

SUMMARY
While signals that activate group 3 innate lymphoid cells (ILC3s) have been described, the factors that negatively regulate these cells are less well understood. Here we found that the tumor necrosis factor (TNF) superfamily member receptor activator of nuclear factor kappa-B ligand (RANKL) suppressed ILC3 activity in the intestine. Deletion of RANKL in ILC3s and T cells increased C-C motif chemokine receptor 6 (CCR6)+ ILC3 abundance and enhanced production of interleukin 17A (IL-17A) and IL-22 in response to IL-23 and during infection with the enteric murine pathogen Citrobacter rodentium. Additionally, CCR6+ ILC3s produced higher amounts of the master transcriptional regulator RORγt at steady state in the absence of RANKL. RANKL-mediated suppression was independent of T cells, and instead occurred via interactions between CCR6+ ILC3s that expressed both RANKL and its receptor, RANK. Thus, RANK-RANKL interactions between ILC3s regulate ILC3 abundance and activation, suggesting that cell clustering may control ILC3 activity.
eTOC Blurb
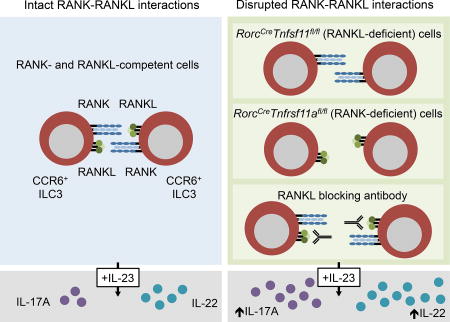
While signals that activate group 3 ILCs (ILC3s) have been described, the factors that negatively regulate these cells are less well understood. Bando et al demonstrate that the TNF superfamily member RANKL suppresses the abundance and effector functions of intestinal CCR6+ ILC3s, and that RANKL-mediated suppression occurs through ILC3-ILC3 interactions.
INTRODUCTION
Group 3 innate lymphoid cells (ILC3s) are innate lymphocytes that share developmental requirements for the transcription factor RORγt and that secrete the cytokines IL-22 and/or IL-17A (Artis and Spits, 2015; Bando and Colonna, 2016; Diefenbach et al., 2014; Eberl et al., 2015). In mice, ILC3s are divided into subsets defined by differential expression of the surface markers CCR6 and NKp46. CCR6+ ILC3s are required for lymph node, Peyer’s patch, and gut tertiary lymphoid tissue organogenesis; NKp46+ ILC3s express the transcription factor T-bet and can secrete interferon γ (IFNγ); and CCR6−NKp46− ILC3s have the capacity to differentiate into NKp46+ ILC3s (Klose et al., 2013; Sawa et al., 2010; van de Pavert and Vivier, 2016). While NKp46− ILC3s secrete both IL-22 and IL-17A, NKp46+ ILC3s secrete IL-22 but not IL-17A. These cells are important sources of effector cytokines under steady state conditions, during recovery after radiation-induced tissue damage, and during infection with pathogens, including C. rodentium (Satoh-Takayama et al., 2008; Zheng et al., 2008). In models of inflammatory bowel disease, ILC3 activation and cytokine secretion can alter intestinal pathology (Buonocore et al., 2010; Cox et al., 2012; Pearson et al., 2016; Song et al., 2015).
Unlike T cells, ILCs are activated by antigen-independent pathways. Group 3 ILCs are stimulated by soluble mediators, including the cytokines IL-23 and IL-1β. Bacterial metabolites and dietary components that engage the aryl hydrocarbon receptor (AHR) promote ILC3 proliferation and cytokine secretion (Kiss et al., 2011; Lee et al., 2012; Qiu et al., 2012). Retinoic acid also enhances the production of IL-22 by ILC3s (Mielke et al., 2013), and regulates the size of the fetal and adult ILC3 pool (Spencer et al., 2014; van de Pavert et al., 2014). More recently, glial cell-derived neurotrophic factor family of ligands (GFL) members (Ibiza et al., 2016) and prostaglandin E2 (PGE2) (Duffin et al., 2016) were shown to drive IL-22 production in ILC3s. The signals in tissue that negatively regulate ILC3 activity are less understood. IL-25, an alarmin secreted by intestinal Tuft cells (Gerbe et al., 2016; Howitt et al., 2016; von Moltke et al., 2016), indirectly suppresses the production of IL-22 by ILC3s via myeloid cells (Sawa et al., 2011). Butyrate has been reported to suppress Peyer’s patch NKp46+ ILC3s (Kim et al., 2017). Determining additional ways these cells are negatively regulated will provide insight into the balance of positive and negative signals that maintain intestinal homeostasis.
Human and mouse ILC3s express the TNF superfamily member RANKL (Cella et al., 2010; Sugiyama et al., 2012), a transmembrane and proteolytically shed homotrimer encoded by the gene Tnfsf11 (Walsh and Choi, 2014). RANKL binds to the signaling receptor RANK and the soluble decoy receptor osteoprotegerin (OPG). Tnfsf11−/− mice have defects in the development of osteoclasts, lactating mammary glands, medullary thymic epithelial cells (mTECs), and intestinal microfold (M) cells (Fata et al., 2000; Hikosaka et al., 2008; Knoop et al., 2009; Kong et al., 1999). Additionally, Tnfsf11−/− mice lack lymph nodes and have reduced tertiary gut lymphoid organs (Kim et al., 2000; Knoop et al., 2009; Kong et al., 1999). Peyer’s patches still develop but are reduced in number in RANK-deficient mice (Dougall et al., 1999). RANKL can promote dendritic cell (DC) survival during colitis in Il2−/− mice (Ashcroft et al., 2003), and is required for T cell localization in the central nervous system (CNS) during experimental autoimmune encephalomyelitis (EAE) (Guerrini et al., 2015). RANKL is also required for regulatory T cell (Treg) generation in pancreatic lymph nodes and islets in a mouse model of diabetes (Green et al., 2002), and exogenous administration of RANKL promotes oral tolerance to ovalbumin (Williamson et al., 2002). The functions of RANKL in adult ILC3s remain unclear.
Using a newly generated Tnfsf11 floxed mouse, here we show that RANKL negatively regulates CCR6+ ILC3s during homeostasis and infection. Genetic deletion of RANKL increased the numbers of CCR6+ ILC3s in the intestine and induced these cells to enter a hyperresponsive state in which they produced elevated amounts of IL-17A and IL-22 in response to IL-23 and during infection with C. rodentium. We further used antibody-mediated blockade to show that RANKL was constitutively required to limit ILC3 hyperresponsiveness. CCR6+ ILC3s express both RANKL and the RANKL receptor, RANK, and suppression occurred through ILC3-ILC3 interactions. Comparison of genome-wide expression data between RANKL-deficient and -sufficient ILC3s revealed that RANKL impacted ILC3 expression of RORγt. Thus, RANK signaling in the intestine negatively regulates CCR6+ ILC3 abundance and effector function, and reduces RORγt expression.
RESULTS
Tnfsf11fl/flRorcCre mice have elevated numbers of CCR6+ ILC3s
To investigate the functions of RANKL in ILC3s, conditionally deficient Tnfsf11fl/flRorcCre mice were generated. In these mice, exons 3 and 4 of Tnfsf11 were excised in ILC3s and T cells as validated by genomic PCR (Supplemental Figure 1). RANKL deficiency in Tnfsf11fl/flRorcCre ILC3s was confirmed at the protein level by cell surface antibody staining (Figure 1A). We observed that Tnfsf11fl/flRorcCre mice had elevated numbers and frequencies of CCR6+ ILC3s in the small intestine, while NKp46+ and CCR6−NKp46− (double negative, or DN) ILC3 numbers were unaffected by RANKL deficiency (Figure 1B and data not shown). Other ILC populations, including Eomes− ILC1s, Eomes+ conventional natural killer (NK) cells, and group 2 ILCs (ILC2s), were unaltered in numbers and frequencies in Tnfsf11fl/flRorcCre mice (Figure 1B and data not shown). The increase in CCR6+ ILC3s in Tnfsf11fl/flRorcCre mice was associated with >5-fold increase in the cell proliferation marker Ki67 (Figure 1C). CCR6+ ILC3s from Tnfsf11fl/flRorcCre mice also expressed more CCR6, CD127, and CD25 compared to cells isolated from Tnfsf11fl/fl mice (Figure 1D). Thus, intestinal CCR6+ ILC3s are numerically expanded and have altered cell surface marker expression in conditional RANKL-deficient mice.
Figure 1. Tnfsf11fl/flRorcCre mice have elevated numbers of CCR6+ ILC3s at steady state.

(A) Cell surface antibody staining for RANKL in ILC3s isolated from Tnfsf11fl/flRorcCre or Tnfsf11fl/fl small intestine lamina propria. (B) ILC gating strategy and cell counts in Tnfsf11fl/flRorcCre and Tnfsf11fl/fl small intestine lamina propria (n=4). (C) Ki67 (n=7), (D) CCR6, CD127, and CD25 (n=3–5) expression in Tnfsf11fl/flRorcCre and Tnfsf11fl/fl small intestine lamina propria CCR6+ ILC3s. Negative staining controls for CD127 and CD25 were isotype control antibodies. Negative staining controls for CCR6 were NKp46+ ILC3s stained with CCR6 antibodies. Bars indicate mean (+/− s.d). **P≤0.01, ***P≤0.001, ****P≤0.0001. Data are representative of three independent experiments (A, B), are pooled from two independent experiments (C), or are representative of two independent experiments (D). Also see Figure S1.
RANKL regulates CCR6+ and DN ILC3 cytokine production
To investigate whether the observed changes in the abundance of Tnfsf11fl/flRorcCre CCR6+ ILC3s were associated with functional changes, intestinal ILC3s were assessed for their ability to respond to the activating cytokine IL-23. In response to varying concentrations of IL-23 in vitro, CCR6+ ILC3s from Tnfsf11fl/flRorcCre small intestine lamina propria produced more IL-17A and IL-22 than Tnfsf11fl/fl cells (Figure 2A, B). CCR6+ ILC3s from Tnfsf11fl/flRorcCre mice were activated by low concentrations of IL-23 that did not enhance effector cytokine production in control cells. DN ILC3s from Tnfsf11fl/flRorcCre mice were also more responsive to IL-23, producing more IL-17A, but not IL-22, than DN Tnfsf11fl/fl ILC3s (Figure 2B). In comparison, activated RANKL-deficient NKp46+ ILC3s did not produce elevated amounts of IL-22 compared to control cells. Thus, CCR6+ and DN ILC3s from Tnfsf11fl/flRorcCre mice respond more robustly to IL-23 than ILC3s from Tnfsf11fl/fl mice by producing increased amounts of IL-17A and/or IL-22.
Figure 2. RANKL regulates CCR6+ and DN ILC3 cytokine production.

(A) Representative intracellular staining for IL-17A and IL-22 in small intestine lamina propria CCR6+ ILC3s stimulated with IL-23 in vitro. (B) IL-17A and IL-22 production in CCR6+, CCR6−NKp46−(DN), and NKp46+ ILC3 subsets in response to IL-23 (n=3–5). (C) Cell counts of small intestine lamina propria IL-17A+ and IL-22+ CCR6+ ILC3s on d 7 of C. rodentium infection (n=7–11). (D) CCR6+ ILC3 cell counts (left) and percent of CCR6+ ILC3s that produced IL-17A and IL-22 on d 7 of infection (center and right) (n=7–11). (E) CCR6 expression and (F) cytokine production in CCR6+ ILC3s after in vivo treatment with a blocking antibody to RANKL (n=4). Bars indicate mean (+/− s.d). *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001. Data are representative of two independent experiments (A, B, E, F), or two independent experiments pooled (C, D).
Similar effects of RANKL deficiency on CCR6+ ILC3s were observed during infection with C. rodentium. At day 7 post-inoculation, more IL-17A+ and IL-22+ CCR6+ ILC3s were present in the small intestines of Tnfsf11fl/flRorcCre mice than in Tnfsf11fl/fl controls (Figure 2C). These differences were due to higher total numbers of CCR6+ ILC3s in Tnfsf11fl/flRorcCre mice as well as higher frequencies of CCR6+ ILC3s that produced IL-17A and IL-22 (Figure 2D). With the exception of moderately increased IL-17A production by DN ILC3s in Tnfsf11fl/flRorcCre mice, NKp46+ and DN ILC3 numbers and cytokine production were not significantly different between the two groups at day 7. Thus, RANKL regulates CCR6+ ILC3 cytokine production, and to a lesser extent, DN ILC3 IL-17A production, during C. rodentium infection.
The hyperresponsive phenotype observed in Tnfsf11fl/flRorcCre CCR6+ ILC3s could be due to either developmental or constitutive requirements for RANKL. To determine whether RANKL is constitutively required for ILC3 suppression, wild type adult mice were treated with either a blocking antibody to RANKL (IK22/5) or isotype control antibody (2A3). Since intestinal CCR6+ ILC3s in adult mice are tissue resident cells maintained numerically by slow cell division in tissue rather than by de novo differentiation from bone marrow precursors (Gasteiger et al., 2015), administration of blocking antibodies to adult mice would be expected to block RANK signaling among mature ILC3s in the tissue, rather than affect ILC3 development. Intestinal CCR6+ ILC3s isolated from mice treated with blocking antibody exhibited increased CCR6 expression (Figure 2E), and produced increased amounts of IL-17A and IL-22 upon stimulation with IL-23 compared to cells isolated from mice treated with isotype control antibody (Figure 2F). Similar to the observations made in Tnfsf11fl/flRorcCre mice, DN ILC3s isolated from IK22/5 antibody-treated mice produced more IL-17A after stimulation with IL-23 than did control cells, while IL-22 production was unaffected in DN and NKp46+ ILC3s (Figure 2F). Thus, RANKL acts constitutively to regulate cytokine production in ILC3s, and antibodies that block RANKL can be used to enhance ILC3 effector functions in vivo.
RANKL-deficient T cells are not necessary or sufficient to drive ILC3 hyperresponsiveness
Autoimmune regulator (Aire)-expressing medullary thymic epithelial cells (mTEC) are required for appropriate negative selection of self-reactive T cells during thymopoiesis, and fewer develop in Tnfsf11−/− and Tnfrsf11a−/− (RANK-deficient) mice (Akiyama et al., 2008; Hikosaka et al., 2008; Rossi et al., 2007). To investigate whether Tnfsf11fl/flRorcCre mice had altered T cell development, TEC populations were characterized in these mice. Tnfsf11fl/flRorcCre mice had reduced numbers of mature mTECs (mTEChi) as well as fewer Aire+ mTECs compared to Tnfsf11fl/fl mice (Figure 3A). However, Tnfsf11fl/flRorcCre mice did not generate greater frequencies of CD44hiCD62Llo activated or memory T cells in lymph nodes (Figure 3B). Additionally, normal frequencies of Treg, T helper 1 (Th1), Th2, and Th17 cells were present in the small intestine lamina propria of Tnfsf11fl/flRorcCre mice (Figure 3C). Thus, although mTEC development is hampered in Tnfsf11fl/flRorcCre mice, these animals do not exhibit increased frequencies of activated T cells at steady state.
Figure 3. RANKL-deficient T cells are not necessary or sufficient to drive ILC3 hyperresponsiveness.

(A) Thymic epithelial cell (TEC) counts in Tnfsf11fl/flRorcCreand Tnfsf11fl/fl control thymi. TECs were identified as Epcam+CD45− cells that either expressed Ly51 (cTEC); low MHCII and CD80 (mTEClo); high MHCII and CD80 (mTEChi); or high MHCII, CD80, and AIRE (AIRE+ mTEC) (n=6–7). (B) Frequencies of CD44hiCD62Llo CD4+ T cells from pooled lymph nodes (n=5–6). (C) Gating strategy and helper T cell frequencies in small intestine lamina propria (n=4). (D) IL-17A and IL-22 production in Rag1−/−Tnfsf11fl/flRorcCre and Rag1−/−Tnfsf11fl/fl mice. (E) CCR6 expression and (F) IL-17A and IL-22 production by in vitro stimulated small intestine lamina propria CCR6+ ILC3s from Rag1−/− mice injected with a blocking antibody to RANKL (n=4). (G) Percent donor- and receipient-derived small intestine lamina propria CCR6+ ILC3s in chimeras. (n=7). (H) IL-17A and IL-22 production in small intestine lamina propria CCR6+ ILC3s from chimeras after in vitro stimulation with IL-23 (n=3–4). (I) C. rodentium in feces of infected mice d 4 and d 5 post-inoculation (n=15–20). Bars indicate mean (+/− s.d). *P≤0.05, **P≤0.01, ***P≤0.001. Data are pooled from two (A, B, G) or three (I) independent experiments, or are representative of two independent experiments (C–F).
To test whether T cells were required for RANKL-dependent ILC3 suppression, Rag1−/−Tnfsf11fl/flRorcCre mice were generated. CCR6+ ILC3s from Rag1−/−Tnfsf11fl/flRorcCre small intestines produced more IL-22 and IL-17A compared to controls, indicating that T cells were not necessary for the hyperresponsive CCR6+ ILC3 phenotype that developed in the absence of RANKL (Figure 3D). In support of these data, administration of a RANKL blocking antibody to Rag1−/− mice induced elevated CCR6 expression (Figure 3E) and increased IL-23-elicited cytokine production in vitro by CCR6+ ILC3s (Figure 3F). To test whether RANKL-deficient T cells were sufficient to induce ILC3 hyperresponsiveness, we used chimeric mice that lacked RANKL in T cells, while retaining RANKL-sufficient CCR6+ ILC3s (Figure 3G). These mice were generated by transferring Tnfsf11fl/flRorcCre bone marrow into sublethally irradiated Rag1−/− mice (chimeric Tnfsf11fl/flRorcCre ➔ Rag1−/−), which allowed for the development of RANKL-deficient donor T cells while retaining RANKL-sufficient receipient-derived ILC3s. Chimeric Tnfsf11fl/flRorcCre ➔ Rag1−/− mice did not develop hyperresponsive CCR6+ ILC3s based on IL-17A and IL-22 production (Figure 3H). Thus, RANKL-deficient T cells are neither required nor sufficient to induce hyperresponsive CCR6+ ILC3s, indicating that RANKL expression by ILC3s contributes to the suppression of CCR6+ ILC3s.
To test whether hyperresponsive ILC3s impacted bacterial clearance, we assessed bacterial colonization during C. rodentium infection in RANKL-deficient mice. Tnfsf11fl/flRorcCreRag1−/−mice were infected to eliminate the contributions of altered thymic T cell selection caused by reduced mTEC maturation. During early infection, C. rodentium CFU in feces were reduced in Rag1−/−Tnfsf11fl/flRorcCre mice compared to Rag1−/−Tnfsf11fl/fl controls (Figure 3I), indicating that enhanced cytokine production by RANKL-deficient CCR6+ ILC3s was sufficient to improve host responses to bacterial infection.
RANKL directly suppresses ILC3 function through ILC3-ILC3 interactions
RANKL binds the signaling receptor RANK. To characterize RANK expression in the gut, small intestines of RANK reporter Tnfrsf11aeGFP-Cre knock-in mice (Maeda et al., 2012) were assessed by flow cytometry. All three ILC3 subsets expressed GFP in the small intestine, with the highest expression in CCR6+ ILC3s and the lowest expression in NKp46+ ILC3s (Figure 4A). Intestinal ILC2s and T cells also expressed GFP, while ILC1s, cNK cells, and IgA+ B cells had low (<2%) or no expression. GFP was also detected in CD11c+MHCII+ DC populations in the small intestine, regardless of CD103 and CD11b expression (Figure 4A and data not shown).
Figure 4. RANKL directly suppresses ILC3 function in vitro.

(A) GFP expression in Tnfrsf11aeGFP-Cre mice (n=10). Shaded histograms represent cells isolated from wild type control mice. (B) RANKL expression in ILC3 subsets and T cells (n=7). Shaded histograms represent RANKL antibody-stained Tnfsf11−/− cells. (C, D) IL-23 and IL-1β-elicited IL-17A and IL-22 production in primary sorted CCR6+ ILC3s pretreated with (C) soluble trimeric RANKL (n=7) or (D) a blocking antibody to RANKL (n=5). Also see Figures S2 and S3.
Since the expression of RANK in each ILC3 subset correlated with the extent each population was functionally affected by RANKL deficiency, we hypothesized that ILC3s were suppressed by directly responding to RANKL. Importantly, CCR6+ ILC3s (1) not only expressed high amounts of RANK, but also expressed more RANKL than other ILC3 subsets (Figure 4B), and (2) reside in cryptopatches, anatomical sites where these cells are closely clustered together and thus have high potential for ILC3-ILC3 interactions (Lügering et al., 2010). Human tonsil CD56+NKp44+CCR6+ ILC3s also expressed both RANK and RANKL (Supplemental Figure 2A), confirming previous reports (Cella et al., 2010; Montaldo et al., 2014). To investigate whether RANKL could directly suppress ILC3s in vitro, we functionally assessed sorted mouse CCR6+ ILC3s in the presence or absence of RANKL-dependent signaling. After pre-treatment with soluble RANKL homotrimers (Lam et al., 2001; Nelson et al., 2012), CCR6+ ILC3s produced less IL-17A and IL-22 in response to IL-23 and IL-1β in comparison to cells cultured in media alone (Figure 4C). In contrast, pre-treatment of purified CCR6+ ILC3s with a RANKL blocking antibody led to increased IL-17A and IL-22 production after IL-23 and IL-1β stimulation, indicating that ILC3s can suppress each other in a RANKL-dependent manner in vitro (Figure 4D). The ILC3 cell line MNK-3, which expresses both RANK and RANKL (Allan et al., 2015), also produced significantly less IL-17A and IL-22 after pretreatment with homotrimeric RANKL compared to control cells, while administration of a RANKL-blocking antibody to MNK-3 cultures elevated IL-17A and IL-22 production compared to isotype control-treated cells (Supplemental Figure 3A–B). Additionally, sorted human CD56+NKp44+CCR6+ tonsil ILC3s pretreated with the souble decoy receptor osteoprotegerin (OPG) produced more IL-22 upon stimulation with IL-23 and IL-1β that control cells (Supplemental Figure 2B). Thus, CCR6+ ILC3s are directly suppressed by RANKL in vitro, and RANKL expressed by ILC3s can cause suppression.
To test whether ILC3 cytokine production is regulated by RANK activation in vivo, Tnfrsf11afl/flRorcCre mice were generated. In these mice, deletion of Tnfrsf11a was confirmed in ILC3s and T cells, while Tnfrsf11a remained intact in DC and B cells (Supplemental Figure 4). Similar to mice conditionally-deficient in RANKL, mice conditionally-deficient in RANK possessed higher numbers of CCR6+, but not NKp46+ or DN, ILC3s in the small intestine compared to control mice (Figure 5A). Additionally, greater frequencies of CCR6+ ILC3s from Tnfrsf11afl/flRorcCre mice expressed Ki67 compared to those from Tnfrsf11afl/fl mice (Figure 5B). RANK-deficient CCR6+ ILC3s also produced more IL-17A and IL-22 upon stimulation with IL-23 compared to control mice (Figure 5C). No reduction in RANKL surface expression was observed in Tnfrsf11afl/flRorcCre ILC3s (Figure 5D). Thus, RANK is required for the suppression of CCR6+ ILC3 proliferation and effector cytokine production.
Figure 5. CCR6+ ILC3s require RANK for suppression in vivo.

(A) ILC3 frequencies and cell counts in Tnfrsf11afl/flRorcCre small intestine lamina propria (n=6–7). (B) Ki67 expression (n=7) and (C) IL-23-elicited IL-17A and IL-22 production (n=4) in small intestine lamina propria CCR6+ ILC3s from Tnfrsf11afl/flRorcCre mice. (D) RANKL expression in small intestine lamina propria CCR6+ ILC3s from Tnfrsf11afl/flRorcCre mice (n=3). Bars indicate mean (+/− s.d). *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001. Data are pooled from two independent experiments (A–C), or are representative of two independent experiments (D). Also see Figures S4 and S5.
Membrane bound RANKL can be proteolytically cleaved and released by osteoblasts (Lum et al., 1999; Nakashima et al., 2000). To determine whether ILC3s also shed RANKL, supernatants from sorted CCR6+ ILC3 cultures were assessed for soluble RANKL by ELISA. Over 3 d, RANKL was undetectable in supernatants but abundant in cell lysates, indicating that RANKL primarily remains bound to the cell surface under non-stimulatory conditions (Supplemental Figure 5A). Upon stimulation with IL-23 and IL-1β, supernatants contained soluble RANKL, while the abundance of membrane bound RANKL was reduced (Supplemental Figure 5B–C). These data suggest that at steady state, RANK-RANKL interactions between CCR6+ ILC3s occur primarily through direct cell-cell contact rather than through soluble RANKL. However, since RANKL may be shed upon stimulation, direct cell contact may not be required for RANK signaling upon cellular activation.
Tnfsf11fl/flRorcCre and Tnfrsf11afl/flRorcCre mice have partial defects in secondary lymphoid tissue organogenesis
Unlike Tnfsf11−/− and Tnfrsf11a−/− mice, which have impaired lymph node organogenesis (Dougall et al., 1999; Kim et al., 2000; Kong et al., 1999), Tnfsf11fl/flRorcCre and Tnfrsf11afl/flRorcCre mice developed normal numbers of cervical, axillary, brachial, inguinal, and mesenteric lymph nodes (Figure 6A). To test the possibility that intact lymph node development in these mice was due to inefficient Cre-mediated deletion during lymph node organogenesis, we generated additional mice expected to have higher RANK or RANKL deletion efficiency by introducing alleles with germline deletion of Tnfsf11 or Tnfrsf11a to individual animals. Conditional RANK mice carrying one germline deleted allele of Tnfrsf11a (Tnfrsf11afl/−RorcCre mice), but not Tnfrsf11afl/− mice, exhibited defects in cervical, brachial, and inguinal lymph node development (Figure 6A). In contrast, conditional RANKL mice carrying one germline deleted allele of Tnfsf11 (Tnfsf11fl/−RorcCre mice) developed lymph nodes normally. These results obtained using more efficient gene deletion demonstrate that RANK expression by fetal ILC3s contributes to lymph node organogenesis, while RANKL expression by other cell types may be sufficient for normal lymph node development to proceed.
Figure 6. Lymphoid tissue development in RANKL- and RANK-conditionally deficient mice.

(A) Percent of lymph nodes present in adult mice of the specified genotypes. (B) Peyer’s patch numbers in adult Tnfsf11fl/flRorcCre and Tnfrsf11afl/flRorcCre mice. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001.
Peyer’s patches were still present, but reduced in number, in Tnfsf11fl/flRorcCre and Tnfrsf11afl/flRorcCre mice (Figure 6B). Peyer’s patches were typically absent from the proximal region of Tnfsf11fl/flRorcCre small intestines (Supplemental Figure 6A). In comparison, Rag1−/−mice exhibited normal Peyer’s patch distribution, indicating that RANKL expression in ILC3s is required for the development of proximal Peyer’s patches (Supplemental Figure 6A). Gut tertiary lymphoid organs were not reduced in Tnfsf11fl/flRorcCre mice (Supplemental Figure 6B). Although M cell development is reduced in Tnfsf11−/− mice (Knoop et al., 2009), normal M cell populations were found within follicle-associated epithelium (FAE) in Peyer’s patches from Tnfsf11fl/flRorcCre mice (Supplemental Figure 6C), consistent with recent findings that RANKL is not required in hematopoietic cells for M cell development (Nagashima et al., 2017). Thus, RANKL deficiency in Tnfsf11fl/flRorcCre mice partially disrupts Peyer’s patch, but not tertiary gut lymphoid tissue, M cell, or lymph node, development.
RANKL deficiency increases RORγt expression in CCR6+ ILC3s
To gain insights into how RANKL-deficiency affects CCR6+ ILC3 transcription, cells from naïve Tnfsf11fl/flRorcCre mice and co-housed Tnfsf11fl/fl littermates were sorted from small intestine lamina propria and subjected to microarray analysis. Genes that were more highly expressed in Tnfsf11fl/flRorcCre CCR6+ ILC3s included the core ILC3 identity transcipts Rorc and Cxcr5, suggesting that RANKL-deficient ILC3s have an enhanced canonical ILC3 phenotype (Figure 7A, B). Cd69, which is induced upon lymphocyte activation, was also more highly expressed in RANKL-deficient cells. In contrast, genes induced by RANK signaling in osteoclasts, including Bmp2 and Nfkb2 (Cappellen et al., 2002; Wittrant et al., 2006), as well as Runx2, a regulator of Bmp2-induced signaling in osteoclasts (Rahman et al., 2015), were more highly expressed in RANKL-sufficient CCR6+ ILC3s.
Figure 7. Gene expression in RANKL-deficient CCR6+ ILC3s.

(A) Volcano plot of genes with 1.5-fold greater expression in Tnfsf11fl/fl CCR6+ ILC3s (green) versus genes with 1.5-fold greater expression in Tnfsf11fl/flRorcCre CCR6+ ILC3s (red). (B) Heatmap of selected genes differentially expressed in Tnfsf11fl/fl and Tnfsf11fl/flRorcCre CCR6+ ILC3s (n=3). (C) Intracellular staining for RORγt in Tnfsf11fl/fl and Tnfsf11fl/flRorcCre CCR6+ ILC3s from small intestine lamina propria (n=3). (D) Intracellular staining for RORγt in CCR6+ ILC3s from anti-RANKL antibody-or isotype control antibody-injected wild type mice (n=4). Bars indicate mean (+/− s.d). ****P≤0.0001. Data are representative of three (C) or two (D) independent experiments. Also see Figure S6.
CCR6+ ILC3s from Tnfsf11fl/flRorcCre and Tnfsf11fl/fl mice also differed in their transcription of cytokines and receptors to soluble molecules. RANKL-deficient ILC3s more highly expressed Tnf and Lif, as well as the semaphorin Sema4b. RANKL-deficient ILC3s also exhibited increased expression of the leukotriene receptors Cysltr2 and Ltb4r1, while control cells had increased expression of Vipr2, which encodes a receptor that binds the peptide hormone vasoactive intestinal peptide (VIP). These data suggest that RANK signaling alters the production of additional cytokines beyond IL-22 and IL-17A, and may induce differential responsiveness to leukotrienes and VIP.
To test whether differences in Rorc transcript expression led to changes in RORγt abundance, RORγt protein levels were assessed in CCR6+ ILC3s ex vivo. By intracellular antibody staining, more RORγt was detected in CCR6+ ILC3s from Tnfsf11fl/flRorcCre mice compared to Tnfsf11fl/fl cells (Figure 7C), as well as in CCR6+ ILC3s from RANKL antibody-treated mice compared to isotype control-treated mice (Figure 7D). Together, our data indicate that RANK-RANKL signaling under homeostasis induces an altered state in CCR6+ ILC3s defined by reductions in both RORγt expression and canonical ILC3 effector function.
DISCUSSION
Here, we demonstrated that RANKL suppressed CCR6+ ILC3 proliferation and activity in the small intestine. Mice conditionally deficient in RANKL in ILC3s and T cells had increased numbers of CCR6+ ILC3s in the small intestinal lamina propria. Additionally, CCR6+ ILC3s in these mice produced elevated amounts of IL-22 and IL-17A after stimulation with IL-23 and during infection with C. rodentium. CCR6+ ILC3s from adult mice injected with a RANKL blocking antibody exhibited similar phenotypes, indicating that RANKL is required constitutively; this eliminates the possibility that these effects are caused by differences in microbial colonization, changes in ILC development, or altered Peyer’s patch organogenesis. Although T cells were also subjected to Cre-mediated gene excision in Tnfsf11fl/flRorcCre mice, we found that T cells were neither sufficient nor required for ILC3s to acquire the hyperresponsive phenotype.
We further demonstrated that RANKL acted directly through the RANKL receptor, RANK, expressed on ILC3s. CCR6+ ILC3s expressed high amounts of RANK compared to other ILC3 populations. Moreover, pre-treatment of purified CCR6+ ILC3s with soluble trimeric RANKL in vitro modulated responsiveness to IL-23. Importantly, Rorc-mediated deletion of RANK in CCR6+ ILC3s replicated phenotypes observed in conditional RANKL-deficient mice, demonstrating that CCR6+ ILC3 suppression requires both RANK and RANKL in vivo. With these data, we propose a model in which RANK-RANKL interactions within the CCR6+ ILC3 population limit cell proliferation and effector cytokine production. In support of this model, we found that culturing purified CCR6+ ILC3s with a blocking antibody to RANKL enhanced responsiveness to IL-23. CCR6+ ILC3s are well positioned for homotypic cell-cell interactions, since they reside in close proximity to each other in cryptopatches and isolated lymphoid follicles. At these sites, ILC3-ILC3 interactions involving membrane bound or proteolytically shed RANKL may modulate ILC3 proliferation and functionality. By this reasoning, CCR6+ ILC3s that leave cryptopatches may have reduced RANK-RANKL signaling, and possess enhanced effector functions. A comparison of CCR6+ ILC3s isolated from tertiary lymphoid tissues and lamina propria will be required to further test this hypothesis. It is noteworthy that in addition to increasing proliferation and IL-17 and IL-22 production in ILC3s, lack of RANK-RANKL signaling resulted in reduced expression of Ebi3, Bmp2, and a receptor for VIP. Thus, RANK-RANKL signaling may also activate alternative functions in CCR6+ ILC3s in cryptopatches, which will require future investigation.
Our analysis of transcripts in RANKL-deficient CCR6+ ILC3s has revealed an inhibitory activity of RANK-RANKL signaling on the expression of Rorc, the key transcription factor driving ILC3 development and function. In support of these data, higher amounts of RORγt protein was present in CCR6+ ILC3s of RANKL-deficient and RANKL antibody-treated mice compared to cells from control mice. Together, these data demonstrate a novel relationship between RANK-RANKL signaling and RORγt expression. Since RORγt controls expression of Il17 and Il22, it is likely that CCR6+ ILC3 hyperresponsiveness is caused by increased amounts of this transcription factor. It remains be determined which of the multiple signaling pathways activated by RANK controls Rorc.
Previous studies using Tnfsf11−/− and Tnfrsf11a−/− mice demonstrated requirements for RANK and RANKL in the genesis of lymph nodes (Dougall et al., 1999; Kim et al., 2000; Kong et al., 1999; Mueller and Hess, 2012). RANKL- and RANK-deficiency in Tnfsf11fl/flRorcCre and Tnfrsf11afl/flRorcCre mice resulted in a reduction of Peyer’s patch numbers in the proximal small intestine but had no detectable impact on lymph node development. However, conditional RANK-deficient mice carrying one germline-deleted allele of Tnfrsf11a (Tnfrsf11afl/−RorcCre mice) and therefore exhibiting a more complete RANK deletion, partially lacked lymph nodes, indicating that RANK is required by fetal ILC3s for lymph node development. In contrast, lymph node development was intact in conditional RANKL-deficient mice carrying one germline-deleted allele of Tnfsf11 (Tnfsf11fl/−RorcCre mice), indicating that other RANKL+ cells likely compensate for RANKL+ ILC3s during lymph node organogenesis. Although RANKL is required for the development of M cells in FAE (Knoop et al., 2009), M cells were maintained in Tnfsf11fl/flRorcCre mice, consistent with the recent report that RANKL-expressing subepithelial mesenchymal cells are required for M cell development in FAE (Nagashima et al., 2017). We conclude that other cell types besides ILC3s provide RANKL during lymph node organogenesis and M cell induction.
In summary, our study demonstrates that RANKL controls CCR6+ ILC3 proliferation and effector cytokine production through ILC3-ILC3 interactions that activate RANK signaling and suppress expression of RORγt. These observations may open new avenues of therapeutic intervention in intestinal infections and inflammatory bowel diseases through the modulation of ILC3 activation.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marco Colonna (mcolonna@wustl.edu).
Experimental Models and Subject Details
Animals
Six- to 12-week-old male and female mice were used in this study. Sex- and age-matched littermates were used within each experiment, and were randomly assigned to experimental groups. All mice were backcrossed to the C57BL/6 background. Rag1−/− and Tnfrsf11afl/fl mice were purchased from Jackson Laboratories. RorcCre/+ mice (Eberl and Littman, 2004) were provided by Gerard Eberl. Tnfrsf11aeGFP-Cre mice (Maeda et al., 2012) were provided by Deborah Novack and Yasuhiro Kobayashi. Tnfsf11fl/fl (Tnfsf11tm1c/tm1c) mice were generated from ES cells purchased from Eucomm (Tnfsf11 EPD0724_4_C08; Tnfsf11tm1a (EUCOMM)Wtsi) and B6.Tg(CAG-FLP3e)36 mice from RIKEN (Kanki et al., 2006) as previously described (Wang et al., 2012). Tnfsf11fl/− and Tnfrsf11afl/− mice were generated from germline deletion that occurred sporatically in progeny of RorcCreTnfsf11fl/fl × Tnfsf11fl/fl or RorcCreTnfrsf11afl/fl × Tnfrsf11afl/fl breeding pairs (Bando et al., 2015). All RorcCreTnfsf11fl/fl and RorcCreTnfrsf11afl/fl mice reported in this study were genotyped against germline deletion of Tnfsf11 or Tnfrsf11a. Mice were maintained in specific pathogen-free facilities at Washington University in Saint Louis. All studies were conducted in accordance with the Washington University Animal Studies Committee.
Human studies
Pediatric tonsils were obtained from tonsillectomies performed at Children’s Hospital in Saint Louis under the approval of institutional review boards of Washington University in Saint Louis and with informed consent. The samples were provided as surgical waste with no identifiers attached. Tonsils were minced, disupted against mesh, and enriched for CD56+ cells using microbeads (Miltenyl biotec). Enriched cells were sorted for NKp44+CCR6+CD103− ILC3s or NKp44+CD103+ ILC1s.
Cell lines and primary cell cultures
Cells were cultured at 37 °C under 5% CO2. MNK-3 cells and primary mouse cells were cultured in RPMI 1640 supplemented with sodium pyruvate, HEPES, kanamycin sulfate, glutamine, nonessential amino acids, β-mercaptoethanol, and 10% BCS. Human primary cells were cultured in RPMI 1640 supplemented with sodium pyruvate, kanamycin sulfate, ciprofloxacin (Bayer), glutamine, nonessential amino acids, β-mercaptoethanol, and 10% FCS (Atlanta Biologics). For 3 d cultures, media was supplemented with 4% in-house generated mouse IL-7 supernatant for mouse cells or 50 ng/ml recombinant human IL-7 for human cells. The sex of MNK-3 cells has not been determined.
Method Details
Tissue dissociation
Small intestines were flushed to remove luminal contents and Peyer’s patches were removed. Intestines were opened lengthwise and gently agitated for 20 min in HBSS containing HEPES, BCS, and EDTA. Intestines were votexed before subjected to a second round of gentle agitation and vortexing in EDTA. The tissue was then rinsed with HBSS prior to digestion with Collagenase IV (Sigma) in complete RPMI-10 for 40 min at 37 deg C under agitation. Digests were filtered through 100 micron mesh and subjected to density gradient centrifugation using 40% and 70% Percoll solutions. To isolate thymic epithelial cells, thymii were mechanically dissociated in RPMI containing 2% BCS. The tissue was then digested with Liberase and Dnase I for 40 min at 37 deg C, and then treated with EDTA for 5 min. Digested samples were vortexed, filtered, and subjected to density gradient centrifugation using media and a 50% Percoll solution. To generate single cell suspensions of peripheral lymph nodes, inguinal, axillary, brachial, and cervical lymph nodes were pooled and mashed against a 70 micron mesh filter.
Cell stimulation and cytokine analysis
Cells from naïve mice were stimulated ex vivo with either 1 ng/ml, 0.1 ng/ml, or 0.01 ng/ml IL-23 for 3.5 h, while cells from C. rodentium-infected mice were cultured for 3.5 h with no additional stimulation. In other experiments, CCR6+ ILC3s were sorted and pre-treated for 3 d with RANKL or media alone in flat bottom wells (6×104 cells/well), or were treated with anti-RANKL or isotype control antibodies in round bottom wells (4×104 cells/well), prior to stimulation with 25 pg/ml IL-23 and 0.6 pg/ml IL-1β. MNK-3 cells were pre-treated with RANKL, anti-RANKL, isotype control antibodies, or media alone for 3 d in round bottom wells (5×104 cells/well), and then stimulated with 0.1 ng/ml IL-23 and 0.04 ng/ml IL-1β to measure IL-17A production, or 25 pg/ml IL-23 and 0.6 pg/ml IL-1β to measure IL-22 production. Human cells were cultured with 50 ng/ml human OPG (Sigma) for 3 d prior to stimulation with 10 ng/ml hIL-23 and 4 ng/ml hIL-1β for 10 h. In experiments requiring intracellular cytokine staining, brefeldin A was present for the last 3 h of culture. In experiments quantifying changes in RANKL cell surface staining upon stimulation, sorted cells were cultured with 10 ng/ml IL-23 and 10 ng/ml IL-1β for 5 hours. In other experiments, culture supernatants from stimulated or unstimulated cells were collected on d 3, or cells were washed and lysed on d3, and subjected to ELISA using polyclonal antibodies that bind the extracellular domain of RANKL.
Flow cytometry
Single cell suspensions were incubated with Fc Block for 10 minutes, and then stained with antibodies and Fc Block for 20 min at 4°C. For dete ction of cell surface RANKL, cells were cultured in complete RPMI at 37°C for 2 h (mouse) o r 1 h (human) prior to staining with anti-RANKL for 20 min at room temperature. Negative staining controls for RANKL stains were RANKL-deficient cells cultured and antibody stained in parallel for mouse cells, or isotype control antibodies for human cells. Dead cells were excluded using either a Live/Dead Fixable Cell Stain Kit (ThermoFisher Scientific), DAPI (Sigma), or 7-AAD. Intracellular proteins were stained using either the BD Biosciences Fixation/Permeabilization Solution Kit or eBioscience Transcription Factor staining kit. Cells were run on a FACSCanto II or LSRFortessa (BD Biosciences), and were analyzed using FlowJo (FlowJo LLC). Cells counts were conducted with counting beads (eBioscience). ILCs were identified as live CD3e−CD5−CD19− lymphocyte-sized cells that were either GATA3hi (ILC2), RORγt+ (ILC3), or RORγt−GATA3intNKp46+ (ILC1+NK). In experiments with intracellular cytokine staining, ILC3 were identified as CD3e−CD5−CD19−CD90.2hiCD45int live lymphocytes, and then were further subdivided using NKp46 and CCR6. CCR6+ ILC3s were purified by sorting live, lymphocyte sized cells that were negative for CD3e, CD5, CD19, CD11b, CD11c, NK1.1, Gr-1, B220, Ter119, KLRG1, and NKp46; positive for CD90.2 and CCR6; and intermediate for CD45.
Bone marrow chimeras
Recipient mice were irradiated once with 550 Rads and 4×106 donor bone marrow cells were injected i.v. Mice were analyzed five weeks after irradiation.
In vivo antibody administration
Rat anti-RANKL (IKK2/5) or isotype control antibodies (2A3) were purchased from Bio X Cell (West Lebanon, NH). 100 or 200 ug of antibody was injected ip on d 0, 3, and 6, or on d 0, 2 and 4. Mice were analyzed on either d 5 or d 7.
Citrobacter rodentium infection
Mice were orally gavaged with 2–5×109 CFU of C. rodentium. Weight was monitored daily. Fecal samples were homogenized and plated overnight on MacConkey agar (Sigma).
M cell staining
Ileal Peyer’s patches were fixed in formalin overnight, and then washed with 70% ethanol and PBS. Fixed tissue was incubated with BSA in 0.1% triton prior to staining with GP-2 antibodies.
Microarray analysis
RNA was extracted from sorted small intestine lamina propria CCR6+ ILC3s with an RNeasy Micro Kit (Qiagen). RNA amplification and hybridization to Affymetrix Mouse Gene 1.0 ST array was conducted by the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine in Saint Louis. Data were analyzed using Multiplot by GenePattern.
Quantification and Statistical Analysis
Data were analyzed with either t-tests or one-way ANOVA and Tukey post hoc tests. Each dot in graphs indicate biological replicates, except where indicated in figure legends. All other statistical details can be found in figure legends. Bars indicate mean (+/− s.d). ns, not significant; *P<0.05; **P≤0.01, ***P≤0.001, ****P≤0.0001.
Data and Software Availability
The microarray data have been deposited in the Gene Expression Omnibus (GEO) respository under accession number GSE112710.
Supplementary Material
Highlights.
-
-
CCR6+ ILC3s from RorcCreTnfsf11fl/fl mice have enhanced IL-17A and IL-22 production
-
-
Loss of Tnfsf11 in ILC3s, and not T cells, leads to hyperresponsive CCR6+ ILC3s
-
-
RANKL directly suppresses ILC3s via RANK, and RANK-deficiency enhances ILC3 activity
-
-
CCR6+ ILC3s lacking RANKL have increased amounts of RORγt
Acknowledgments
The authors thank Deborah Novack, Anna Ballard, and Jesse Gibbs for providing sRANKL and RANK-eGFP mice. Daved Fremont and Chris Nelson developed the sRANKL reagent. Gerard Eberl generously provided RorcCre/+ mice. Daved Fremont, Michael Patnode, Blanda Di Luccia, Luisa Cervantes-Barragan, Alexander Barrow, and Victor Cortez provided helpful discussion. Tyler Ulland provided help with bone marrow chimera experiments. The Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine assisted with microarray experiments. All flow cytometry work was conducted in the Flow Cytometry and Fluorescence Activated Cell Sorting Core in the Department of Pathology and Immunology at Washington University School of Medicine. This work was supported by the NIH grants DE025884, DK103039, and AI120606 (to MCo), CA176695 (to MCe), and DK097317 (to RN). This work was also supported by the Digestive Diseases Research Core Center (P30 DK52574); and the Crohn's and Colitis Foundation of America. JKB is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
J.K.B and M.Co. conceived experiments. J.K.B. conducted experiments and wrote the manuscript. S.G, C.S, K.G.M, and R.D.N conducted experiments. S.G, S.C-C.H, Y.K., D.S.J.A, J.R.C, and M. Ce. provided reagents. S.G., M.Ce., and M.Co. provided expertise and feedback.
Declaration of Interests
The authors declare no competing interests.
References
- Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, Asaumi Y, Kitazawa J, Takayanagi H, Penninger JM, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. doi: 10.1016/j.immuni.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Allan DS, Kirkham CL, Aguilar OA, Qu LC, Chen P, Fine JH, Serra P, Awong G, Gommerman JL, Zúñiga-Pflücker JC, Carlyle JR. An in vitro model of innate lymphoid cell function and differentiation. Mucosal Immunol. 2015;8:340–351. doi: 10.1038/mi.2014.71. [DOI] [PubMed] [Google Scholar]
- Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- Ashcroft AJ, Cruickshank SM, Croucher PI, Perry MJ, Rollinson S, Lippitt JM, Child JA, Dunstan C, Felsburg PJ, Morgan GJ, Carding SR. Colonic dendritic cells, intestinal inflammation, and T cell-mediated bone destruction are modulated by recombinant osteoprotegerin. Immunity. 2003;19:849–861. doi: 10.1016/s1074-7613(03)00326-1. [DOI] [PubMed] [Google Scholar]
- Bando JK, Colonna M. Innate lymphoid cell function in the context of adaptive immunity. Nat Immunol. 2016;17:783–789. doi: 10.1038/ni.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bando JK, Liang HE, Locksley RM. Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat Immunol. 2015;16:153–160. doi: 10.1038/ni.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D, Luong-Nguyen NH, Bongiovanni S, Grenet O, Wanke C, Susa M. Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NFkappa B. J Biol Chem. 2002;277:21971–21982. doi: 10.1074/jbc.M200434200. [DOI] [PubMed] [Google Scholar]
- Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A. 2010;107:10961–10966. doi: 10.1073/pnas.1005641107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JH, Kljavin NM, Ota N, Leonard J, Roose-Girma M, Diehl L, Ouyang W, Ghilardi N. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 2012;5:99–109. doi: 10.1038/mi.2011.54. [DOI] [PubMed] [Google Scholar]
- Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. 2014;41:354–365. doi: 10.1016/j.immuni.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffin R, O'Connor RA, Crittenden S, Forster T, Yu C, Zheng X, Smyth D, Robb CT, Rossi F, Skouras C, et al. Prostaglandin E2 constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science. 2016;351:1333–1338. doi: 10.1126/science.aad9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB, Lacey DL, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50. doi: 10.1016/s0092-8674(00)00103-3. [DOI] [PubMed] [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–985. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, Cesses P, Garnier L, Pouzolles M, Brulin B, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature. 2016;529:226–230. doi: 10.1038/nature16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green EA, Choi Y, Flavell RA. Pancreatic lymph node-derived CD4(+)CD25(+) Treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity. 2002;16:183–191. doi: 10.1016/s1074-7613(02)00279-0. [DOI] [PubMed] [Google Scholar]
- Guerrini MM, Okamoto K, Komatsu N, Sawa S, Danks L, Penninger JM, Nakashima T, Takayanagi H. Inhibition of the TNF Family Cytokine RANKL Prevents Autoimmune Inflammation in the Central Nervous System. Immunity. 2015;43:1174–1185. doi: 10.1016/j.immuni.2015.10.017. [DOI] [PubMed] [Google Scholar]
- Hikosaka Y, Nitta T, Ohigashi I, Yano K, Ishimaru N, Hayashi Y, Matsumoto M, Matsuo K, Penninger JM, Takayanagi H, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. 2008;29:438–450. doi: 10.1016/j.immuni.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, Gallini CA, Redding K, Margolskee RF, Osborne LC, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science. 2016;351:1329–1333. doi: 10.1126/science.aaf1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibiza S, García-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, Misic AM, Bartow-McKenney C, Larson DM, Pavan WJ, et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature. 2016;535:440–443. doi: 10.1038/nature18644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki H, Suzuki H, Itohara S. High-efficiency CAG-FLPe deleter mice in C57BL/6J background. Exp Anim. 2006;55:137–141. doi: 10.1538/expanim.55.137. [DOI] [PubMed] [Google Scholar]
- Kim D, Mebius RE, MacMicking JD, Jung S, Cupedo T, Castellanos Y, Rho J, Wong BR, Josien R, Kim N, et al. Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J Exp Med. 2000;192:1467–1478. doi: 10.1084/jem.192.10.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Cho BH, Kiyono H, Jang YS. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer's patches. Sci Rep. 2017;7:3980. doi: 10.1038/s41598-017-02729-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, Diefenbach A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d'Hargues Y, Göppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature. 2013;494:261–265. doi: 10.1038/nature11813. [DOI] [PubMed] [Google Scholar]
- Knoop KA, Kumar N, Butler BR, Sakthivel SK, Taylor RT, Nochi T, Akiba H, Yagita H, Kiyono H, Williams IR. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J Immunol. 2009;183:5738–5747. doi: 10.4049/jimmunol.0901563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- Lam J, Nelson CA, Ross FP, Teitelbaum SL, Fremont DH. Crystal structure of the TRANCE/RANKL cytokine reveals determinants of receptor-ligand specificity. J Clin Invest. 2001;108:971–979. doi: 10.1172/JCI13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, Colonna M. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2012;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum L, Wong BR, Josien R, Becherer JD, Erdjument-Bromage H, Schlöndorff J, Tempst P, Choi Y, Blobel CP. Evidence for a role of a tumor necrosis factor-alpha (TNF-alpha)-converting enzyme-like protease in shedding of TRANCE, a TNF family member involved in osteoclastogenesis and dendritic cell survival. J Biol Chem. 1999;274:13613–13618. doi: 10.1074/jbc.274.19.13613. [DOI] [PubMed] [Google Scholar]
- Lügering A, Ross M, Sieker M, Heidemann J, Williams IR, Domschke W, Kucharzik T. CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin Exp Immunol. 2010;160:440–449. doi: 10.1111/j.1365-2249.2010.04103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, Kikuchi Y, Takada I, Kato S, Kani S, et al. Wnt5a–Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18:405–412. doi: 10.1038/nm.2653. [DOI] [PubMed] [Google Scholar]
- Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, Misiak A, Dungan LS, Sutton CE, Streubel G, et al. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. 2013;210:1117–1124. doi: 10.1084/jem.20121588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaldo E, Teixeira-Alves LG, Glatzer T, Durek P, Stervbo U, Hamann W, Babic M, Paclik D, Stölzel K, Gröne J, et al. Human RORγt(+)CD34(+) cells are lineage-specified progenitors of group 3 RORγt(+) innate lymphoid cells. Immunity. 2014;41:988–1000. doi: 10.1016/j.immuni.2014.11.010. [DOI] [PubMed] [Google Scholar]
- Mueller CG, Hess E. Emerging Functions of RANKL in Lymphoid Tissues. Front Immunol. 2012;3:261. doi: 10.3389/fimmu.2012.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima K, Sawa S, Nitta T, Tsutsumi M, Okamura T, Penninger JM, Nakashima T, Takayanagi H. Identification of subepithelial mesenchymal cells that induce IgA and diversify gut microbiota. Nat Immunol. 2017;18:675–682. doi: 10.1038/ni.3732. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275:768–775. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- Nelson CA, Warren JT, Wang MW, Teitelbaum SL, Fremont DH. RANKL employs distinct binding modes to engage RANK and the osteoprotegerin decoy receptor. Structure. 2012;20:1971–1982. doi: 10.1016/j.str.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, West N, Tung S, Seddon BP, Uhlig HH, Powrie F. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. 2016;5:e10066. doi: 10.7554/eLife.10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MS, Akhtar N, Jamil HM, Banik RS, Asaduzzaman SM. TGF-β/BMP signaling and other molecular events: regulation of osteoblastogenesis and bone formation. Bone Res. 2015;3:15005. doi: 10.1038/boneres.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi SW, Kim MY, Leibbrandt A, Parnell SM, Jenkinson WE, Glanville SH, McConnell FM, Scott HS, Penninger JM, Jenkinson EJ, et al. RANK signals from CD4(+)3(-) inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J Exp Med. 2007;204:1267–1272. doi: 10.1084/jem.20062497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP, Eberl G. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. 2010;330:665–669. doi: 10.1126/science.1194597. [DOI] [PubMed] [Google Scholar]
- Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Bérard M, Kleinschek M, Cua D, Di Santo JP, Eberl G. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, Mack M, Cella M, Colonna M. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J Exp Med. 2015;212:1869–1882. doi: 10.1084/jem.20151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, Nutman TB, Urban JF, Wang J, Ramalingam TR, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science. 2014;343:432–437. doi: 10.1126/science.1247606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama M, Nakato G, Jinnohara T, Akiba H, Okumura K, Ohno H, Yoshida H. Expression pattern changes and function of RANKL during mouse lymph node microarchitecture development. Int Immunol. 2012;24:369–378. doi: 10.1093/intimm/dxs002. [DOI] [PubMed] [Google Scholar]
- van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, Almeida FF, Ibiza S, Barbosa I, Goverse G, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. 2014;508:123–127. doi: 10.1038/nature13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Vivier E. Differentiation and function of group 3 innate lymphoid cells, from embryo to adult. Int Immunol. 2016;28:35–42. doi: 10.1093/intimm/dxv052. [DOI] [PubMed] [Google Scholar]
- von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. 2016;529:221–225. doi: 10.1038/nature16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front Immunol. 2014;5:511. doi: 10.3389/fimmu.2014.00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13:753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson E, Bilsborough JM, Viney JL. Regulation of mucosal dendritic cell function by receptor activator of NF-kappa B (RANK)/RANK ligand interactions: impact on tolerance induction. J Immunol. 2002;169:3606–3612. doi: 10.4049/jimmunol.169.7.3606. [DOI] [PubMed] [Google Scholar]
- Wittrant Y, Lamoureux F, Mori K, Riet A, Kamijo A, Heymann D, Redini F. RANKL directly induces bone morphogenetic protein-2 expression in RANK-expressing POS-1 osteosarcoma cells. Int J Oncol. 2006;28:261–269. [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.