to following equation:
| (2) |
where m0 = initial slope of the stress-strain curve.
Swelling
Specimens (0.56 ± 0.15 g) were incubated in methylene chloride for 24 h. Wet mass was measured immediately upon removal from the solvent. % swelling was calculated as:
| (3) |
Contact angle and protein adsorption
Cured speciments were cut into chips with ~1 mm thickness and cross-sectional area compatible with a 24-well tissue culture plate. The surface of the chips was polished with a series of wet silicon carbide papers using K1000 roughness for 10 s, K1200 roughness for 10 s, and K4000 roughness for 60 s. Additionally, LTI-poly(ester-urethane) (LTI-PEUR) without nHA was prepared as a polymer control by mixing and curing LTI with poly(ε-caprolactone triol) and FeAA catalyst at the same conditions described for the nHA-PEUR composites above. The cured LTI-PEUR samples were polished using the same procedure as other samples. Water contact angle was measured using a Rame-Hart (Moutain Lakes, NJ) Model A-100 goniometer. Protein adsorption was measured by Pierce BCA kit after incubating samples in fibronectin or vitronectin solutions (5 μg ml−1) at 37°C overnight.
Cell culture
MC3T3 cells were maintained in α-MEM with FBS (10%) and antibiotic-antimycotic (1%). Cells were detached at sub-confluency by trypsin EDTA (0.25%) and re-suspended (5x104 cells mL−1) in complete medium and seeded on substrates pre-soaked in fibronectin solution (4 μg mL−1). Cell proliferation and metabolism were measured by total protein (BCA Protein Assay Reagent, Thermo) and MTS assay (CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay, Promega), respectively.
Mineralization
Human mesenchymal stem cells (hMSCs) were maintained in mesenchymal stem cell growth medium with antibiotic-antimycotic (1%). At sub-confluency, cells were detached and seeded on substrates pre-coated with fibronectin solution as described above. After 3 days of culture on substrates, mesenchymal stem cell osteogenic differentiation medium was added to induce differentiation. After 7 days from induction, samples were washed with DPBS, fixed with formalin (10%), and stained with Alizarin Red S solution (2%). Acellular control substrates showed negligible Alizarin red staining. Stained cell layers were removed from the substrates and washed with distilled water before imaging under light microscopy. Mineralization was quantified by counting the % stained area using Image J software. After imaging under light microscopy, alizarin red was extracted by SDS (5%), and absorbance at 550 nm and 405 nm was measured.
Osteoclastogenesis Assays
Dentin was cut into chips (~1 mm thick) and polished following the same prodedure described for nHA-PEUR above as a positive control with a chemical composition comparable to bone.53,54 MC3T3 cells were seeded on the substrates and cultured in osteoinductive medium supplemented with 1α,25-Dihydroxyvitamin D3 (10 nM) for two days before RAW 264.7 cells were added. MC3T3 and RAW cells were co-cultured in osteoinductive medium supplemented with 1α,25-Dihydroxyvitamin D3 (10 nM) for up to 28 days. At day 15, cells were washed with PBS, fixed in formalin (10%), permeabilized in Triton-X-100 (0.1%), stained with rhodamine phalloidin (Life Technologies) and DAPI, and imaged under a Zeiss LSM 710 confocal microscope. At day 28, samples were sonicated in water for 10 min, air-dried, and sputter-coated for imaging using a Zeiss Merlin SEM.
Statistical Analysis
The statistical significance between experimental groups was determined by Student’s t test or by a two-way ANOVA. Tukey’s multiple comparison test was conducted as post-hoc test to determine statistical differences following ANOVA. Graphs show mean and S.D., and p < 0.05 was considered statistically significant.
Results and discussion
Prepolymer synthesis and characterization
To synthesize the injectable prepolymer, nHA (65 wt%) and LTI (35 wt%) were mixed with iron acetylacetonate (FeAA) catalyst (0.027 wt%) at 50°C for 3 h. The reactive mixture was initially granular (comparable to wet sand). After adding FeAA catalyst and mixing for 1 minute, the mixture transformed from a granular to a viscous dispersion of surface-grafted nHA (nHA-LTI) in LTI (referred to as the nHA-LTI/LTI prepolymer). LTI was grafted to nHA through reaction of a primary NCO group with a P-OH group on the surface of nHA (Figure 1A). To confirm the presence of grafted LTI, the %NCO of the catalyzed mixture (open black circles in Figure 1B) was measured by titration as a function of nHA concentration. Theoretical values of % NCO (filled black circles in Figure 1B) were calculated based on dilution assuming no reaction. The conversion of NCO groups to phosphate urethane groups is
Figure 1.

Synthesis of the nHA-LTI prepolymer. (A) Schematic of the urethane reaction between the P-OH groups on surface of nHA particles and the isocyanate groups of LTI. (B) The theoretical %NCO of the nHA/LTI mixture assuming no reaction occurred (filled black circles) was higher than the actual %NCO of the catalyzed nHA-LTI mixture (open black circles), and NCO conversion increased with nHA loading (red line). (C) FTIR spectra of LTI (black) and uncatalyzed (red) and catalyzed (blue) mixtures of nHA and LTI.
| (4) |
where (%NCO)0 and (%NCO) represent the theoretical (calculated assuming dilution) and experimental (meaured for the catalyzed mixture) NCO content, respectively. ξNCO (red line in Figure 1B) increased from 3.6 to 14.6% with increasing nHA concentration. FTIR analysis of the catalyzed nHA-LTI/LTI dispersion showed a reduction in the N=C=O peak at 2260 cm−1 and an increase in the P-O-C peak at 1140 cm−1 compared to the uncatalyzed dispersion, suggesting that N=C=O groups in LTI were consumed and new P-O-C groups formed in the presence of catalyst (Figure 1C). These observations further confirm the reaction of LTI with nHA in the presence of catalyst.38
Surface characterization of grafted nHA
The nHA particles grafted with LTI (nHA-LTI) were recovered from the catalyzed nHA-LTI/LTI prepolymer and their surface chemistry characterized using XPS. The increased C 1s peak (blue arrow) in the survey spectrum of the nHA-LTI particles (Fig. 2B) compared to that in the survey spectrum of the nHA particles (Fig. 2A), as well as the presence of the N1s peak (red arrow Fig. 2B) in the nHA-LTI survey spectrum but not the nHA survey spectrum, are consistent with LTI grafting to the nHA particles. High-resolution spectra (Figure 2C–F) of the Ca 2p, P 2p, and C 1s peaks confirm differences in the chemical bonding states of these elements between the nHA and nHA-LTI samples. The binding energies of both the Ca 2p (346.5 eV) and P 2p (132.6 eV) transitions on the nHA-LTI surface are lower than the expected (and observed) binding energies of Ca 2p and P 2p electrons on the nHA surface (347.1 eV and 133.0 eV, respectively). This binding energy shift is indicative of a change in chemical bonding of these elements in the nHA particles due to LTI grafting. The carbon-oxygen type bonding evident in the C 1s spectrum from the nHA sample (Fig. 2F) is typical of adventitious hydrocarbon adsorption due to atmospheric exposure and is attributed to residual chemicals from the nHA manufacturing process. The C 1s spectrum of the nHA-LTI sample contains contributions from -CH2- type bonding (284.8 eV) as well as O–C=O- type bonding (≈ 288 eV), which are consistent with the structure of grafted LTI. Quantitative analysis showed that Ca/P = 1.7 for both nHA-LTI and nHA, consistent with the stoichiometry of HA, while the C:P and N:P ratios increased after grafting (Figure 2 Table). The conversion of OH groups (ξOH) is calculated from:
| (5) |
where C:P100% = 5.5 and N:P100% = 1.5 are the atomic ratios assuming complete reaction of the OH groups with the primary NCO groups in LTI (Figure 1A). Thus, the conversion of OH groups was 40%. At higher conversions, gelation of the nHA-LTI/LTI prepolymer was observed, which is conjectured to result from either urea formation due to water or allophonate crosslinking reactions between grafted phosphate urethane groups and LTI in the liquid phase.55
Figure 2.

Surface characterization of grafted nHA particles. (A–B) XPS survey spectra of (A) nHA and (B) nHA-LTI particles recovered from the prepolymer. (C–F) XPS high-resolution spectra of (C) Ca, (D) N, (E) P, and (F) C peaks measured for nHA (open circles) and nHA-LTI (filled circles) particles.
Effects of surface grafting on nHA size and crystallinity
The effects of LTI grafting on nHA particle size and crystallinity were assessed by SEM and x-ray diffraction (XRD). The particle size distribution measured from SEM images (Figure 3A–B) showed no difference in the mean size of nHA (45 ± 16 nm) and nHA-LTI (45 ± 15 nm) particles. Similarly, grafting did not alter nHA crystallinity (Figure 3C). The grain size determined from the XRD spectra using Scherrer equation was 36 nm (0 0 2 Miller’s plane family) for both nHA and nHA-LTI.
Figure 3.

Effects of surface grafting on nHA properties. (A) SEM images and (B) particle size distribution of nHA and nHA-LTI particles measured from counts of >500 particles. (C) XRD spectra of nHA and nHA-LTI particles. (D) Viscosity of nHA-LTI/LTI prepolymer (red) was almost two orders of magnitude lower than that of the uncatalyzed nHA/LTI mixture (black).
Both nHA and nHA-LTI nanoparticles (65 wt%) were dispersed in LTI to prepare nHA/LTI and nHA-LTI/LTI prepolymers. The resulting dispersions were shear-thinning, as evidenced by the decrease in viscosity with increasing shear rate (Figure 3D). Furthermore, the viscosity of nHA-LTI/LTI was almost two orders of magnitude lower than that of nHA/LTI, which is consistent with the notion that grafting LTI to the nHA increases colloidal stability, resulting in a more homogeneous dispersion.37,39 At shear rates relevant to injectable bone cements (1 – 10 s−1), the nHA-LTI/LTI prepolymer (65 wt% nHA) exhibited kinematic viscosity < 20,000 cSt, which enabled it to be injected.2
Effects of surface grafting on dispersion in nHA/PEUR nanocomposites
To demonstrate the injectability of the nHA-LTI nanocomposite, nHA-LTI/LTI prepolymer and PCL triol were mixed and injected through a double barrel syringe fitted with a static mixer (MedMix, Figure 4A). The NCO groups in LTI and nHA-LTI react with hydroxyl groups in the PCL triol to form crosslinked organic-inorganic hybrid polymers (Figure 4B). Dispersion of nHA and nHA-LTI in the nanocomposites was evaluated by SEM (Figure 4C). The area% of nHA-LTI aggregates was 5 times smaller than that measured for nHA (Figure 4D), which is consistent with the rheology data (Figure 3D) finding that nHA-LTI is more effectively dispersed in the reactive nanocomposite. Swelling (assessed by incubating the nanocomposites in dichloromethane for 24 h) decreased significantly with LTI grafting and increasing isocyanate index (Figure 4E), which further suggests that surface grafting enhanced dispersion and crosslinking. These findings agree with a recent study reporting that the colloidal stability of nHA grafted with lactic acid oligomers increased with increasing polymer concentration on the surface.37
Figure 4.
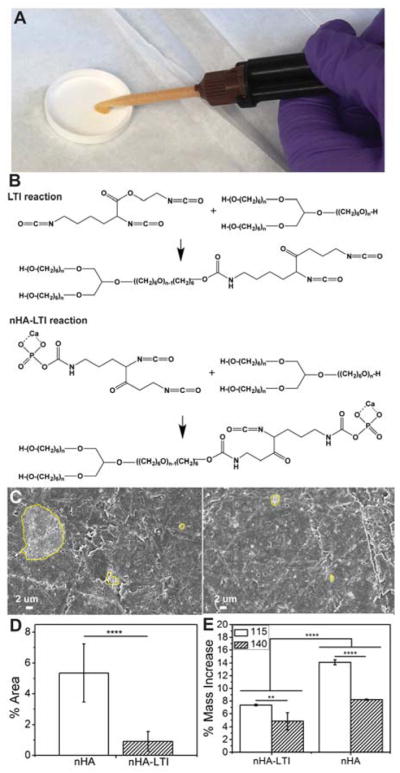
Effects of surface grafting on dispersion in nHA/PEUR composites. (A) Mixing and injection of nHA-LTI/LTI prepolymer with PCL triol in a double-barrel syringe fitted with a static mixture. (B) Schematic illustrating the reaction between NCO groups in LTI and nHA-LTI with hydroxyl groups in poly(caprolactone) (PCL) triol to form crosslinked nanocomposites. (C) SEM images of aggregated particles (>1 μm, yellow lines) in nHA (left) and nHA-LTI (right) nanocomposites. (D) Area% of aggregates in nHA nanocomposites was 5 times larger than that measured for nHA-LTI nanocomposites. (E) Swelling of nHA and nHA-LTI nanocomposites after incubating in dichloromethane for 24 h decreased significantly with surface grafting and increasing index from 115 (white) to 140 (hatched).
Mechanical properties of nHA/PEUR composites
Four-point bending properties of nHA and nHA-LTI nanocomposites were measured using the apparatus described in ISO 5833 (Figure 5A), the international standard for PMMA, at Index 115 and 140.3,4 The effects of the isocyanate index on bending strength and modulus were significant for nHA but not nHA-LTI nanocomposites. Surface grafting significantly increased nanocomposite bending modulus and strength by 20 – 50% at both indices compared to no grafting (Figure 5B). For quasi-static compression testing, nanocomposites were cured in 6-mm cylindrical tubes, cut to 12 mm, and soaked in PBS at 37 °C for 24 hours prior to testing. The yield strength of nHA-LTI nanocomposite increased with nHA-LTI loading up to 52 wt% (65 wt% nHA-LTI in nHA-LTI/LTI prepolymer) (Figure 5C). Furthermore, nHA-LTI nanocomposites with > 26 wt% nHA exhibited higher compressive strength than nHA nanocomposites with 52 wt% nHA. Similar trends were observed for Young’s modulus. The mechanical properties of nHA-LTI nanocomposite exceeded the standard requirements for non-resorbable PMMA, including compressive strength of 70 – 90 MPa, compressive modulus of 2000 – 3000 MPa, and bending strength >80 MPa.3,4
Figure 5.

Mechanical properties of nHA/PEUR nanocomposites. (A) Experimental setup for the four-point bending test. (B) Four-point bending modulus and strength of nHA-LTI nanocomposites were significantly higher than that of nHA nanocomposites at index 115 (white) and 140 (hatched). (C) Compression modulus and strength of nHA-LTI nanocomposites (red) increased with nHA loading and were significantly higher than that of nHA nanocomposites (black).
Protein adsorption on nHA/PEUR composites
To evaluate the effect of nHA-LTI grafting on the biological properties of the nanocomposites, water contact angle (Figure 6A) and adsorption of fibronectin and vitronectin (Figure 6B, C) were measured. LTI-poly(ester urethane) (LTI-PEUR) without nHA had a contact angle θ = 50°. With 30 vol% (52 wt%) nHA loading, nHA-LTI nanocomposite (θ = 25.2° ± 4.9°) was more hydrophilic than nHA nanocomposite (θ = 32.2° ± 5.8°), suggesting that that the more homogenous dispersion of nHA-LTI rendered the surface more like HA (θ=10°).56 Specimens were also incubated in 5 μg/ml fibronectin or vitronectin solutions at 37°C and protein adsorption measured using a Pierce BCA kit. The nanocomposites exhibited a two-fold increase in fibronectin and vitronectin adsorption compared to the LTI-PEUR control. Fibronectin adsorption was comparable to that reported for pure HA with similar grain size, while vitronectin adsorption was lower than that reported for HA.57
Figure 6.

Protein adsorption and osteoblast proliferation on nHA/PEUR nanocomposites. (A) Water contact angle measured for nHA-LTI and nHA nanocomposites and LTI-PEUR control. (B–C) Protein adsorption of (B) fibronectin and (C) vitronectin on nHA-LTI and nHA nanocomposites were significantly higher than that on LTI-PEUR. (D) Total protein measurement at day 1, 4 and 7 showed insignificant differences in proliferation of MC3T3 pre-osteoblast cells between nHA and nHA-LTI nanocomposites. Total protein was significantly higher on LTI-PEUR controls on days 4 and 7. (E) MTS assay showed significant differences between groups on day 7.
Cell proliferation and osteoblast mineralization on nHA/PEUR nanocomposites
Cell viability, proliferation, and differentiation of osteoblasts on the nanocomposites were also assessed. Mouse MC3T3 cells were suspended in complete medium and seeded on the substrates. Total protein increased from Day 1 to 7 for all groups, thereby indicating that cells proliferated on the surface (Figure 6D). Proliferation was significantly higher on the LTI-PEUR control compared to the nanocomposites, but differences in proliferation between the nHA and nHA-LTI groups were insignificant. This observed reduction in proliferation on nHA and nHA-LTI substrates is in agreement with increased differentiation and mineralization of hMSCs on these surfaces compared to the nHA-PEUR control (Figure 7), since induction of osteoblast gene induction is linked to down-regulation of proliferation.58 Cell proliferation assessed by the MTS assay showed significant differences between groups on day 7, with the highest cell numbers on the nHA nanocomposite (Figure 6E). This observation suggests that the more rapidly differentiating cells cultured on nHA-LTI and nHA substrates have higher metabolic activity than the more proliferative cells cultured on the LTI-PEUR polymeric control.
Figure 7.

Osteoblast mineralization on nHA/PEUR nanocomposites. (A) Alizarin red staining of human mesenchymal stem cells (hMSCs) cultured on nHA-LTI, nHA and LTI-PEUR. Substrates were stained on day 7. Mineralization (red staining) was observed as early as day 7 on nHA and nHA-LTI nanocomposites but not on LTI-PEUR controls. (B–C) Quantification of alizarin red staining (day 7) by (B) extraction of the dye and (C) area % stained using ImageJ showed that mineralization was most extensive on nHA-LTI nanocomposites. (D) SEM images of the substrates after 7 days of culture with hMSCs. Mineralized nodules (yellow arrows) were observed on nHA-LTI and nHA but not on LTI-PEUR.
Grain sizes less than 100 nm enhance osteogenic differentiation compared to micron-scale HA.23 However, the effects of surface grafting on nHA activity have not been extensively investigated. Mineralization of human mesenchymal stem cells (hMSCs) cultured on the nanocomposites was assessed by Alizarin Red staining and SEM to assess the effects of surface grafting on nHA activity. Human MSCs were cultured in osteogenic medium, and mineralization was assessed by Alizarin Red staining on day 7 (Figure 7A). Staining was quantified by extraction of Alizarin Red from the substrates and also by measuring the area% of stained surface (Figure 7B, C). On day 7, the LTI-PEUR control showed minimal staining. In contrast, nHA-LTI showed significantly higher absorption and area% stained compared to nHA, while SEM images revealed evidence of mineralized nodules on the nanocomposites on day 7 (yellow arrows in Figure 7D). This enhanced mineralization of hMSCs on nHA-LTI substrates is attributed to the improved dispersion of nHA (Figure 4C–D), which is consistent with the notion that grain sizes less than 100 nm enhance osteogenic differentiation compared to micron-scale HA.23 Similarly, a recent study has reported that grafting L-lactic acid oligomer to nHA enhanced mineralization and bone healing of scaffolds fabricated from nHA/poly(lactic-co-glycolic acid) blends.37 Taken together, these findings suggest that surface grafting organic molecules to nHA enhances its dispersion in polymeric nanocomposites, thereby preserving its <100 nm feature size and enhancing mineralization.
Osteoclast-mediated resorption of nHA/PEUR nanocomposites
To evaluate osteoclast-mediated resorption of the nanocomposites, MC3T3 cells were co-cultured with RAW 264.7 cells in osteogenic medium supplemented with 10 nM Vitamin D3 to stimulate their differentiation to osteoclasts. Actin (red)/DAPI (blue nucleus) staining was performed on day 15. Osteoclasts were identified as multi-nucleated cells with an actin ring (Figure 8A, top row). Resorption pits on the surface of nHA and nHA-LTI nanocomposites as well as the dentin control were observed by SEM on day 28 (Figure 8A, bottom row). No evidence of osteoclasts or resorption was observed for LTI-PEUR. The in vitro degradation rate of the nHA-LTI nanocomposite was assessed by immersion in PBS or oxidative medium (20% H2O2 + CoCl2) at 37°C. While the nHA-LTI nanocomposite was hydrolytically stable in PBS, it exhibited >35% mass loss in oxidative medium after 21 days and fully degraded within 48 days (Figure 8B). These findings agree with another study reporting that the LTI-PEUR polymer showed minimal (< 5%) degradation in PBS after 8 months. In oxidative medium, the nHA-LTI nanocomposite degraded faster than the LTI-PEUR polymer, which degraded <10% after 21 days.59,60 The faster rate of degradation in oxidative compared to hydrolytic medium is consistent with the observed resorption of nHA-LTI by osteoclasts, which secrete reactive oxygen species (ROS) during bone remodeling.61–64 The combination of enhanced mineralization by osteoblasts and osteoclast-mediated resorption is anticipated to minimize resorption and fibrous scar formation in vivo by aligning the rates of new bone formation and graft resorption.12,65
Figure 8.

Osteoclast-mediated resorption of nHA-PEUR nanocomposites. (A) Actin (red)/nuclear (blue) staining of osteoclasts (white arrows) on day 15 (top row). Resorption pits formed on nHA and nHA-LTI nanocomposites and the dentin control at day 28 (bottom row). Osteoclasts did not form on PEUR controls. (B) nHA-LTI nanocomposites were hydrolytically stable but degraded in oxidative (20% H2O2 + CoCl2) medium.
Injectable and settable bone grafts may present potentially harmful effects on host tissue, such as heat released from exothermic chemical reactions or reaction of monomers with host tissue.66 Non-lysine-derived polyisocyanates have shown cytotoxic effects on cells, including fibroblasts and lymphocytes.67–69 While the biocompatibility of injectable nHA-PEUR nanocomposites was not directly assessed in this study, previous work from our group and others’ has reported that reactive lysine-derived polyurethanes do not release cytotoxic components or large amounts of heat that damage cells and host tissue.48,60,70–74
Conclusions
This study highlights the potential of nHA-PEUR nanocomposites as a new approach for promoting bone healing at weight-bearing sites. These materials set within 5 – 10 minutes after injection, exhibit strength comparable to non-resorbable PMMA bone cement, stimulate osteogenic differentiation of endogenous cells, and resorb at a rate aligned with patient biology. This ideal combination of properties, which is essential for treating weight-bearing bone defects, cannot be achieved using other biomaterials. Surface grafting of LTI to nHA enhanced the dispersion of nHA in the nanocomposite, resulting in compression and bending strengths exceeding that of PMMA (the only biomaterial indicated for structural repair of bone) as well as enhanced mineralization of osteoprogenitor cells. While nHA-PEUR nanocomposites were hydrolytically stable, they degraded in response to ROS and osteoclasts, which are associated with physiological bone remodelling.61–64 These proof-of-concept findings highlight the potential of nHA-PEUR nanocomposites for repair and restoration of bone defects at weight-bearing sites.
Acknowledgments
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01AR064304. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Confocal images of osteoclasts were obtained through the use of the VUMC Cell Imaging Shared Resource, supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
Notes and references
- 1.Ramakrishna S, Mayer J, Wintermantel E, Leong KW. Compos Sci Technol. 2001;61:1189–1224. [Google Scholar]
- 2.Bohner M. Eur Cells Mater. 2010;20:1–12. doi: 10.22203/ecm.v020a01. [DOI] [PubMed] [Google Scholar]
- 3.ISO. Implants for Surgery – Acrylic Resin Cements. 2002. International Standard 5833. [Google Scholar]
- 4.Kuehn K-D. Bone Cements. Springer; Berlin: 2000. [Google Scholar]
- 5.Aamodt A, Nordsletten L, Havelin LI, Indrekvam K, Utvag SE, Hviding K. Acta Orthop Scand. 2004;75:663–676. doi: 10.1080/00016470410004021. [DOI] [PubMed] [Google Scholar]
- 6.Bettencourt A, Calado A, Amaral J, Alfaia A, Vale FM, Monteiro J, Montemor MF, Ferreira MGS, Castro M. Int J Pharm. 2004;278:181–186. doi: 10.1016/j.ijpharm.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Savarino L, Stea S, Ciapetti G, Paganetto G, Donati ME, Alvergna P, Pizzoferrato A. J Biomed Mater Res. 1995;29:701–705. doi: 10.1002/jbm.820290605. [DOI] [PubMed] [Google Scholar]
- 8.Berry DJ, Harmsen WS, Cabanela ME, Morrey BF. J Bone Joint Surg-Am Vol. 2002;84A:171–177. doi: 10.2106/00004623-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Jasty M, Maloney WJ, Bragdon CR, Oconnor DO, Haire T, Harris WH. J Bone Joint Surg-Br Vol. 1991;73:551–558. doi: 10.1302/0301-620X.73B4.2071634. [DOI] [PubMed] [Google Scholar]
- 10.Frankenburg EP, Goldstein SA, Bauer TW, Harris SA, Poser RD. J Bone Joint Surg-Am Vol. 1998;80A:1112–1124. doi: 10.2106/00004623-199808000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Harmata AJ, Uppuganti S, Granke M, Guelcher SA, Nyman JS. J Mech Behav Biomed Mater. 2015;51:345–355. doi: 10.1016/j.jmbbm.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dumas JE, Prieto EM, Zienkiewicz KJ, Guda T, Wenke JC, Bible J, Holt GE, Guelcher SA. Tissue Eng, Part A. 2014;20:115–129. doi: 10.1089/ten.tea.2012.0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gay S, Arostegui S, Lemaitre J. Mater Sci Eng C. 2009;29:172–177. [Google Scholar]
- 14.Dumas JE, Davis TE, Yoshii T, Nyman J, Holt GE, Perrien DS, Boyce TM, Guelcher SA. Acta Biomater. 2010;6:2394–2406. doi: 10.1016/j.actbio.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 15.Kruger R, Groll J. Biomaterials. 2012;33:5887–5900. doi: 10.1016/j.biomaterials.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 16.Kruger R, Seitz JM, Ewald A, Bach FW, Groll J. J Mech Behav Biomed Mater. 2013;20:36–44. doi: 10.1016/j.jmbbm.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Mahony O, Tsigkou O, Ionescu C, Minelli C, Ling L, Hanly R, Smith ME, Stevens MM, Jones JR. Adv Funct Mater. 2010;20:3835–3845. [Google Scholar]
- 18.Valliant EM, Romer F, Wang DM, McPhail DS, Smith ME, Hanna JV, Jones JR. Acta Biomater. 2013;9:7662–7671. doi: 10.1016/j.actbio.2013.04.037. [DOI] [PubMed] [Google Scholar]
- 19.Yu BB, Turdean-Ionescu CA, Martin RA, Newport RJ, Hanna JV, Smith ME, Jones JR. Langmuir. 2012;28:17465–17476. doi: 10.1021/la303768b. [DOI] [PubMed] [Google Scholar]
- 20.Valliant EM, Jones JR. Soft Matter. 2011;7:5083–5095. [Google Scholar]
- 21.Poologasundarampillai G, Yu BB, Tsigkou O, Wang DM, Romer F, Bhakhri V, Giuliani F, Stevens MM, McPhail DS, Smith ME, Hanna JV, Jones JR. Chem-Eur J. 2014;20:8149–8160. doi: 10.1002/chem.201304013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahony O, Yue S, Turdean-Ionescu C, Hanna JV, Smith ME, Lee PD, Jones JR. J Sol-Gel Sci Technol. 2014;69:288–298. [Google Scholar]
- 23.Webster TJ, Ergun C, Doremus RH, Siegel RW, Bizios R. Biomaterials. 2000;21:1803–1810. doi: 10.1016/s0142-9612(00)00075-2. [DOI] [PubMed] [Google Scholar]
- 24.Laurencin CT, Kumbar SG, Nukavarapu SP. Wiley Interdiscip Rev-Nanomed Nanobiotechnol. 2009;1:6–10. doi: 10.1002/wnan.25. [DOI] [PubMed] [Google Scholar]
- 25.Sun F, Zhou H, Lee J. Acta Biomater. 2011;7:3813–3828. doi: 10.1016/j.actbio.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Ngiam M, Liao SS, Patil AJ, Cheng ZY, Chan CK, Ramakrishna S. Bone. 2009;45:4–16. doi: 10.1016/j.bone.2009.03.674. [DOI] [PubMed] [Google Scholar]
- 27.Sato M, Webster TJ. Expert Rev Med Devices. 2004;1:105–114. doi: 10.1586/17434440.1.1.105. [DOI] [PubMed] [Google Scholar]
- 28.Tran PA, Sarin L, Hurt RH, Webster TJ. J Mater Chem. 2009;19:2653–2659. [Google Scholar]
- 29.Meirelles L, Melin L, Peltola T, Kjellin P, Kangasniemi I, Currie F, Andersson M, Albrektsson T, Wennerberg A. Clin Implant Dent Relat Res. 2008;10:245–254. doi: 10.1111/j.1708-8208.2008.00089.x. [DOI] [PubMed] [Google Scholar]
- 30.Meirelles L, Arvidsson A, Andersson M, Kjellin P, Albrektsson T, Wennerberg A. J Biomed Mater Res Part A. 2008;87A:299–307. doi: 10.1002/jbm.a.31744. [DOI] [PubMed] [Google Scholar]
- 31.MacMillan AK, Lamberti FV, Moulton JN, Geilich BM, Webster TJ. Int J Nanomed. 2014;9:5627–5637. doi: 10.2147/IJN.S66852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webster TJ, Ergun C, Doremus RH, Siegel RW, Bizios R. Biomaterials. 2001;22:1327–1333. doi: 10.1016/s0142-9612(00)00285-4. [DOI] [PubMed] [Google Scholar]
- 33.Harmata AJ, Ward CL, Zienkiewicz KJ, Wenke JC, Guelcher SA. J Mater Res. 2014;29:2398–2407. doi: 10.1557/jmr.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Supova M. J Mater Sci: Mater Med. 2009;20:1201–1213. doi: 10.1007/s10856-009-3696-2. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka H, Watanabe T, Chikazawa M, Kandori K, Ishikawa T. J Coll Interfac Sci. 1998;206:205–211. doi: 10.1006/jcis.1998.5634. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q, de Wijn JR, De Groot K, van Blitterswijk CC. Biomaterials. 1998;19:1067–1072. doi: 10.1016/s0142-9612(98)00033-7. [DOI] [PubMed] [Google Scholar]
- 37.Wang ZL, Xu Y, Wang Y, Ito Y, Zhang PB, Chen XS. Biomacromolecules. 2016;17:818–829. doi: 10.1021/acs.biomac.5b01543. [DOI] [PubMed] [Google Scholar]
- 38.Dong GC, Sun JS, Yao CH, Jiang GJ, Huang CW, Lin FH. Biomaterials. 2001;22:3179–3189. doi: 10.1016/s0142-9612(01)00070-9. [DOI] [PubMed] [Google Scholar]
- 39.Lee HJ, Kim SE, Choi HW, Kim CW, Kim KJ, Lee SC. Euro Polym J. 2007;43:1602–1608. [Google Scholar]
- 40.Jiang LY, Jiang LX, Xiong CD, Xu LJ, Li Y. J Biomate Appl. 2016;30:750–758. [Google Scholar]
- 41.Dong SJ, Yu T, Wei JC, Jing XB, Zhou YM, Zhang PB, Chen XS. Chem J Chin Univ. 2009;30:1018–1023. [Google Scholar]
- 42.Fan RR, Zhou LX, Song W, Li DX, Zhang DM, Ye R, Zheng Y, Guo G. In J Biol Macromol. 2013;59:227–234. doi: 10.1016/j.ijbiomac.2013.04.051. [DOI] [PubMed] [Google Scholar]
- 43.Jiang LY, Xiong CD, Chen DL, Jiang LX, Pang XB. Appl Surf Sci. 2012;259:72–78. [Google Scholar]
- 44.Jiang LX, Jiang LY, Xu LJ, Han CT, Xiong CD. Express Polym Lett. 2014;8:133–141. [Google Scholar]
- 45.Qiu XY, Chen L, Hu JL, Sun JR, Hong ZK, Liu AX, Chen XS, Jing XB. J Polym Sci, Part A: Polym Chem. 2005;43:5177–5185. [Google Scholar]
- 46.Hafeman AE, Zienkiewicz KJ, Zachman AL, Sung HJ, Nanney LB, Davidson JM, Guelcher SA. Biomaterials. 2011;32:419–429. doi: 10.1016/j.biomaterials.2010.08.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin JR, Gupta MK, Page JM, Yu F, Davidson JM, Guelcher SA, Duvall CL. Biomaterials. 2014;35:3766–3776. doi: 10.1016/j.biomaterials.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Page JM, Prieto EM, Dumas JE, Zienkiewicz KJ, Wenke JC, Brown-Baer P, Guelcher SA. Acta Biomater. 2012;8:4405–4416. doi: 10.1016/j.actbio.2012.07.037. [DOI] [PubMed] [Google Scholar]
- 49.ASTM D2572-97. Standard Test Method for Isocyanate Groups in Urethane Materials for Prepolymers. ASTM International; West Conshohocken, PA: 2010. [Google Scholar]
- 50.WFS, Moulder John F, Sobol Peter E, Bomben Kenneth D. Handbook of X-ray photoelectron spectroscopy. Physical Electronics USA, Inc; Chanhassen, Minnesota: 1995. [Google Scholar]
- 51.Yubao L, Klein CPAT, DeWijn J, Meer SVd, DeGroot K. J Mater Sci: Mater Med. 1994;5:263–268. [Google Scholar]
- 52.ASTM D6272-10. Standard Test Method for Flexural Properties of Unreinforced and Reinforced Plastics and Electrical Insulating Materials by Four-Point Bending. ASTM International; West Conshohocken, PA: 2010. [Google Scholar]
- 53.Sidqui M, Collin P, Vitte C, Forest N. Biomaterials. 1995;16:1327–1332. doi: 10.1016/0142-9612(95)91048-4. [DOI] [PubMed] [Google Scholar]
- 54.Schilling AF, Linhart W, Filke S, Gebauer M, Schinke T, Rueger JM, Amling M. Biomaterials. 2004;25:3963–3972. doi: 10.1016/j.biomaterials.2003.10.079. [DOI] [PubMed] [Google Scholar]
- 55.Kopusov LI, Zharkov VV. J Appl Spectrosco. 1966;5:95–97. [Google Scholar]
- 56.Aronov D, Rosen R, Ron EZ, Rosenman G. Process Biochem. 2006;41:2367–2372. [Google Scholar]
- 57.Webster TJ, Ergun C, Doremus RH, Siegel RW, Bizios R. J Biomed Mater Res. 2000;51:475–483. doi: 10.1002/1097-4636(20000905)51:3<475::aid-jbm23>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 58.Stein GS, Lian JB, Stein JL, Van Wijnen AJ, Frenkel B, Montecino M. In: Principles of Bone Biology. Bilezikian JP, Raisz LG, Rodan GA, editors. Academic Press; San Diego: 1996. p. 71. c1996. [Google Scholar]
- 59.Guelcher SA, Srinivasan A, Dumas JE, Didier JE, McBride S, Hollinger JO. Biomaterials. 2008;29:1762–1775. doi: 10.1016/j.biomaterials.2007.12.046. [DOI] [PubMed] [Google Scholar]
- 60.McEnery MAP, Lu S, Gupta MK, Zienkiewicz KJ, Wenke JC, Kalpakci KN, Shimko d, Duvall CL, Guelcher SA. RSC Adv. 2016;6:109414–109424. doi: 10.1039/c6ra24642g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheweita SA, Khoshhal KI, Baghdadi H. In: Systems Biology of Free Radicals and Antioxidants. 1. Laher I, editor. ch 132. Springer, Verlag; Berlin Heidelberg: 2014. pp. 2973–2995. [DOI] [Google Scholar]
- 62.Garrett IR, Boyce BF, Oreffo ROC, Bonewald L, Poser J, Mundy GR. J Clin Invest. 1990;85:632–639. doi: 10.1172/JCI114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Key LL, Wolf WC, Gundberg CM, Ries WL. Bone. 1994;15:431–436. doi: 10.1016/8756-3282(94)90821-4. [DOI] [PubMed] [Google Scholar]
- 64.Banfi G, Iorio EL, Corsi MM. Clin Chem Lab Med. 2008;46:1550–1555. doi: 10.1515/CCLM.2008.302. [DOI] [PubMed] [Google Scholar]
- 65.Lorden ER, Miller KJ, Bashirov L, Ibrahim MM, Hammett E, Jung Y, Medina MA, Rastegarpour A, Selim MA, Leong KW, Levinson H. Biomaterials. 2015;43:61–70. doi: 10.1016/j.biomaterials.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Guelcher SA. In: Injectable Biomaterials: Science and Applications. 1. Vernon B, editor. Woodhead Publishing; Philadelphia: 2011. [Google Scholar]
- 67.Ertel SI, Ratner BD, Kaul A, Schway MB, Horbett TA. J Biomed Mater Res. 1994;28:667–675. doi: 10.1002/jbm.820280603. [DOI] [PubMed] [Google Scholar]
- 68.Pons F, Fischer A, Frossard N, Lugnier A. Cell Biol Toxicol. 1999;15:333–340. doi: 10.1023/a:1007671903406. [DOI] [PubMed] [Google Scholar]
- 69.Mishra PK, Panwar H, Bhargava A, Gorantla VR, Jain SK, Banerjee S, Maudar KK. J Biochem Mol Toxicol. 2008;22:429–440. doi: 10.1002/jbt.20260. [DOI] [PubMed] [Google Scholar]
- 70.Hafeman AE, Li B, Yoshii T, Zienkiewicz K, Davidson JM, Guelcher SA. Pharm Res. 2008;25:2387–2399. doi: 10.1007/s11095-008-9618-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adhikari R, Gunatillake PA, Griffiths I, Tatai L, Wickramaratna M, Houshyar S, Moore T, Mayadunne RTM, Field J, McGee M, Carbone T. Biomaterials. 2008;29:3762–3770. doi: 10.1016/j.biomaterials.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 72.Buckley MJ, Beckman EJ. Oral Maxillofac Surg Clin N Am. 2010;22:195–199. doi: 10.1016/j.coms.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 73.Zhang JY, Beckman EJ, Hu J, Yang GG, Agarwal S, Hollinger JO. Tissue Eng. 2002;8:771–785. doi: 10.1089/10763270260424132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bennett S, Connolly K, Lee DR, Jiang Y, Buck D, Hollinger JO, Gruskin EA. Bone. 1996;19:S101–S107. doi: 10.1016/s8756-3282(96)00130-5. [DOI] [PubMed] [Google Scholar]