

Abstract
Rationale:
Holoprosencephaly is a structural malformation of the brain that results from the complete or incomplete noncleavage of the forebrain of the embryo into 2 hemispheres. We report a severe case of alobar holoprosencephaly diagnosed at 38 weeks, associated with cebocephaly, microcephaly, and craniosynostosis.
Patient concern:
The main knowledge added by this case is the late ultrasound diagnosis and chromosomal analysis that revealed a very rare abnormality (45X/46,XX/47,XX) with mosaicism at chromosome 18.
Diagnoses:
Investigation of the mother revealed nothing remarkable from clinical point of view and on laboratory tests. Ultrasonography identified a fetal biometry appropriate for gestational age, except for the head biometry and abdominal circumference, that were appropriate for less than the fifth percentile. Microcephaly, a large midline monoventricle, absent midlinestructures, cleft lip, cebocephaly (hypotelorism, single-nostril nose), ethmocephaly (hypotelorism, interorbital proboscis) and craniosynostosis, were also present. Fetal magnetic resonance imaging of fetus revealed an absent midline structure, a central monoventricle, abnormal corpus calosum, and abnormal gyri.
Interventions:
A cesarean section at 38 weeks was indicated for fetal bradycardia and a female baby was delivered, with Apgar score 6, weight 2290g. After birth, the diagnosis of the fetus confirmed holoprosencephaly with facial anomalies and demonstrated repeated tonic-clonic seizure, severe respiratory failure, cyanosis, decreased muscle tone, palor, and apnea. Laboratory examination of the newborn revealed acidosis and a prolonged of prothrombin time. The neonate was treated for severe respiratory distress syndrome, with immediate intubation and resuscitation. Vitamin K, fresh frozen plasma, and antibiotics were also administered.
Outcomes:
After delivery, exitus of the fetus occurred at 3 days and 18hours due to massive pulmonary hemorrhage.
Lessons:
We described a case of alobar holoprosencephaly diagnosed at 38 weeks of gestation and associated with a rare chromosomal abnormality (45X/46,XX/47,XX) with mosaicism at chromosome 18. Emotional implications could have been less severe if the patient underwent regular ultrasonography allowing a diagnosis in the first or early second trimester.
Keywords: cebocephaly, facial abnormalities, holoprosencephaly, hypotelorism, microcephaly, single nostril nose
1. Introduction
Holoprosencephaly (HPE) is a complex brain malformation resulting from the incomplete midline cleavage of the prosencephalon and occurring between the 18th and the 28th day of gestation,[1,2] indicating that HPE is a disorder of gastrulation. HPE is a rare structural abnormality of the brain and is associated with neurologic impairment and facial dysmorphism.[3–5] Demyer and Zeman[2] suggested this resulted from a defect in the ventral induction and from the patterning of the rostral neural tube by the prechordal mesenchyma. As ventral induction is related to facial development, many HPE cases also demonstrate craniofacial abnormalities, leading to a so-called “holoprosencephaly sequence.”[1] HPE is estimated to occur in 1 of 16,000 live births.[1] Cyclopia or synophthalmia, severe ocular hypotelorism with divided orbits, and a proboscis-like nasal structure are mostly associated with alobar HPE.[2,5] Cebocephaly and median cleft lip are also seen in alobar HPE. Less severe facial dysmorphisms, including ocular hypotelorism, iris coloboma, absence of nasal bones, single central incisor, unilateral or bilateral cleft lip/palate, and midface hypoplasia may be seen in any of the anatomic forms.[3,4] The most widely used classification is the one proposed by DeMyer and Zeman in 1963.[2] They outlined 3 types of HPE alobar, semilobar, and lobar, depending on the degree of separation of the hemispheres and the presence or absence of the interhemispheric fissure.[2,5] This classification is also useful from a prognostic point of view, as it offers information about the severity and neurological impact of the malformation. In the most severe form, alobar HPE, there is a complete lack of cleavage of the cortex, while in the mildest form, lobar HPE, only a part of the frontal lobes and ventricular horns are fused.[6] In 1993, a new variant of HPE called “middle interhemispheric” (MIH) variant or syntelencephaly was described by Barkovich and Quint[7]; the main characteristic of their cases, presented by them, was the existence of a degree of middle interhemispheric fusion. [7]
We present a rare case of alobar HPE diagnosed at 38 weeks of gestation associated with a special and rare chromosomal abnormality, a (45X/46,XX/47,XX) with mosaicism at chromosome 18, combined with the previous literature review.
2. Case report
A 22-year-old pregnant woman (Caucasian, gravida 4, para 3, 38 weeks pregnant) was admitted in August 2016 to the Department of Obstetrics Gynecology of our unit, from a Clinical Emergency Hospital in Bucharest, for false labor to establish therapeutic specialist management. A written informed consent form signed by the patient at admission included the permission to present her case and images for publication if necessary and the study was approved by the Ethics Committee of Sf Pantelimon Clinical Emergency Hospital in accordance to the ethical guidelines of the Declaration of Helsinki.
The patient's history showed that she had 2 normal births, 5 years and 3 years ago, with normal children and 1 miscarriage. She was a nonsmoker, without diabetes mellitus, without teratogenic potential disease, nor exposure to radiation or toxic substances abuse in the first and second trimesters of pregnancy. Moreover, there was no personal family history of congenital malformations. The patient did not undergo screening for chromosomal abnormalities in the first trimester or any other tests to assess fetal DNA. A combined first-trimester nuchal translucency with biochemistry screening, a second-trimester maternal serum screening and ultrasonography for fetal anomalies, and combined ultrasonography for second-trimester maternal serum markers with risk assessment were not performed because the patient did not go to the clinic for monitoring during the pregnancy. The patient saw the maternal-fetal specialist at 11 weeks; when the ultrasound was normal, subsequently, she did not see a specialist again until 38 weeks of pregnancy. On admission, at 38 weeks gestation, the patient had irregular painful uterine contractions. The general clinical examination was normal. The laboratory examination with complete blood counts was within normal limits. Screenings for HIV, syphilis, toxoplasmosis, cytomegalovirus, hepatitis B, hepatitis C, and group B streptococcus infection were negative. Tests for blood group and rhesus D status revealed A group and Rh positivity.
A detailed ultrasound examination was performed and revealed a biometry appropriate for the gestational age, with the exception of head biometry. The examination was conducted with transabdominal transducer from GE Voluson 730 Expert Ultrasound Equipement (GE Medical Systems, Zipf, Austria), equipped with a 4 to 8-MHz volumetric convex transducer.
Several abnormalities were noted on ultrasound. The fetal head was microcephalic with the biparietal diameter (BPD) and head circumference (HC) measuring less than the fifth percentile, the BPD expected for the gestational age was 95 mm, but the measured value was 85 mm and the HC expected for the gestational age was 328 mm, but the measured value was 302 mm. Abdominal circumference was 290.2 mm, less than the fifth percentile. Also, other biometric parameters such as HC/abdominal circumference and HC/femur length were less than the fifth percentile, 0.90 and 4.0, respectively. Length of femur, tibia, fibula, humerus, radius, and ulna were appropriate for the 50 percentile. The cerebral hemispheres were completely fused into a holosphere and the interhemispheric fissure was completely absent, such that the resulting brain was smaller than normal. The monoventricle communicated posteriorly with a dorsal cyst (Fig. 1).
Figure 1.
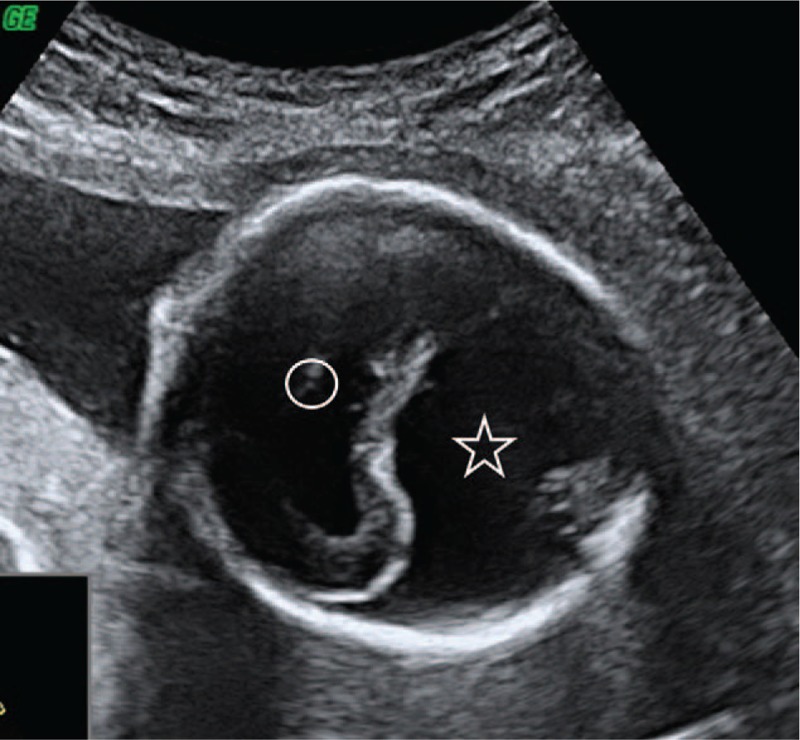
Cerebral hemispheres fussed with a single midline ventricle in the middle (circle). In the occipital region, a large dorsal cyst is present (star).
No midline structures were present, including the falx cerebri, interhemispheric fissure, cavum septum pellucidum, or corpus callosum (Fig. 2).
Figure 2.
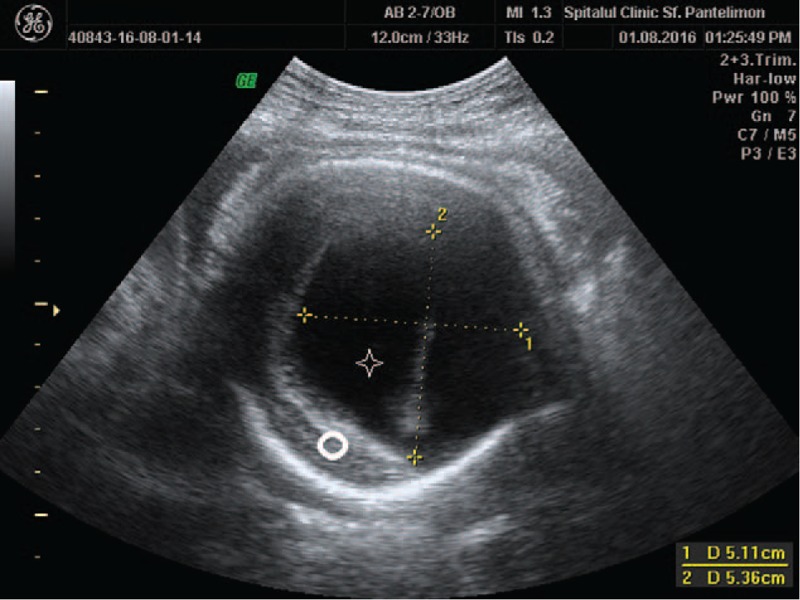
Single midline monoventricle (star) and absent midline structures (circle).
The nuclei of the basal ganglia, hypothalamus, and thalamus were fused in the midline, so the third ventricle was not visible (Fig. 3).
Figure 3.
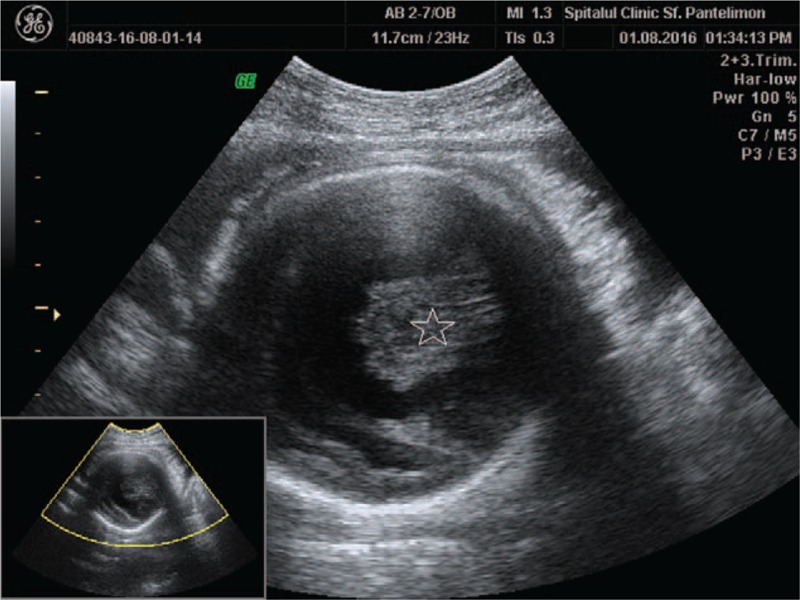
Hypothalamus and thalamus fused in midline (star).
The gross appearances of the brainstem and cerebellum were normal, but a single cerebral peduncle was noted (Fig. 4). Deep-gray nuclei appeared to be incompletely separated. Facial anomalies of cebocephaly (hypotelorism with a single-nostril nose) and ethmocephaly (hypotelorism, interorbital proboscis) were present and were suggestive of alobar HPE.
Figure 4.
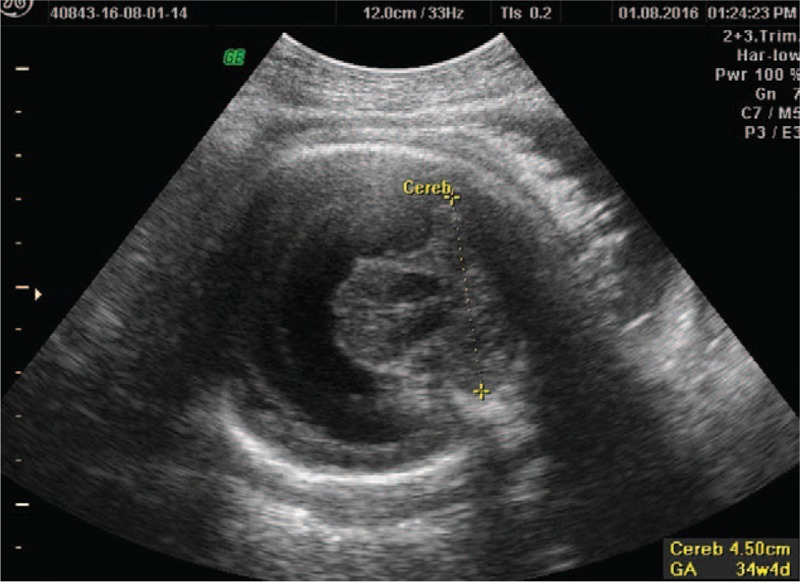
Normal ultrasound appearance of cerebellum.
The fetal MRI from the coronal view or in axial view (Fig. 5) shows the posterior part of the cerebrum, which is shaped like a horse-shoe with the posterior-dorsal rim formed by the membrane covering the dorsal cyst. From the sagittal view, depending on the degree to which the cerebrum surrounds the dorsal membranous roof of the ventricle, the brain is shaped like a cup where the monoventricle is not completely encircled by the cortex (Fig. 6).
Figure 5.
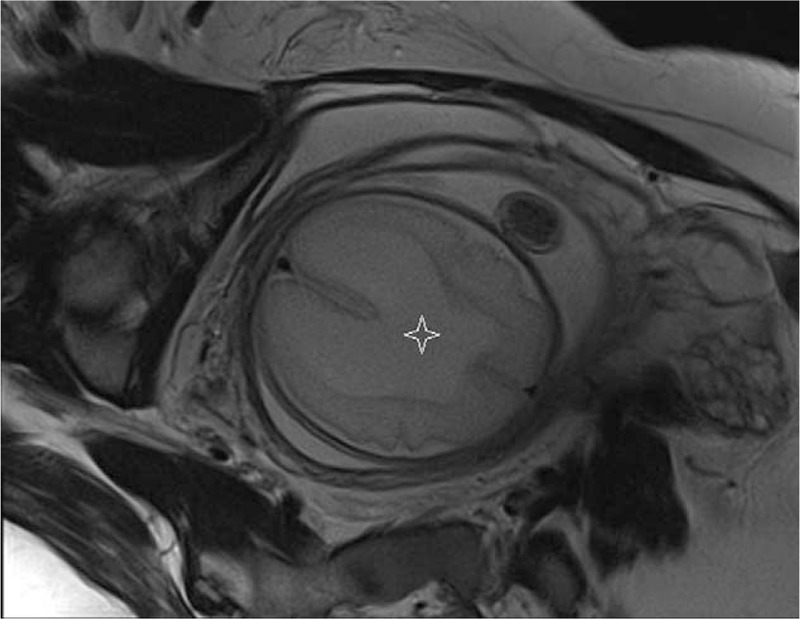
Fetal MRI (axial view). Posterior part of the cortex that surrounds the monoventricle (star).
Figure 6.
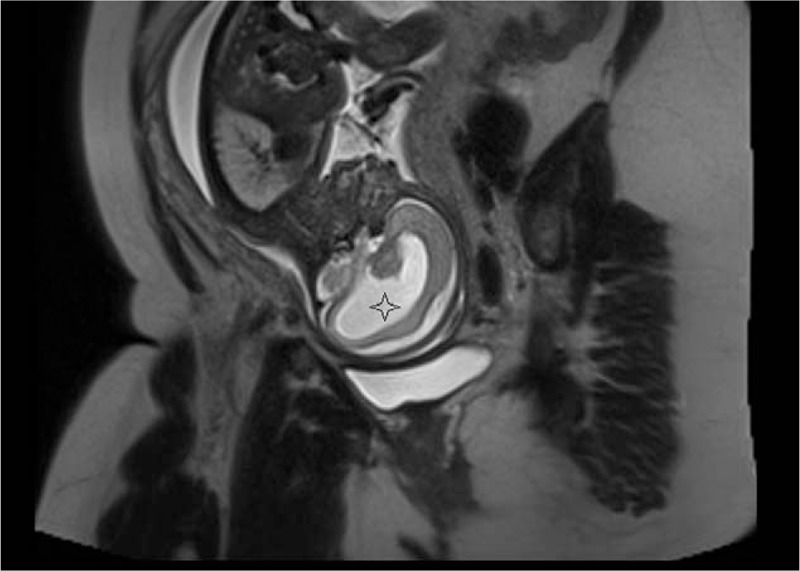
Fetal MRI (sagittal view). Cortex is like a cup and encircles the monoventricle (star).
At 38 weeks gestation, 1 day after admission, labor began with spontaneously ruptured membranes and painful uterine contractions. Fetal bradycardia (90 bpm) was diagnosed via cardiotocography at the beginning of the labor. Because of the fetal bradycardia, a female fetus (weight 2290 g, length 46 cm) was delivered by cesarean section (CS) under spinal anesthesia. The APGAR score of the fetus was 6 (Activity: 2, Pulse:1, Grimace:2, Respiratory effort: 1), 5 (Activity: 2, Pulse:2, Grimace:0, Respiratory effort: 1), and 5 (Activity: 2, Pulse:2, Grimace:1, Respiratory effort: 0) at1, 5, and 10 minutes, respectively. Blood gas analysis showed a pH of 7.149, pCO2 of 57.9 mm Hg, pO2 of 30 mm Hg, and BE of -4.2 mmol/L. Immediate intubation and resuscitation were necessary. The female fetus showed poor respiratory effort, decreased muscle tone, and pallor. She was transferred to the neonatal intensive care unit for further treatment. The measurement of the fetus revealed a small-for-gestational age fetus, with a fronto-occipital HC of 24.5 cm, secondary to microcephalia. Closed cranial sutures without a fontanelle, heart rate of 179 bpm, and blood pressure of 68/40 mm Hg and no external signs of spontaneous hemorrhage were noted. The corneal, Moro, and rooting reflexes were absent. Hepatomegaly and splenomegaly were not found. Laboratory tests revealed a hemoglobin level of 74 g/L, platelet count of 254 × 109/L, and white blood cell count of 15.57 × 109/L. Prothrombin time exceeded 70 seconds (upper limit of reference), and the activated partial thromboplastin time was greater than 150 seconds (upper limit of reference). Thrombin time (16.0 seconds) and fibrinogen (3.00 g/L) were normal. Serology assay showed the following values: albumin 28 g/L; alanintransaminase 5 U/L; T-bilirubin 27.4 mmol/L; creatine-kinase-muscle/brain 6.3 mg, and creatine-kinase 353 U/L.
The mother's progress was favorable and without complications, under antibiotic prophylaxis and anti-inflammatory drugs. Phenotypically, the fetus had a short neck, triangular face, low-set ears, small and protruded chin, hypotelorism, a single-nostril nose, and ogival palate (Fig. 7). Fetal progress was poor from the onset, with repeated tonic-clonic seizures and severe respiratory failure, cyanosis, and apnea. Fetal exitus occurred after 3 days and 18 hours of life due to massive pulmonary hemorrhage and cardiorespiratory arrest. A blood sample had been collected on the first day of life for fetal karyotyping. The result was a very rare chromosomal abnormality (45X/46,XX/47,XX) with mosaicism at chromosome 18. Usually, a fetus with this chromosomal abnormality has a very poor prognosis.
Figure 7.
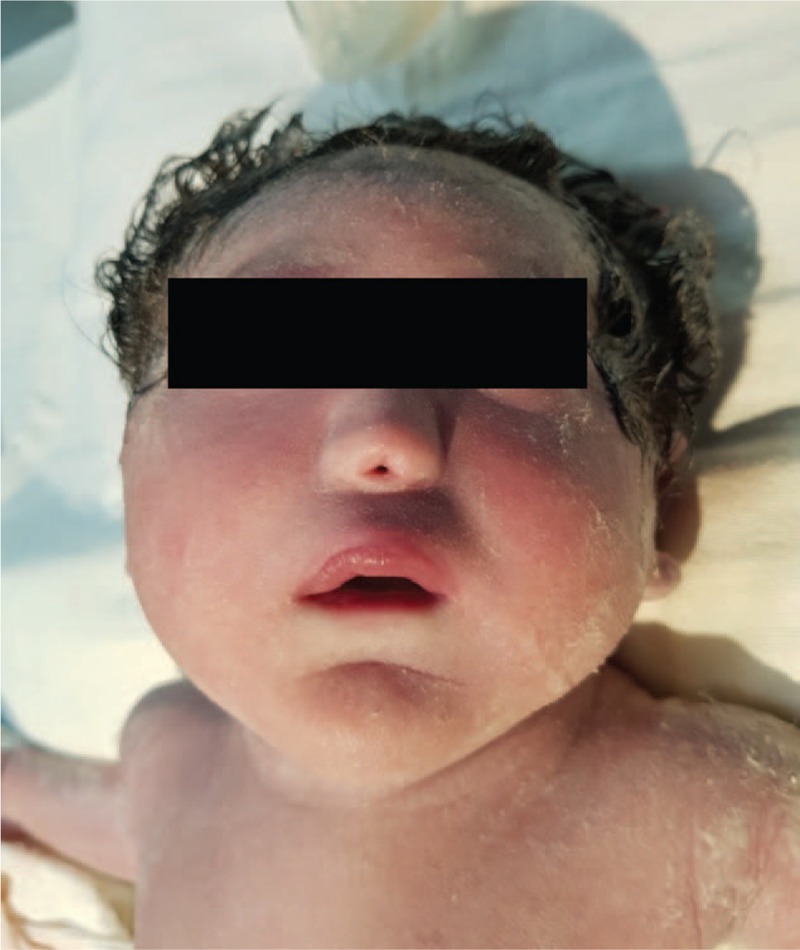
Single nostril and ethmocephaly.
A pathologic examination of the fetus was performed and revealed single nostril and ethmocephaly (Fig. 7). The cranial sutures were closed (Fig. 8) with brain weighing 120 g. Additionally noted were a single cerebral ventricle with the absence of midline structures (Fig. 9) and the absence of the corpus callosum and separate cerebral hemispheres.
Figure 8.
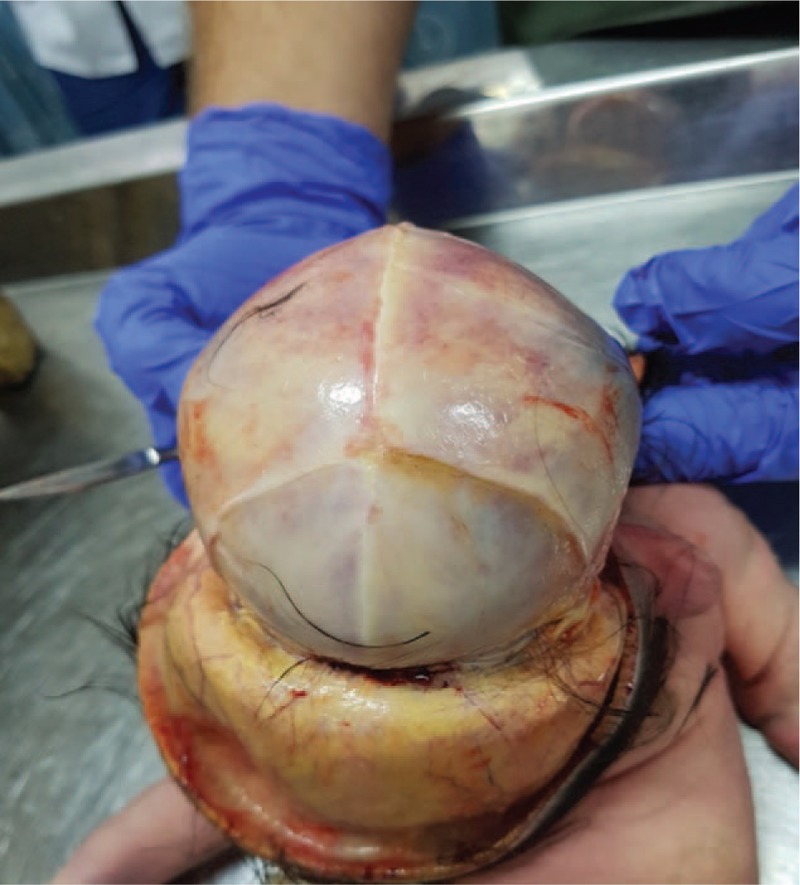
Closed cranial sutures without fontanels.
Figure 9.
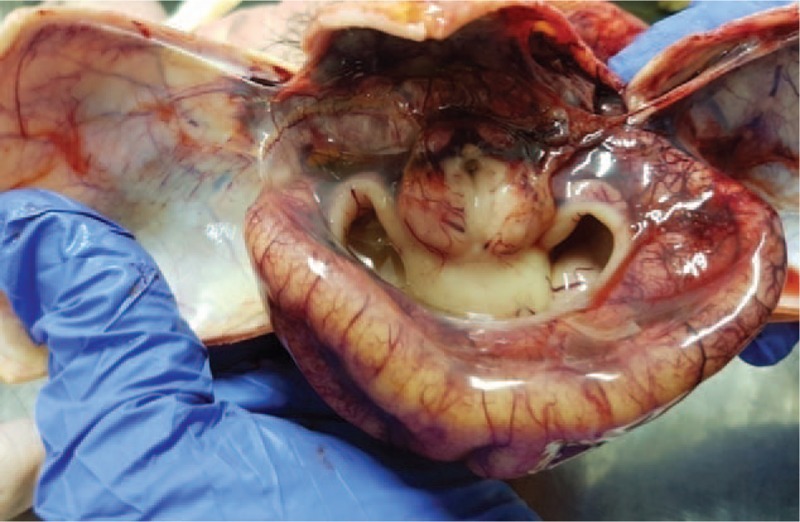
Single cerebral ventricle and absence of separate brain hemispheres.
3. Discussion
The etiology of HPE is notably heterogeneous and unclear. Incriminated factors include maternal diabetes, ethanol, cytomegalovirus infection, salicylates, antiepileptic medications, retinoic acid, aspirin, misoprostol, methotrexate, and cholesterol-lowering agents.[8–10] Association of HPE with vascular cerebral anomalies is not a rule.[8] Genetic causes have also been implicated. Approximately 18% to 25% of HPE cases have a recognizable monogenic syndrome and up to 45% of live births with HPE have nonrandom chromosomal abnormalities.[11] Other authors have shown the presence of monogenic syndrome, including the Smith–Lemli–Opitz, Pallister–Hall, and Rubinstein–Taybi syndromes and most frequently numeric anomalies in chromosomes 13 (Patau syndrome), 18 (Edwards syndrome), and 21 (Down syndrome) and structural anomalies involving 13q, 18p,7q36, 3p24-pter, 2p21, and 21q22.3.[12]
Studying HPE on a molecular level has led to the identification of the HPE genes: Sonic Hedgehog (SHH), ZIC2, and SIX3, in addition to several candidate genes.[13] To date, at least 12 loci located on 11 different chromosomes contain genes involved in HPE. For 5 of them, a minimal critical region has been identified: HPE1 at 21q22.3, HPE2 at 2p21, HPE3 at 7q36, HPE4 at 18p, and HPE5 at 13q32.[14] In 3 of the loci (HPE2, HPE3, HPE5), an HPE gene has been identified, and in a fourth loci (HPE4), a possible candidate gene is currently being studied. SHH was the first HPE gene identified by this approach. SHH was found to map to 7q36, a critical region where deletions and translocations had been reported in association with HPE.[14] About 75% of chromosomally normal patients with HPE do not have identifiable mutations in any screened genes, indicating the need to identify an additional susceptibility gene.[15]
The first prenatal diagnosis of HPE was performed by Kurtz et al in 1980.[16] Since then, a few studies and some case reports have described the different ultrasound findings of HPE.[17] The ultrasound findings of alobar HPE are characterized by a complete failure of cleavage of the prosencephalon, resulting in a single midline forebrain with a single forebrain monoventricle with cerebral hemispheres, which are completely fused into a holosphere. Usually, the interhemispheric fissure and third ventricle are absent, often resulting in a smaller than normal brain. The monoventricle usually communicates posteriorly with a dorsal cyst.[17] Microcephaly is usually present, but it must be differentiated from primary microcephaly.[18]
Fetal magnetic resonance imaging (MRI) was first described in 1990, but because of the relatively long acquisition time, its use for fetal examination was initially discouraged. With the acquisition of the ultrafast method of examination, fetal MRI has been introduced into clinical practice.[19] Fetal MRI is generally indicated when fetal ultrasound is suggestive of fetal abnormality or if the fetal abnormality requires further assessment.[19] In alobar HPE, the diagnosis can be made by ultrasonography, but a fetal MRI can be requested by a neurosonographer for a second-level examination if deemed necessary to identify other cerebral anomalies.[20]
Our case is notable, as it was diagnosed at 38 weeks of gestation because the patient did not see the doctor since 11 weeks of gestation. To our knowledge, there are only a few cases of alobar HPE diagnosed at 37 weeks gestation from different case series.[21] Moreover, the alobar HPE in our case was associated with a special and rare chromosomal abnormality, a (45X/46,XX/47,XX) with mosaicism at chromosome 18. Usually, fetuses with alobar HPE demonstrate mainly trisomy 13 and triploidy.[21]
The frequency of HPE has been estimated to be between 1:5200 and 1:16,000 live births.[1–3] Assuming a frequency of at least 1:5200 for HPE and 1:1000 for other causes of fetal ventricular dilatation, HPE represents 16% or more of all cases of fetal “hydrocephalus” detected prenatally.[12,13] A perinatal study from California detected 121 cases of HPE in 1,035,386 live births and fetal death deliveries, registered from more than 20 weeks of gestational age to 1 year after delivery with a prevalence of 1.2/10,000.[12] The study reported 56 (46%) alobar, 24 (20%) semilobar, and 11 (9%) lobar HPE cases, whereas 30 specimens had an undetermined HPE type. Another study noted that from a total of 1750 cases with fetal anomalies detected prenatally by ultrasound, 360 had anomalies of the central nervous system. Thirty fetuses were included in the study group, of HPE associated with facial anomalies. There were 18 alobar (60%), 5 semilobar (17%), and 2 lobar (6.7%) cases of HPE in this study.[21] One of the largest series included 38 cases; however, Berry et al[22] did not specify the type of HPE. Since the introduction of transvaginal ultrasound, early diagnosis of HPE in the postembryonic and embryonic phases has become more common. The earliest diagnosis of alobar HPE was made at 9 weeks gestational age, and the earliest reported diagnosis of semilobar HPE was made at 13 weeks gestational age.[23]
HPE is classified into 4 types based on the degree of nonseparation of the prosencephalon: the alobar form, characterized by diffuse cortical nonseparation; the semilobar form characterized by nonseparation of the frontal lobes; the lobar form characterized by nonseparation of the basal aspect of the frontal lobes; and the middle interhemispheric variant, characterized by nonseparation of the posterior frontal and the parietal lobes.[4,24] In our case, the fetus associated alobar HPE with cebocephaly, craniosynostosis, and microcephaly. Some of differential diagnoses considered at ultrasound were semilobar HPE and lobar HPE. Usually, in semilobar HPE, there is some degree of separation of the posterior cerebral hemispheres and a complete fusion of the anterior half of the lateral ventricles; midline structures such as the corpus callosum are visible. Lobar HPE is characterized by a hypoplastic interhemispheric fissure and the falx cerebri partially separates the 2 cerebral hemispheres. The frontal horns are hypoplastic and are partially fused at the level where the cavum septum pellucidum should be found. Moreover, alobar HPE must be distinguished from hydranencephaly, the presence of the fused thalami of a dorsal sac, and the evidence of a very thin cerebral cortex may establish a diagnosis of HPE.
The exact causes of HPE are yet to be determined. Mutations in the gene encoding SHH, which is involved in the development of the central nervous system, can cause HPE.[13,14] Increases in the expression of genes such as Pax-2, as well as the inhibition of Pax-6, from the notochord, have been implicated in the normal differentiation of cephalic midline structures. Inappropriate expression of any of these genes may result in mild to severe forms of HPE.[15] The SHH, which maps to the region 7q36, and ZIC2, which maps to the critical region in chromosome band 13q32, are genes in which mutations can cause HPE.[25] While SHH functions in the ventral neural tube, the ZIC2 message is seen in the dorsal neural tube. In SHH mutations (HPE 3), patients with ZIC2 mutations (HPE 5) do not usually demonstrate major malformations of the face.[15,17]
Examples of syndromes and associations in which HPE have been described are Meckel, Martin, Fitch, Pallister–Hall, Steinfeld, hypertelorism and ectrodactyly, velocardiofacial, pseudotrisomy, Lambotte, Genoa, Goldenhar, acrocallosal, Smith–Lemli–Opitz, Down, Patau, Edwards, Cri du chat (5p deletion), Williams (7q11.23 deletion), Di George (22q11 deletion), and Cornelia de Lange syndrome.[3,22]
The most severe of facial anomalies is cyclopia, an abnormality characterized by a single eye located in the area normally occupied by the root of the nose and a missing nose or a proboscis (a tubular-shaped nose) located above the eye. The least common facial anomaly is ethmocephaly, in which a proboscis separates closely set eyes.[2,5] The association between facial anomalies and HPE has led to the well-known phrase, “the face predicts the brain.”[26]
There are several clinical manifestations commonly observed in children with HPE who survive after birth, including the developmental delay. The degree of delay is variable, correlating with the severity of the brain malformation, but tends to be severe. Seizures are common and may be difficult to control. Approximately half of the children with HPE in a cohort study had at least 1 seizure.[27] Microcephaly is usually defined as a HC more than 2 standard deviations below the mean for age and gender. The disorder may stem from a wide variety of conditions that cause abnormal growth of the brain, or from syndromes associated with chromosomal abnormalities.[16,18] Hydrocephalus can also occur, and may result in macrocephaly, rather than the more commonly observed microcephaly. Hahn and Plawner [27] indicated that microcephaly was present in a greater proportion of patients with semilobar and lobar HPE when compared with alobar HPE, because when microcephaly was not present, hydrocephalus was usually the underlying problem and hydrocephalus was more common in alobar patients.
Hypothalamic and brain stem dysfunction may lead to swallowing difficulties and instability of temperature, heart rate, and respiration. Pituitary dysfunction may manifest by partial or complete panhypopituitarism with abnormal function of any or all of the anterior or posterior pituitary hormones, diabetes insipidus, short stature and failure to thrive, and growth hormone deficiency.[26,27] Endocrinopathies contribute significantly to the morbidity and mortality in HPE. Diabetes insipidus, owing to posterior pituitary dysfunction, is a common problem in HPE.[27] Anterior pituitary dysfunction, such as growth hormone deficiency, hypocortisolism, and hypothyroidism, are also observed, but less frequently.
Children with cleft lip and/or palate often have additional difficulties with oral feeding. Feeding and swallowing difficulties may be observed in HPE with or without cleft lip/palate. These include choking episodes and gagging during feedings, slowness in eating, and vomiting.[28] In classic HPE, the severity of the feeding difficulties correlated with the grade of HPE. Approximately two-thirds of patients with alobar and semilobar HPE required a gastrostomy tube.[28]
As the majority of cases of HPE are sporadic, routine obstetric sonography is a potentially important means of prenatal diagnosis. While karyotyping is not necessary for diagnosing HPE, knowledge of a chromosomal anomaly may influence patient counseling. The identification of chromosomal translocation is also important for further genetic evaluation of the patients. Infants with HPE who survive the newborn period have a uniformly poor outcome.
Fetal MRI provides more sensitive diagnosis for milder forms of HPE during the third trimester.[18] In a retrospective study that assessed the accuracy of neurosonography compared with MRI for major fetal nervous system anomalies, with respect to the final diagnoses, neurosonography and MRI were concordant and accurate in 109 of 126 (86.5%) cases. Additional clinically relevant findings were evident on MRI in 10 (7.9%) cases and on neurosonography in 6 (4.8%) cases.[28] Thus, this suggests that MRI had a very limited contribution to the final diagnosis if it had been requested as confirmation of the neurosonography finding, but was more valuable for additional subtle abnormalities that are more evident at MRI than at neurosonography. Also, ultrasound examination performs well for providing the correct diagnosis, but MRI might help in the diagnosis of abnormalities associated with septal agenesis. Although there is no consensus concerning the mode of delivery in cases of fetuses with HPE above 32 weeks gestation, macrocephaly could be a reason for CS.[28]
The early diagnosis of HPE is also possible; the characteristic finding in the first trimester, when evaluating the fetal head, is the presence of a midline “monoventricle” and the absence of the typical echogenic “butterfly” sign corresponding to the choroid plexuses.[29] If sonographers are unable to demonstrate this butterfly image, HPE should be highly suspected. Moreover, in specialized centers using 3D rendering inversion mode, the diagnosis can be established from 9 weeks of gestation.[29]
The management of HPE cases is challenging. Treatment by a multidisciplinary team is recommended and may include hormone replacement therapy for pituitary dysfunction, antiepileptic drugs for seizures, gastrostomy tube or fundoplication for feeding difficulties, special feeding devices and surgical repair of cleft lip and/or palate, ventriculo-peritoneal shunt placement for hydrocephalus, special attention to fluid and electrolyte balance during surgery, and parental support and counseling.[28]
In conclusion, HPE is a complex developmental brain malformation commonly associated with facial anomalies, in the past considered to be lethal but today the prognosis depends on the type of HPE and of facial anomalies associated. The best diagnostic procedure is ultrasound examination, completed eventually by MRI. There are a number of causes of HPE, including genetic alterations and environmental effects. In our case, the mother did not visit the obstetrician from 11 weeks until 38 weeks of gestation. This led to the late establishment of a severe diagnosis of alobar HPE associated with cebocephaly, microcephaly, and craniosynostosis in which the newborn postnatal survival was only 90 hours. Moreover, chromosomal analysis revealed a rare chromosomal abnormality (45X/46,XX/47,XX) with mosaicism at chromosome 18. Emotional implications would probably have been less severe if the mother underwent regular obstetric examination and diagnosis established in the first trimester.
Author contributions
Conceptualization: Cringu Antoniu Ionescu, Catalin Herghelegiu, Liana Ples.
Formal analysis: Dan Calin, Dan Navolan, Catalin Herghelegiu.
Investigation: Cringu Antoniu Ionescu, Dan Calin, Dan Navolan, Alexandra Matei, Mihai Dimitriu, Catalin Herghelegiu, Liana Ples.
Methodology: Cringu Antoniu Ionescu, Dan Calin, Dan Navolan, Alexandra Matei, Mihai Dimitriu, Catalin Herghelegiu, Liana Ples.
Supervision: Cringu Antoniu Ionescu.
Validation: Cringu Antoniu Ionescu, Dan Calin, Dan Navolan, Alexandra Matei, Mihai Dimitriu, Catalin Herghelegiu, Liana Ples.
Visualization: Cringu Antoniu Ionescu, Dan Calin.
Writing – original draft: Cringu Antoniu Ionescu, Dan Calin.
Writing – review & editing: Cringu Antoniu Ionescu.
Footnotes
Abbreviations: BPD = biparietal diameter, CS = cesarean section, HC = head circumference, HPE = holoprosencephaly, MRI = magnetic resonance imaging.
CAI and DN contributed equally to this article.
The authors report no conflicts of interest.
References
- [1].Orioli IM, Castilla EE. Epidemiology of holoprosencephaly: prevalence and risk factors. Am J Med Genet Part C Semin Med Genet 2010;154C:13–21. [DOI] [PubMed] [Google Scholar]
- [2].Demyer W, Zeman W. Alobar holoprosencephaly (arhinencephaly) with median cleft lip and palate: clinical, electroencephalographic and nosologic considerations. Confin Neurol 1963;23:1–36. [DOI] [PubMed] [Google Scholar]
- [3].Raam MS, Solomon BD, Muenke M. Holoprosencephaly: a guide to diagnosis and clinical management. Indian Pediatr 2011;48:457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Matsunaga E, Shiota K. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology 1977;16:261–72. [DOI] [PubMed] [Google Scholar]
- [5].O’Rahilly R, Müller F. Interpretation of some median anomalies as illustrated by cyclopia and symmelia. Teratology 1989;40:409–21. [DOI] [PubMed] [Google Scholar]
- [6].Winter TC, Kennedy AM, Woodward PJ. Holoprosencephaly: a survey of the entity, with embryology and fetal imaging. Radiographics 2015;35:275–90. [DOI] [PubMed] [Google Scholar]
- [7].Barkovich AJ, Quint DJ. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol 1993;14:431–40. [PMC free article] [PubMed] [Google Scholar]
- [8].Herghelegiu D, Ionescu CA, Pacu I, et al. Antenatal diagnosis and prognostic factors of aneurysmal malformation of the vein of Galen: a case report and literature review. Medicine (Baltimore) 2017;96:e7483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Edison RJ, Muenke M. Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N Engl J Med 2004;350:1579–82. [DOI] [PubMed] [Google Scholar]
- [10].Edison RJ, Muenke M. Gestational exposure to lovastatin followed by cardiac malformation misclassified as holoprosencephaly. N Engl J Med 2005;352:2759. [DOI] [PubMed] [Google Scholar]
- [11].Nanni L, Schelper R, Muenke M. Molecular genetics of holoprosenchepaly. Front Biosci 2000;5:334–42. [DOI] [PubMed] [Google Scholar]
- [12].Croen LA, Shaw GM, Lammer EJ. Holoprosencephaly:epidemiologic and clinical characteristics of a California population. Am J Med Genet 1996;64:465–72. [DOI] [PubMed] [Google Scholar]
- [13].Siegel-Bartelt J, Frumkin A, Mitchell HF, et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet 1996;14:353–6. [DOI] [PubMed] [Google Scholar]
- [14].Brown S, Warburton A, Brown D, et al. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet 1998;20:180–3. [DOI] [PubMed] [Google Scholar]
- [15].Roessler E, Ward D, Gaudenz K, et al. Cytogenetic rearrangements involving the loss of the Sonic Hedgehog gene at 7q36 cause holoprosencephaly. Hum Genet 1997;100:172–81. [DOI] [PubMed] [Google Scholar]
- [16].Kurtz AB, Wapner RJ, Rubin CS, et al. Ultrasound criteria for in utero diagnosis of microcephaly. J Clin Ultrasound 1980;8:11–6. [DOI] [PubMed] [Google Scholar]
- [17].Joó GJ, Beke A, Papp C, et al. Prenatal diagnosis, phenotypic and obstetric characteristics of holoprosencephaly. Fetal Diagn Ther 2005;20:161–6. [DOI] [PubMed] [Google Scholar]
- [18].Oprescu DN, Fetecau A, Moldoveanu A, et al. Primary microcephaly: difficulties in prenatal diagnosis and parental counselling. Proceedings 5th Romanian Congress of Ultrasound in Obstetrics Gynecology, Edited by: Vladareanu S; Marginean C; Vladareanu R, Filodiritto Editore Proceedings; Tg-Mures, Apr 20–22, 2017: 464–470. [Google Scholar]
- [19].Yuh WT, Nguyen HD, Fisher DJ, et al. MRI of fetal central nervous system abnormalities. AJNR Am J Neuroradiol 1994;15:459–64. [PMC free article] [PubMed] [Google Scholar]
- [20].Paladini D. Additional value of fetal magnetic resonance imaging in the prenatal diagnosis of central nervous system anomalies: a systematic review of the literature and related correspondence. A plea to assess oranges only. Ultrasound Obstet Gynecol 2015;45:625–6. [DOI] [PubMed] [Google Scholar]
- [21].Blass HG, Eriksson AG, Salvesen K, et al. Brains and faces in holoprosencephaly: pre- and postnatal description of 30 cases. Ultrasound Obstet Gynecol 2002;19:24–38. [DOI] [PubMed] [Google Scholar]
- [22].Berry SM, Gosden C, Snijders RJM, et al. Fetal holoprosencephaly. Associated malformations and chromosomal defects. Fetal Diagn Ther 1990;5:92–9. [DOI] [PubMed] [Google Scholar]
- [23].Blaas H-GK, Eik-Nes SH, Vainio T, et al. Alobar holoprosencephaly at 9 weeks gestational age visualized by two- and threedimensional ultrasound. Ultrasound Obstet Gynecol 2000;15:62–5. [DOI] [PubMed] [Google Scholar]
- [24].Simon EM, Hevner RF, Pinter JD, et al. The middle interhemispheric variant of holoprosencephaly. AJNR Am J Neuroradiol 2002;23:151–6. [PMC free article] [PubMed] [Google Scholar]
- [25].Gurrieri F, Trask BJ, van den Engh G, et al. Physical mapping of the holoprosencephaly minimal critical region on chromosome 7q36. Nat Genet 1993;3:247–51. [DOI] [PubMed] [Google Scholar]
- [26].DeMyer W, Zeman W, Palmer CG. The face predicts the brain: diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly). Pediatrics 1964;34:256–63. [PubMed] [Google Scholar]
- [27].Hahn JS, Plawner LL. Evaluation and management of children with holoprosencephaly. Pediatr Neurol 2004;31:79–88. [DOI] [PubMed] [Google Scholar]
- [28].Barr M, Cohen MM. Holoprosencephaly survival and performance. Am J Med Genet 1999;89:116–20. [PubMed] [Google Scholar]
- [29].Timor-Trisch IE, Monteguado A, Santos R. Three-dimensional inversion rendering in the first- and early second-trimester fetal brain: its use in holoprosencephaly. Ultrasound Obstet Gynecol 2008;32:744–50. [DOI] [PubMed] [Google Scholar]