

SUMMARY
Cellular reprogramming converts differentiated cells into induced pluripotent stem cells (iPSCs). However, this process is typically very inefficient, complicating mechanistic studies. We identified and molecularly characterized rare, early intermediates poised to reprogram with up to 95% efficiency, without perturbing additional genes or pathways, during iPSC generation from mouse embryonic fibroblasts. Analysis of these cells uncovered transcription factors (e.g., Tfap2c, Bex2), which are important for reprogramming but dispensable for pluripotency maintenance. Additionally, we observed striking patterns of chromatin hyperaccessibility at pluripotency loci, which preceded gene expression in poised intermediates. Finally, inspection of these hyperaccessible regions revealed an early wave of DNA demethylation, which is uncoupled from de novo methylation of somatic regions late in reprogramming. Our study underscores the importance of investigating rare intermediates poised to produce iPSCs, provides insights into reprogramming mechanisms, and offers a valuable resource for the dissection of transcriptional and epigenetic dynamics intrinsic to cell fate change.
IN BRIEF
Cellular reprogramming to induced pluripotent stem cells (iPSCs) is typically inefficient, complicating mechanistic analyses. Schwarz et al. use cell surface marker combinations to identify and molecularly characterize early intermediates poised to reprogram with up to 95% efficiency.
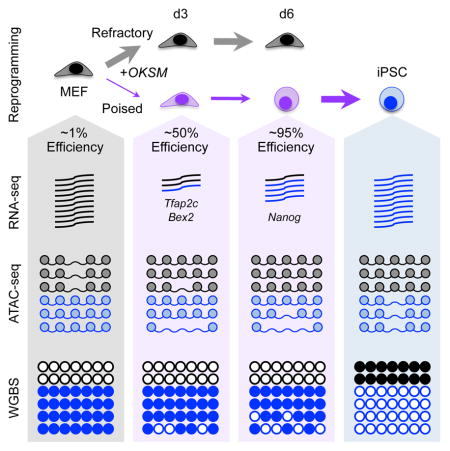
INTRODUCTION
Cellular reprogramming refers to the process by which differentiated somatic cells are converted into induced pluripotent stem cells (iPSCs) upon ectopic expression of defined transcription factor (TF) combinations, typically Oct4 (Pou5f1), Klf4, Sox2, and Myc (OKSM) (Takahashi and Yamanaka, 2006). This technology has enormous potential for regenerative medicine, disease modeling, and drug screening, as well as the study of cell fate change following forced redirection of cellular identity (Takahashi and Yamanaka, 2016). However, reprogramming is generally slow (>2 weeks) and inefficient (<1%), complicating mechanistic studies. Most cells fail to reprogram, indicating the existence of epigenetic barriers as well as the requirement for additional facilitators of this process, which remain largely unidentified. Since cells that do not contribute to successful reprogramming dominate assays that rely on bulk reprogramming cultures, it is imperative to identify and examine those select cells poised to generate iPSCs in order to gain insights into the underlying mechanisms.
Our laboratory and others previously characterized intermediate stages of reprogramming from mouse embryonic fibroblasts (MEFs) to iPSCs using surface markers (Brambrink et al., 2008; Polo et al., 2012; Stadtfeld et al., 2008). Briefly, we have shown that Thy1 is expressed by MEFs and intermediates that are refractory to reprogramming but lost by early intermediates with iPSC potential. A subset of Thy1− intermediates then upregulates SSEA-1 before activating an Oct4-GFP reporter at later time points, which coincides with the acquisition of a stable, transgene-independent pluripotent state. Although SSEA-1 expression significantly enriches for intermediates that successfully progress towards iPSCs, this population remains heterogeneous and inefficient in conventional reprogramming assays (5–10% reprogramming efficiency).
Recently, the utility of SSEA-1 as a prospective reprogramming marker has been challenged and other marker combinations such as CD44 and ICAM1 (O’Malley et al., 2013) or CD49d and CD73 (Lujan et al., 2015) have been proposed. However, the relevance of ICAM1 has only been demonstrated at a late time point using a highly efficient reprogramming system, whereas CD49d and CD73 were exclusively tested at early stages of reprogramming using an inefficient and heterogeneous retroviral reprogramming system. Thus, there is no current consensus on which markers are the most useful for isolating cells poised to produce iPSCs. Additionally, no previously reported enrichment protocol has achieved reprogramming efficiencies of greater than 10–15% for early intermediates (Lujan et al., 2015; Polo et al., 2012). In order to resolve these discrepancies, it will be critical to compare published markers and identify additional markers using the same reprogramming conditions. Furthermore, it will be important to account for the differential ability of sorted cells to survive and adhere to cell culture plates (plating efficiency) as this could profoundly skew the measure of reprogramming efficiency.
Here we validate that SSEA-1 is an early predictive marker of reprogramming progression when adjusting for plating efficiency. By systematically testing over a dozen additional markers, we found that Sca-1 and either CD73 (at d3) or EpCAM (at d6) subdivides the SSEA-1+ population and allows for the enrichment of early intermediates poised to produce iPSCs with unparalleled efficiencies of up to 95%. We finally exploit this approach to define the dynamics of transcription, chromatin accessibility, and DNA methylation patterns in those rare cells poised to produce iPSCs, revealing unexpected principles about the process of TF-induced cell fate change.
RESULTS
Plating Efficiency Profoundly Impacts Measures of Reprogramming Potential
“Three/four” (¾) MEFs were derived from mice heterozygous for (i) Col1a1-tetO-OKSM, a tetracycline inducible polycistronic 4-factor vector; (ii) Col1a1-tetO-OKSmCherry, a tetracycline inducible polycistronic 3-factor vector with a fluorescent reporter; (iii) Rosa26-M2rtTA; and (iv) an Oct4-GFP knock-in allele (Figure 1A) (Bar-Nur et al., 2014; Stadtfeld et al., 2010). This system ensures near homogenous OKSM expression, is highly reproducible, and allows for temporal control of reprogramming by adding or removing doxycycline (dox). Furthermore, the mCherry allele allows us to track expression of the OKS transgene and to differentiate reprogramming cells from mCherry− feeders. Unless otherwise specified, all reprogramming assays were performed in the presence of 15% serum, 1,000 U/mL LIF, and 50 ug/mL ascorbic acid (AA).
Figure 1. SSEA-1 Identifies Progressing Reprogramming Intermediates.

(A) Experimental Scheme.
(B) Flow analysis of ¾ MEF reprogramming. Shown are representative plots with the percentage of cells in the Thy1+ and SSEA-1+ gates.
(C) Reprogramming efficiency of Thy1+, Thy1−SSEA-1−, and SSEA-1+ populations. Shown are representative AP stains of wells exposed to dox for 9 additional days followed by a period of dox withdrawal.
(D) Reprogramming efficiency was calculated by dividing the number of AP+ dox-independent iPSC colonies by the number of cells plated for each of the indicated time points. Results are shown as the mean of 3 experiments ±1 S.D.
(E) Adjusted reprogramming efficiency was calculated by dividing reprogramming efficiencies (Figure 1D) by plating efficiencies (Figure S1B–S1F). Results are shown as the mean of 3 experiments ±1 S.D.
Following dox exposure, a subset of ¾ MEFs rapidly lose Thy1 and gain SSEA-1 expression (Figure 1B). To determine the functional significance of these markers, we employed fluorescence-activated cell sorting (FACS) to isolate Thy1+, Thy1−SSEA-1−, and SSEA-1+ intermediates. Sorted cells were then re-plated on feeders and allowed to continue reprogramming for additional days on dox. After a period of dox withdrawal, the numbers of iPSC colonies were determined by alkaline phosphatase (AP) staining to calculate reprogramming efficiencies (Figure 1A, top). We confirmed that all dox-independent AP+ colonies are Oct4-GFP+ (Figure S1A), and have previously demonstrated that AP+ colonies obtained with this system are Nanog+, and support the development of germ line chimeras (Bar-Nur et al., 2014), indicating that they represent bona fide iPSCs. Consistent with our prior results (Polo et al., 2012; Stadtfeld et al., 2008), Thy1+ cells had poor reprogramming potential at every time point and their ability to form iPSCs progressively decreased during the reprogramming time course (Figure 1C and 1D). However, contrary to our previous findings, SSEA-1+ cells were no better than Thy1−SSEA-1− intermediates until late in reprogramming.
In order to measure reprogramming efficiencies, we have to disrupt the reprogramming process by dissociating plate-adherent cells, exposing them to the high pressures of cell sorting, and then re-plate them. Few cells survive this process, which could explain our low measure of reprogramming efficiency. Furthermore, if different intermediates exhibit differential survival rates, this could greatly bias our results. In order to account for these important variables, we devised a plating efficiency assay. Briefly, defined numbers of cells were sorted onto feeders in 96-well plates and the limiting dilution (LD) of cells required to detect mCherry+ and/or Oct4-GFP+ progeny was determined for each intermediate population (Figure S1B–F). We chose LD over a single cell assay as LD is more robust, thus allowing us to more precisely assess a wider range of possible plating efficiencies, using fewer plates. Nevertheless, we confirmed that plating efficiencies calculated by LD were equivalent to those determined by sorting single cells for d3 intermediates (Figure S1G). 10,000 cells from the same sort were also transferred to 6-well plates to determine reprogramming efficiencies (Figure 1A). Adjusted reprogramming efficiencies were calculated by dividing reprogramming efficiencies by plating efficiencies for each sorted population. Correcting for plating efficiency improved the overall efficiency of live cells from ~1% to ~7% at d3 and from ~1% to ~12% at d6 of OKSM induction (Figure S1H). Critically, by accounting for plating efficiency, SSEA-1 emerges as an important marker of reprogramming progression at every examined time point (Figure 1E), confirming previous observations by our group. Furthermore, studies that concluded that SSEA-1 was not an early predictive marker of reprogramming did not assess differential plating (Lujan et al., 2015; O’Malley et al., 2013). Finally, the actual reprogramming potential of SSEA-1+ cells is remarkably high (~40% at d3 and d6). We conclude that any accurate measure of reprogramming potential must account for plating.
Systematic Analysis of Surface Markers
Next, we set out to identify additional markers that could be used in conjunction with Thy1, SSEA-1, and Oct4-GFP to define stages of reprogramming and further enrich for subsets poised to form iPSCs. For this analysis we used Col1a1-tetO-OKSMhet Rosa26-rtTAhet (het/het) MEFs with an endogenous Oct4-GFP reporter. To identify candidate markers, we performed RNA-seq on FACS-purified SSEA-1+ and SSEA-1−intermediates. We identified a number of genes encoding for cell surface proteins whose expression changes during reprogramming and which are differentially expressed between SSEA-1+ and SSEA-1− cells. We selected 16 antigens, including previously published markers, for further analysis based on the availability of commercial antibodies for flow cytometry (Figures 2A and S2A). We observed 3 major patterns of marker expression: MEF markers, which are expressed by MEFs and down-regulated in SSEA-1+ intermediates; transient markers, which are specifically induced during reprogramming but silenced in iPSCs; and iPSC markers, which are gradually induced during reprogramming and expressed by iPSCs.
Figure 2. Identification and Characterization of Reprogramming Markers.

(A) Transcriptional profiles of candidate cell-surface markers. SSEA-1− (red) and SSEA-1+ intermediates (blue) derived from het/het MEFs were analyzed by RNA-seq. Results are shown as the mean of 3 replicates.
(B) Flow analysis of het/het MEF reprogramming. Representative dot plots are gated on total live cells (red), SSEA-1+ cells (blue), or Oct4-GFP+ cells (green). Vertical lines indicate the cutoff between negative and positive staining based on an isotype-matched negative control.
(C) Quantification of the plots in Figure 2B. Lines show the percentage of total live cells (red), SSEA-1+ cells (blue), and Oct4-GFP+ cells (green) positive for the indicated markers.
(D) Schematic of reprogramming marker expression based on the results above and Figure S2.
Surface protein expression, assessed by flow cytometry, mirrored RNA expression (Figures 2A–2C and S2A–S2C). With respect to MEF markers, PDGFRβ is rapidly down-regulated in all intermediates, demonstrating that cells uniformly respond to reprogramming factors. VCAM1 and CD44 are gradually and specifically down-regulated in SSEA-1+ intermediates, with VCAM1 being lost before CD44 (Figure 2B and 2C, left). Regarding transient markers, CD73 (Nt5e), CD49d (Itga4), and Sca-1 (Ly6a) are rapidly induced and then silenced prior to Oct4-GFP induction. CD73 and Sca-1 are expressed by both SSEA-1+ and SSEA-1− cells, whereas CD49d is mainly restricted to SSEA-1+ intermediates and is more rapidly silenced (Figure 2B and 2C, center). For iPSC markers, CD71 (Tfrc) is initially induced by all cells and then becomes restricted to SSEA-1+ intermediates. EpCAM is first induced at d6 specifically in SSEA-1+ intermediates. Unlike other iPSC markers, ICAM1 is first expressed by Thy1+ cells and only late in reprogramming by Oct4-GFP+ intermediates (Figure 2B and 2C, right). Altogether, these markers identify cellular transitions during the reprogramming process and define the heterogeneity of SSEA-1+ progressing intermediates (Figure 2D).
In order for markers to have general utility, they must be applicable to other reprogramming systems and conditions. We first analyzed the effects of small molecules that increase reprogramming efficiency (AA, GSKi, Alk5i) (Bar-Nur et al., 2014; Vidal et al., 2014) (Figure S3). SSEA-1 and EpCAM gain, as well as VCAM1, Sca-1, CD24 and Podxl loss correlate with reprogramming progression, whereas other markers did not. Interestingly, ICAM1 expression appears dependent on AA. We next evaluated tail tip fibroblasts (TTFs) derived from neonatal het/het mice. Marker expression is essentially the same as that for MEFs (Figure S4A). Expression of all markers is similar between het/het MEFs and ¾ MEFs, with the exception of CD49d that is expressed more highly and for a prolonged period of time in the ¾ system (Figure S4B). These systems are similar in that they share the Stemcca (polycistronic OKSM) allele. We therefore tested reprogramming of MEFs infected with individual dox-inducible lentiviruses (LV) for the 4 factors. Remarkably, all progression markers had similar expression patterns in this system compared to het/het MEFs (Figure S4C). Finally, we analyzed another secondary system, “OSKM (Jae)”, which differs from Stemcca in the stoichiometry of the 4 factors and the Klf4 isoform (Carey et al., 2011; Kim et al., 2015). These MEFs reprogrammed more slowly and with lower efficiency. They respond to dox induction early by losing PDGFRβ and gaining Podxl, CEACAM1, and Sca-1 (Figure S4D). SSEA-1 is first detectable after d10 of reprogramming followed by subsequent expression of CD71, EpCAM, and finally c-Kit. This order of marker expression within the SSEA-1+ subset is the same as that in het/het MEFs. We conclude that progression markers are conserved among all examined systems and conditions and thus have general applicability.
Revisiting Published Markers
Next, we revisited published markers using the same reprogramming system (¾ MEFs) and accounting for plating. We first sorted d3 CD73+ or CD73− and CD49d+ or CD49d− intermediates and assessed reprogramming and adjusted reprogramming efficiencies (Figure 3A and 3B). Consistent with Lujan et al., CD73 and CD49d positivity correlate with reprogramming, yielding plating-adjusted efficiencies of 20–30%. However, this effect was less striking than that of SSEA-1+ cells, which reached efficiencies of up to 40%. Both CD73 and CD49d subdivide the SSEA-1+ population during early reprogramming. We therefore asked whether the CD73+ or CD49d+ subsets within the d3 SSEA-1+ populations further enrich for reprogramming potential. Indeed, SSEA-1+CD73+ and SSEA-1+CD49d+ subsets had higher reprogramming efficiencies than their SSEA-1+ marker-negative counterparts at d3 (Figures 3C, S5A, and S5B). However, while these marker combinations were predictive of successful reprogramming early, they tracked with cells that failed to reprogram at later time points.
Figure 3. Functional Evaluation of Published Markers.

(A) ¾ MEFs were reprogrammed for 3 days and then sorted for CD73 or CD49d. Shown are AP stains of representative wells exposed to dox for 9 additional days followed by a period of dox withdrawal.
(B) Adjusted reprogramming efficiencies of d3 CD73 and CD49d subsets derived from ¾ MEFs. Results are shown as the mean of 3 experiments ±1 S.D.
(C) Adjusted reprogramming efficiencies of the indicated SSEA-1+ subsets derived from ¾ MEF reprogramming. Results are shown as the mean of 3 experiments ±1 S.D.
(D) Flow analysis for mCherry reporter expression at d3 of ¾ MEF reprogramming. Histograms are gated on all cells (gray), marker− cells (blue), or marker+ cells (red).
(E) Adjusted reprogramming efficiencies of mCherrylow (lowest 25% of mCherry expression) and mCherryhigh (highest 25% of mCherry expression) SSEA-1+ subsets derived from ¾ MEFs. Results are shown as the mean of 3 experiments ±1 S.D.
(F) MEFs of the indicated genotypes were reprogrammed for 3 days and then analyzed by flow cytometry for SSEA-1 and CD49d expression. Results are shown as the mean of 3 experiments ±1 S.D.
(G) MEFs of the indicated genotypes were reprogrammed for 3 days and then analyzed by qRT-PCR. Results are shown as the mean of 3 experiments ±1 S.D.
(H) MEFs were infected with individual LVs for OKSM. At d3 of reprogramming, intermediates were sorted for CD49d and analyzed by qRT-PCR. Results are shown as the mean of 3 experiments ±1 S.D.
Surprisingly, CD49d and to a lesser extent CD73 correlate with mCherry levels, suggesting that these markers, unlike Thy1 and SSEA-1, predominantly reflect expression strength of the OKS transgene, similar to CD24 (Shakiba et al., 2015) (Figure 3D). To determine if transgene expression correlates with reprogramming efficiency, we sorted SSEA-1+ mCherrylow and mCherryhigh cells and measured adjusted reprogramming efficiencies (Figure 3E). At d3, higher mCherry levels tracked with increased reprogramming efficiency. However, at d6 this correlation was gone, similar to our results for CD49d and CD73 (Figure 3C and 3E). CD49d, but not CD73, was more highly expressed during the reprogramming of ¾ MEFs compared to het/het MEFs (Figures 2B, 2C, and S4B), suggesting that the higher transgene levels in the ¾ system induce more CD49d expression. To confirm this, we generated MEFs heterozygous or homozygous for Col1a1-OKSM and R26-rtTA and determined the fraction of CD49d+ cells at d3 by flow cytometry. Indeed, CD49d expression correlated with expression of the reprogramming factors (Figure 3F and 3G). Finally, we reprogrammed MEFs using individual LVs. At d3, CD49d expression again corresponded with increased reprogramming factor expression (Figure 3H). Thus, CD49d and CD73 appear to be early predictive markers of reprogramming as they enrich for cells with high OKSM levels. Consistent with this, CD73 and CD49d have been shown to be highly expressed by partially reprogrammed iPSCs, which remain addicted to high levels of exogenous reprogramming factors (Lujan et al., 2015). This property makes these markers most useful for systems exhibiting heterogeneous expression of OKSM.
ICAM1 expression has also been suggested to correlate with reprogramming potential (O’Malley et al., 2013). However, ICAM1 is expressed predominantly by Thy1+ cells, which are refractory to reprogramming (Figure 2A–C). While ICAM1 is first expressed at d6 within the SSEA-1+ population, its expression negatively correlates with adjusted reprogramming efficiencies (Figure 3C, S5A, S5B). ICAM1 is likely a late marker of reprogramming as Oct4-GFP+ intermediates are ICAM1+ (Figure 2A–C). Altogether, we have systematically compared previously described markers under identical conditions and find that the SSEA-1+ population contains the largest fraction of cells poised to form iPSCs at each time point.
Early Reprogramming Intermediates Poised to Become iPSCs with High Efficiency
The SSEA-1+ population is heterogeneous for VCAM1, Sca-1, CD71, and EpCAM expression early in reprogramming (Figure 2). Consistent with its expression pattern, VCAM1 loss within the SSEA1+ population correlates with increased adjusted reprogramming efficiency (Figure 4A, S5A, S5B). Sca-1 is a transient marker suggesting that its upregulation might enrich for cells poised to form iPSCs. Unexpectedly, SSEA-1+Sca-1+ cells are less efficient at reprogramming at every time point analyzed, implying that Sca-1 expression marks an alternative reprogramming route that fails to reach the iPSC fate whereas Sca-1− further enriches for cells poised to become iPSCs (Figure 4A, S5A, S5B). CD71 and EpCAM are both iPSC markers that correlate with reprogramming progression at each time point with EpCAM+ cells being more efficient than CD71+ cells at d6 (Figure 4A, S5A, S5B). Together, these data demonstrate that our markers enable further enrichment for cells poised to become iPSCs when combined with SSEA-1.
Figure 4. Functional Evaluation of Surface Marker Combinations.

(A) Adjusted reprogramming efficiencies of the indicated SSEA-1+ subsets derived from ¾ MEF reprogramming. Results are shown as the mean of 3 experiments ±1 S.D.
(B) Flow analysis of d3 (top) and d6 (bottom) intermediates derived from het/het MEFs. Shown are representative pseudocolor plots gated on SSEA-1+ cells for the indicated markers. Numbers indicate the percentage of cells in each quadrant.
(C) Adjusted reprogramming efficiencies of SSEA-1+ intermediates sorted for the indicated marker combinations at d3 or d6 of ¾ MEF reprogramming. Results are shown as the mean of 3 experiments ±1 S.D.
(D) Overlays of d3 (top) and d6 (bottom) Eff cells (purple) on total live cells (gray) derived from het/het MEF reprogramming. Numbers indicate the percentage of Eff cells.
(E) The indicated d6 SSEA-1+ subsets, derived from ¾ MEFs, were sorted and replated without dox. Adjusted reprogramming efficiencies are shown as the mean of 3 experiments ±1 S.D.
(F) Summary of the marker combinations used to define Eff and Ineff SSEA-1+ subsets.
We next tested combinations of the aforementioned markers. At d3 Sca-1, VCAM1, CD73, and CD49d are heterogeneously expressed within the SSEA-1+ population (Figure 4B, top). Critically, SSEA-1+CD73+Sca-1− emerged as the most efficient combination, with the robust reprogramming potential of ~50% (Figure 4C, left and S5C). At d6 many more markers are differentially expressed within the SSEA-1+ population and there are clear correlations between some of these markers (Figure 4B, bottom). For example, VCAM1+ cells are Sca-1+ and EpCAM−. Therefore, it was not necessary to sort every possible combination. Instead, we focused on EpCAM and Sca-1 expression. Remarkably, the combination of SSEA-1+EpCAM+Sca-1− resulted in an unprecedented adjusted reprogramming efficiency of ~95%, whereas the other combinations of EpCAM and Sca-1 all had comparatively low reprogramming potentials (Figure 4C, right). Both d3 efficient (SSEA-1+CD73+Sca-1−) and d6 efficient (SSEA-1+EpCAM+Sca-1−) intermediates, referred to as “Eff”, are extremely rare comprising ~0.3% of all cells at d3 and ~0.1% of total cells at d6 (Figure 4D).
To determine if d3 Eff cells preferentially give rise to d6 Eff cells, we sorted d3 Eff and d3 “Ineff” cells (SSEA-1+CD73−Sca-1+) and analyzed them after 3 additional days on dox. Indeed, d3 Eff cells gave rise to ~10 times more EpCAM+Sca-1− cells than d3 Ineff cells (Figure S5D), suggesting a direct progression from d3 Eff to d6 Eff cells. At d6, Eff cells are the only intermediates that can generate iPSCs without further dox exposure (Figure 4E); however, this efficiency is extremely low, indicating that the majority of these cells are not yet stably reprogrammed. Finally, all the reprogramming systems we analyzed converge on this SSEA-1+EpCAM+Sca-1− intermediate (Figure S5E). These cells arise prior to endogenous Oct4-GFP or Sox2-GFP expression and almost all GFP+ cells are EpCAM+Sca-1−. In summary, we have dissected the heterogeneity of SSEA-1+ intermediates using additional markers and identified the most critical subsets at d3 and d6 (Figure 4F). These intermediates are poised to undergo successful reprogramming at unparalleled efficiencies of up to 50% at d3 and 95% at d6.
Somatic Extinction Precedes Pluripotency Induction in Poised Intermediates
Poised intermediates provides a unique tool to dissect the mechanisms of successful reprogramming. We therefore compared d3 Eff and d6 Eff cells molecularly to corresponding d3 Ineff and d6 Ineff (SSEA-1+EpCAM−Sca-1+) intermediates as well as the starting MEFs and resulting iPSCs by RNA-seq, Assay for Transposase-Accessible Chromatin (ATAC)-seq, and whole genome bisulfite sequencing (WGBS) (Figure 4F). We initially focused on transcriptional analyses to define gene expression patterns that may account for the striking differences in reprogramming potential. Multidimensional scaling (MDS) illustrates a clear trajectory of transcriptional changes that delineates the successful path to reprogramming (Figure 5A). Of note, d6 Ineff intermediates appear stalled and fail to progress beyond d3 intermediates, whereas d6 Eff cells proceed towards iPSCs. Consistent with Polo et al., we observed two waves of transcriptional changes: from MEFs to d3 SSEA-1+ cells and from d6 SSEA-1+ cells to iPSCs (Figure 5B and Table S1). Importantly, d3 Eff cells were more different from MEFs than d3 Ineff cells, whereas d6 Eff cells were more similar to iPSCs than d6 Ineff cells, suggesting that Eff intermediates are more effective at silencing the somatic program at d3 and activating the pluripotency program at d6.
Figure 5. Transcriptional Analysis of Eff Intermediates Reveals Unexplored Regulators of Reprogramming.

(A) MDS analysis of global RNA-seq data for the indicated populations, derived from ¾ MEFs, in duplicate. Numbers indicate the reprogramming time point of intermediates. The dotted arrow shows the proposed trajectory of transcriptional changes.
(B) Quantification of DEGs that are up-regulated (red) or down-regulated (green) for the indicated transitions (Table S1).
(C) Venn diagram showing the overlap of d3 DEGs (comparing d3 Ineff and d3 Eff cells) and d6 DEGs (comparing d6 Ineff and d6 Eff cells) (Table S2).
(D) Heatmaps with hierarchical clustering of the 6 populations in duplicate are shown for d3 DEGs and d6 DEGs.
(E) Gene expression levels determine by RNA-seq for TFs more highly differentially expressed by Eff compared to Ineff cells at both d3 and d6. Results are shown as the mean of two replicates.
(F) The indicated genes were targeted by siRNA transfection of ¾ MEFs at d0, d3, d6, and d9 of reprogramming, followed by 3–5 days of dox withdrawal. Results are shown as the mean of 5 experiments ±1 S.D. and normalized to a Luciferase (Luc) siRNA control. Statistical differences compared to Luc were determined by the unpaired Student’s t-test, * p<0.05, ** p<0.005, *** p<0.0005.
(G) Tfap2cfl/fl MEFs were infected with LV-Stemcca, rtTA, and either Cre or a Puromycin (Puro) control vector. After 15 days of reprogramming, cells were withdrawn from dox for 3–5 days and then stained for AP. Results are shown as the mean of 3 experiments ±1 S.D. Statistical differences were determined by the unpaired Student’s t-test, *** p<0.0005.
(H) Het/het MEFs were infected with LV-Tfap2c, Bex2, or a Puro control. After 12 days of reprogramming, cells were withdrawn from dox for 3–5 days and then stained for AP. Results are shown as the mean of 3 experiments ±1 S.D. Statistical differences were determined by the unpaired Student’s t-test, ** p<0.005, *** p<0.0005.
(I) Flow analysis for reprogramming of Tfap2c OE and uninfected control MEFs. Shown are representative pseudocolor plots for d6. The percentages of cells in the indicated regions are shown.
(J) Schematic of the targeting strategy to generate Utf1-GFP ESCs.
(K) Flow analysis for reprogramming of Utf1-GFP reporter MEFs. Shown are representative pseudocolor plots gated on total live cells (top) and GFP− or GFP+ intermediates (bottom). The percentages of cells in the indicated regions are shown.
(L) Flow analysis for reprogramming of Bex2-GFP reporter MEFs. Shown are representative pseudocolor plots for d6. Plots are gated on total live cells (top) and GFP− or GFP+ intermediates (bottom). The percentages of cells in the indicated regions are shown.
(M) Reprogramming efficiency of SSEA-1+Bex2-GFP− and Bex2-GFP+ intermediates.
Comparing Eff with Ineff intermediates, we detected 264 differentially expressed genes (DEGs) at d3 and 2,209 DEGs at d6 (Figure 5B, 5C, and Table S2). Most d3 DEGs (190) overlap with d6 DEGs, suggesting a progression of transcriptional changes. Hierarchical clustering based on d3 DEGs segregates MEFs from all other samples, indicating that d3 DEGs are driven by genes that distinguish reprogramming intermediates and iPSCs from MEFs (Figure 5D, left). By contrast, d6 DEGs cluster d6 Eff intermediates with iPSCs, indicating that d6 Eff but not Ineff cells have initiated an iPSC-specific transcriptional program (Figure 5D, right).
Cell surface markers differentially expressed between Eff and Ineff intermediates (Figure S6A) include genes for CD73, EpCAM, and Sca-1, validating our sorting strategy. Although Ineff and Eff cells are both SSEA-1+, Fut9, which encodes for the enzyme that produces SSEA-1, was more highly expressed by Eff intermediates. Consistent with this observation, SSEA-1 levels positively correlate with adjusted reprogramming efficiencies (Figure S6B). Collectively, these results demonstrate that early transcriptional changes in the select cells poised to successfully reprogram are driven first by effective extinction of the somatic program at d3 and subsequently by activation of pluripotency loci at d6.
Identification of TFs Important for the Acquisition of Pluripotency
We next focused on differentially expressed TFs as these may drive differences in reprogramming potential. Several known pluripotency factors were more highly expressed by d6 Eff compared to d6 Ineff cells including Nanog, Prdm14, Lin28, Zscan10, Zfp42, etc. (Table S2). However, only 11 TFs were more highly expressed in Eff relative to Ineff cells at both d3 and d6. We selected 8 for siRNA suppression (Figure 5E and 5F). Small interfering RNAs targeting Myb, Utf1, Bex2, Tfap2c, and Nr0b1 significantly reduced reprogramming efficiencies compared to Luciferase control. Nr5a2, which can replace Oct4 in reprogramming (Heng et al., 2010), had no effect in our assay, which may be due to functional redundancy. Nr0b1 and Utf1 have established roles in reprogramming (Buganim et al., 2012; Lujan et al., 2015), whereas Bex2, Tfap2c, and Myb may be novel regulators of induced pluripotency.
Tfap2c (aka Tcfap2c) encodes for AP-2γ. To validate its functional importance, we reprogrammed Tfap2cfl/fl MEFs (Schemmer et al., 2013) with LV-Stemcca. Deletion of Tfap2c with LV-Cre resulted in a profound reduction in reprogramming potential (Figure 5G). Conversely, overexpression (OE) of Tfap2c increased reprogramming efficiency (Figure 5H, left) and this effect was most pronounced in the absence of AA (Figure S6C) (Polo et al., 2012). Furthermore, Tfap2c OE resulted in a striking increase in the fraction of Eff intermediates as well as Oct4-GFP+ cells at d6 (Figure 5I). To corroborate the functional role of Bex2 in reprogramming, we infected Bex2 KO or littermate control MEFs (Ito et al., 2014) with LV-Stemcca. Contrary to our siRNA results, we found no difference in the number of iPSC-like colonies (Figure S6D). We surmise that the high levels of OKSM delivered by LV-Stemcca compensate for the lack of Bex2 during reprogramming, but cannot exclude other possibilities including compensation by other BEX factors in the knockout or off-target effects of the siRNA. In further agreement with a functional role of Bex2 during reprogramming, its OE in het/het MEFs greatly improved reprogramming efficiency (Figure 5H, right). Finally, we confirmed that established Tfap2c and Bex2 KO iPSC clones appear normal (Figure S6E), consistent with reports indicating that both genes are dispensable for embryonic stem cell (ESC) maintenance (Auman et al., 2002; Ito et al., 2014; Schemmer et al., 2013). We conclude that Tfap2c is critical for reprogramming whereas Bex2 enhances reprogramming but is not absolutely required.
To assess whether expression of these TFs corresponds with the emergence of rare intermediates identified by our surface markers, we analyzed reporters for Utf1 and Bex2. Using CRISPR/Cas9 targeting, we generated Utf1-GFP knock-in reporter ESCs and derivative het/het reprogrammable MEFs (Figure 5J). Although there were no GFP+ cells at d3 of reprogramming, we detected rare GFP+ cells at d6 all of which had the immunophenotype of d6 Eff cells (Figure 5K). We next reprogrammed Bex2-GFP MEFs (Ito et al., 2014) and observed a sizable fraction of GFP+ cells by d6, most of which exhibited the surface markers of d6 Eff (Figure 5L). Significantly, SSEA-1+Bex2-GFP+ cells were more efficient than GFP− cells at generating iPSCs (Figure 5M), confirming that Bex2 expression marks cells poised to reprogram. In summary, we have identified regulators of reprogramming by comparing the transcriptional profiles of Eff and Ineff SSEA-1+ subpopulations. The majority of these genes were either not detected at all or only at later stages of reprogramming when analyzing bulk cultures or enriching intermediates solely based on SSEA-1 (Mikkelsen et al., 2008; Polo et al., 2012), highlighting the importance of our high-resolution characterization of progressing intermediates.
Rapid Rewiring of Chromatin States During Reprogramming
OKS act as pioneer factors that initiate cellular reprogramming by binding to closed regions of chromatin resulting in nucleosome displacement and chromatin remodeling (Soufi et al., 2012). However, as previous studies analyzed bulk cultures which are dominated by cells that fail to reprogram, chromatin changes specific to those rare cells poised to form iPSCs remain unknown. We therefore performed ATAC-seq, which globally quantifies chromatin accessibility (Buenrostro et al., 2013), using our highly enriched intermediates (Figure 4F). Principle component analysis (PCA) reveals a rapid change in chromatin structure following OKSM induction, evident in both d3 Ineff and Eff cells relative to MEFs, whereas d6 intermediates are more closely related to iPSCs (Figure 6A). This implies that OKSM facilitates transient changes to chromatin accessibility regardless of whether cells progress or stall during reprogramming.
Figure 6. Eff Intermediates Rapidly Acquire Regions of Chromatin Hyperaccessibility.

(A) ATAC-seq was performed on the indicated populations in duplicate. Shown is a PCA based on peaks identified in MEFs. Numbers indicate the reprogramming time point of intermediates. The dotted arrow indicates the proposed trajectory of chromatin accessibility changes during reprogramming.
(B) Overlap of ATAC-seq peaks between populations. The percentage of total peaks for every possible combination of overlap is shown. Blue bars represent the presence of peaks in the indicated population(s), whereas white denotes the absence of peaks.
(C) Venn diagram showing the overlap of d3 DARs (comparing d3 Ineff and d3 Eff cells) and d6 DARs (comparing d6 Ineff and d6 Eff cells) and DARs between MEFs and iPSCs (Table S3).
(D) Heatmaps with hierarchical clustering shown for the indicated sets of DARs from Figure 6C. White bars indicate regions of closed chromatin and darker bars indicate regions with greater chromatin accessibility.
(E) Correlation between gene expression, chromatin accessibility, and TF binding. Regions were selected from Intersection #2 (576 ATAC-seq peaks, Figure 6C) and associated with the closest gene (Table S3). Shown are heatmaps for RNA-seq (left); ATAC-seq (middle); and published ChIP-seq for OKS binding in ESCs (Chronis et al., 2017) (right). For genes highlighted in blue, chromatin accessibility precedes transcription.
(F) Genome browser view of RNA-seq (red) ATAC-seq (blue) and ChIP-seq (orange) for the region adjacent to Mybl2. Boxes denote areas that are more differentially open by ATAC-seq in d3 Eff, d6 Eff, and iPSCs compared to d3 Ineff, d6 Ineff, and MEFs, respectively.
We next analyzed the overlap of ATAC-seq peaks between different populations (Figure 6B). MEF-specific regions are rapidly closed following reprogramming initiation, whereas a large fraction of ectopic peaks (not open in MEFs or iPSCs) are induced in all intermediates. While most iPSC-specific regions remain closed, “early iPSC” regions are rapidly induced. Significantly, we detected more of these regions in Eff than in Ineff intermediates at both d3 and d6 (Figure 6B, inset). Furthermore, d3 Eff cells undergo chromatin closure for a greater number of MEF regions than d3 Ineff cells (Figure S6F, left). By contrast, d6 Eff cells have more open iPSC regions than d6 Ineff intermediates (Figure S6F, right). We conclude that early changes in chromatin accessibility are driven by silencing of the somatic program at d3 followed by induction of pluripotency regions at d6, in agreement with our transcriptional analysis.
Critical Pluripotency Regions are Hyperaccessible in Poised Intermediates
To narrow down ATAC-seq sites of biological relevance, we considered only differentially accessible regions (DARs) that are open in MEFs (MEF>iPSCs) and remain open in d3 Ineff cells (d3 Ineff>d3 Eff) and d6 Ineff cells (d6 Ineff>d6 Eff), yielding 98 regions (Figure 6C, left, and Table S3). Notably, these regions are more open in d3 Ineff intermediates relative to MEFs and are completely closed in d6 Eff cells and iPSCs, suggesting that their closure is critical for successful reprogramming (Figure 6D, left). Alternatively, 576 DARs are more open in iPSCs (iPSC>MEF), d3 Eff cells (d3 Eff>d3 Ineff), and d6 Eff cells (d6 Eff>d6 Ineff) (Figure 6C, right, and Table S3). Surprisingly, Eff intermediates showed increased accessibility compared to iPSCs for these regions (Figure 6D, right), suggesting that a transient hyperaccessible chromatin state at these sites is important for successful reprogramming. Hyperaccessibility had not been previously detected in the analysis of bulk reprogramming cultures or SSEA-1+ intermediates (Chronis et al., 2017; Knaupp et al., 2017; Li et al., 2017) highlighting the importance of our high-resolution characterization of poised reprogramming subsets.
The 576 DARs more open in iPSCs and Eff intermediates include hyperaccessible peaks adjacent to a number of key pluripotency genes (Figure 6E and Table S3). These encompass genes more highly expressed by Eff cells at both d3 and d6 (Bex2, Tfap2c, Nr0b1, Nr5a2, Mycn) as well as genes not expressed until d6 or later (Prdm14 and Zscan10). Of particular interest was the region adjacent to Dppa3 (Figure S7A), which maps to a known super-enhancer (−45 SE) that controls the expression of both Dppa3 and Nanog (Blinka et al., 2016). Despite the apparent activation of this enhancer as early as d3, Nanog is not expressed until d6, specifically by Eff intermediates, and Dppa3 is not expressed until even later. This observation therefore implies that changes to chromatin accessibility, particularly hyperaccessibility, can precede and may be causal to changes in gene expression.
To ascertain which TFs might bind to the 576 DARs more accessible in Eff intermediates and iPSCs, we performed TF motif enrichment analysis. Top hits included Klf4 (p=7.16×10−60) and Oct4/Sox2 (p=1.09×10−53), suggesting that these regions are enriched for direct binding sites of the reprogramming factors. Thus, hyperaccessibility might be due to superphysiological levels of OKS. However, Eff intermediates expressed less or equal amounts of OKS compared to endogenous levels in iPSCs, arguing against this possibility (Figure S6G). To verify OKS binding to these hyperaccessible loci, we analyzed ESC ChIP-seq data for Oct4, Sox2 and Klf4 (Chronis et al., 2017). Most of the hyperaccessible sites were indeed bound by OKS (Figures 6E and S6H). Furthermore, 70% of these sites were occupied by a combination of Oct4 and Sox2 ± Klf4, suggesting that cooperative binding may be responsible for hyperaccessibility. Examples include two peaks upstream of Mybl2, the peak between Dppa3 and Nanog, and a peak adjacent to Tfap2c (Figures 6F, S7A, and S7B). However, we also identified rare DARs not bound by any reprogramming factors, such as the promoter region of Bex2 (Figures 6E, S6H, and S7C). Motif analysis for this peak suggested a Tfap2c binding site, which was confirmed by published ChIP-seq data (Park et al., 2015). We conclude that regions of hyperaccessibility, revealed only by dissecting the rare intermediates poised to reprogram, identify key cis-regulatory elements critical for reprogramming.
DNA Methylation and Demethylation are Uncoupled During Reprogramming
DNA methylation provides another layer of epigenetic regulation. Reprogramming requires demethylation of pluripotency promoters and enhancers, while MEF-specific cis-regulatory elements are remethylated (Koche et al., 2011). Previous studies on bulk or SSEA-1+ intermediates suggest that DNA methylation changes occur late in reprogramming (Knaupp et al., 2017; Lee et al., 2014; Mikkelsen et al., 2008; Milagre et al., 2017; Polo et al., 2012). To evaluate the dynamics of DNA methylation in our poised intermediates, we performed WGBS (Figure 4F). Globally, MEFs and intermediates have similar DNA methylation levels, whereas iPSCs are relatively hypermethylated and cluster separately (Figure 7A), consistent with previous observations (Lee et al., 2014; Milagre et al., 2017; Polo et al., 2012). For regions that become methylated specifically in iPSCs, there were only subtle differences among early intermediates (Figure 7A, clusters 1–8), confirming that de novo DNA methylation is a late event. Consistent with this, DNMT3A/B, which catalyze de novo DNA methylation, are not required for reprogramming (Pawlak and Jaenisch, 2011). For regions that are hypomethylated in iPSCs relative to MEFs, we detected two clusters that undergo either immediate or delayed demethylation (Figure 7A, clusters 9 and 10), implying two separable waves of demethylation. Cluster 9 contains DNA regions that are demethylated late in reprogramming and includes regions closest to the promoters of Zfp42, Dppa2, and Dppa4, whereas Cluster 10 is composed of regions that are rapidly demethylated including areas adjacent to Oct4, Klf4, and Nanog, revealing a previously unexplored early wave of demethylation (Table S4). Importantly, d6 Eff cells were more demethylated than the other reprogramming intermediates (Figure 7A), linking DNA demethylation of specific loci with increased reprogramming efficiency. In support of this observation, PCA for regions in these two clusters demonstrates a clear trajectory of DNA methylation changes during reprogramming (Figure 7B).
Figure 7. DNA Methylation and Demethylation are Uncoupled During Reprogramming.

(A) WGBS was performed on the indicated populations. Shown is a heatmap of 1 kb tiling regions that show a change of at least 30% between MEF and iPSCs as measured by WGBS. Regions were grouped into ten clusters based on hierarchical clustering.
(B) PCA of WGBS data using 1 kb DNA tiles that are hypomethylated in iPSCs relative to MEFs (Clusters 9 and 10, Figure 7A). Numbers indicate the reprogramming time point of intermediates. The dotted arrow indicates the proposed trajectory of DNA methylation changes during reprogramming.
(C) Violin plots of global DNA methylation for the indicated regions. Plots show the distribution of methylation values for each sample. White circle: median methylation value. Black box: interquartile range. Whisker extend to the most extreme data point which is no more than 1.5 times the interquartile range from the box.
(D) Heatmaps showing DNA methylation levels at ESC enhancers (Shen et al., 2012) that are hypermethylated in MEFs compared to iPSCs (left) and MEF enhancers that are hypomethylated in MEFs compared to iPSCs (right).
(E) DNA methylation levels for key ESC enhancers differentially methylated at d3 (top) or d6 (middle) of reprogramming (Table S5).
(F) Gene expression levels, determine by RNA-seq, for Tet1 and Tet2. Results are shown as the mean of two replicates (Red line: Ineff; Purple line: Eff).
(G) ¾ MEFs were transfected with siRNA targeting Tet2 or a Luc control at d0 and d3 of reprogramming. At d3 (orange) or d6 (green) the percentage of SSEA-1+ cells was determined by flow cytometry. Results are shown as the mean of 3 experiments ±1 S.D. Statistical differences were determined by the unpaired Student’s t-test, ** p<0.005.
(H) Model of chromatin changes observed in Eff and Ineff intermediates during reprogramming.
We assumed the earliest regions of chromatin opening would be at areas of DNA hypomethylation in the starting MEFs, allowing for rapid binding of reprogramming factors (Koche et al., 2011). Surprisingly, DARs more accessible in d3 Eff, d6 Eff, and iPSCs are hypermethylated in MEFs yet rapidly demethylated at d3, further demethylated at in d6 Eff cells, and remain demethylated in iPSCs (Figure 7C, middle). DARs more open in MEFs, d3 Ineff, and d6 Ineff cells are hypomethylated in MEFs and undergo methylation only late in reprogramming (Figure 7C, right). Next, we analyzed sets of annotated enhancers defined in ESCs or MEFs (Shen et al., 2012). For ESC enhancers, d3 intermediates remain predominantly hypermethylated and cluster with MEFs, whereas d6 intermediates are hypomethylated and cluster with iPSCs, with d6 Eff being the most demethylated (Figure 7D, left). Specific differentially methylated regions (DMRs) less methylated in d3 Eff compared to Ineff cells include enhancers for Oct4, Lin28b, Nodal, Tfcp2l1, and Tfap2c, whereas DMRs less methylated in d6 Eff compared to Ineff cells include enhancers for Epcam, Cdh1, Sall1, Sall4, and Tfap2c (Figure 7E and Table S5). Of note, endogenous Oct4 is not expressed until d9 (Figure 2B and 2C), implying that specific demethylation of enhancers in Eff intermediates can precede transcription. For MEF enhancers that are methylated in iPSCs, all intermediates were hypomethylated, further demonstrating that de novo methylation is not required for the silencing of MEF genes early in reprogramming (Figure 7D, right).
DNA demethylation is mediated, in part, by TET enzymes (Koh et al., 2011). Tet2, but not Tet1, is upregulated early in reprogramming specifically in Eff intermediates (Figure 7F). To determine whether Tet2 plays a functional role in the formation of Eff intermediates, we transfected cells with siRNA targeting Tet2. While we observed no effect at d3, we detected a significant reduction in SSEA-1+ reprogramming intermediates at d6, implying an important role for Tet2 in reprogramming between d3 and d6, coinciding with the early wave of DNA demethylation we detected (Figure 7G). We conclude that DNA methylation and demethylation are uncoupled during reprogramming. Additionally, our analysis of highly enriched poised reprogramming intermediates has allowed us to uncover a previously unobserved early wave of DNA demethylation.
DISCUSSION
Here we describe cell surface marker combinations, allowing us to prospectively isolate and characterize rare intermediates poised to generate iPSCs with unprecedented efficiencies in the absence of additional treatments. Molecular dissection of these key intermediates elucidates transcriptional and epigenetic events specific to early stages of TF-induced pluripotency (Figure 7H). Altogether, our study provides a valuable resource of surface markers to delineate the various stages of cellular reprogramming and associated transcriptional, chromatin accessibility, and DNA methylation patterns. Our results highlight the importance of controlling for differential plating. We found that reprogramming intermediates are particularly sensitive to the stress of dissociation and cell sorting, resulting in very poor re-plating potentials. This most likely masked the importance of EpCAM as a marker of reprogramming progression in prior studies (Lujan et al., 2015; Polo et al., 2012). Remarkably, our data show that EpCAM expression, in combination with SSEA-1 expression and Sca-1 loss, enriches for select intermediates poised to reprogram with nearly 95% efficiency. Furthermore, our study resolves controversies regarding the utility of various cell surface markers to track reprogramming intermediates (Brambrink et al., 2008; Lujan et al., 2015; O’Malley et al., 2013; Stadtfeld et al., 2008). We surmise that these discrepancies are likely due to the use of distinct reprogramming systems. Importantly, our study compares markers in parallel across distinct reprogramming systems and conditions. We confirm that SSEA-1 is an early and robust marker of reprogramming progression. Likewise, we corroborate CD73 and CD49d as early markers. However, instead of signifying progression, these markers correlate with exogenous reprogramming factor expression. Furthermore, CD73 and CD49d switch to negatively predictive markers later in reprogramming, limiting their general utility. Surprisingly, we find that expression of ICAM1 is dependent on AA and is first induced in reprogramming-refractory intermediates and thus may not be a predictive marker until late time points.
Transcriptional analysis reveals that Tfap2c and Bex2 are induced specifically in Eff intermediates by d3, representing two of the earliest transcriptional regulators of successful reprogramming. Intriguingly, both genes are adjacent to cis-elements that become hyperaccessible exclusively in Eff intermediates. Furthermore, enhancers of Tfap2c become specifically demethylated in Eff cells. Consistent with a functional role, Tfap2c KO MEFs are significantly impaired in the formation of iPSCs whereas Tfap2c OE augments reprogramming. Bex2, on the other hand, is dispensable for reprogramming with high levels of OKSM, but OE increases reprogramming potential. Additionally, activation of endogenous Bex2 correlates well with cells poised to produce iPSCs. Although Bex2 and Tfap2c are both expressed in pluripotent cell lines, neither is required for ESC self-renewal (Auman et al., 2002; Ito et al., 2014; Schemmer et al., 2013). Our observations provide the basis for future studies aimed at dissecting the mechanisms by which these underexplored TFs specifically facilitate the acquisition but not maintenance of pluripotency.
Our study uncovers that ectopic chromatin accessibility as well as chromatin hyperaccessibility are previously unrecognized hallmarks of successful reprogramming. These data suggest that adoption of an ESC-like chromatin state is insufficient to acquire a stable iPSC state. Instead, a genome-wide increase in chromatin accessibility, combined with focused hyperaccessibility at critical pluripotency loci, correlates with successful reprogramming in our system. Further work remains to determine how these changes in chromatin accessibility correlate with remodeling of histone marks and rewiring of 3D chromatin architecture, both of which can precede transcription (Apostolou et al., 2013; Polo et al., 2012), similar to what we found for hyperaccessibility.
Finally, our study redefines the molecular dynamics of DNA methylation changes during TF-induced cellular reprogramming. Global analysis of our highly enriched intermediates revealed no overall change in DNA methylation levels. However, when focusing specifically on regions that are demethylated in iPSCs, we were able to uncover two distinct waves of demethylation, including a previously unappreciated early wave most evident in our Eff intermediates. Alternatively, de novo methylation of somatic regions occurs only late, demonstrating that demethylation and remethylation are uncoupled during reprogramming. Furthermore, a comparison of ATAC-seq with WGBS data revealed that regions of chromatin that are hyperaccessible in Eff intermediates are targeted for rapid DNA demethylation, directly linking these two epigenetic processes. We conclude that key molecular changes occur very early in reprogramming but are typically obscured by the overwhelming majority of cells that do not effectively contribute to the generation of iPSCs.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Konrad Hochedlinger (hochedlinger@molbio.mgh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Care
All mice used in the study were housed and bred in the Center for Comparative Medicine at Massachusetts General Hospital AAALAC-accredited mouse facility in Specific Pathogen Free (SPF) rooms. All procedures involving mice adhered to the guidelines of the approved Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC) protocol #2006N000104.
Fibroblast Derivation
Col1a1-tetO-OKSMhomo Oct4-GFPhomo mice were mated with either R26-rtTAhomo or Col1a1-tetO-OKSmCherryhomo R26-rtTAhomo mice to generate Col1a1-tetO-OKSMhet R26-rtTAhet Oct4-GFPhet (het/het) or Col1a1-tetO-OKSM/OKSmCherry R26-rtTAhet Oct4-GFPhet (¾) embryos, respectively. Het/het mice were mated to each other to generate Col1a1-tetO-OKSMhomo R26-rtTAhomo and Col1a1-tetO-OKSMhet R26-rtTAhomo embryos. Col1a1-tetO-OSKM(Jae)homo R26-rtTAhomo mice (Carey et al., 2011) were mated with Oct4-GFPhomo mice to generate Col1a1-tetO-OSKM(Jae)het R26-rtTAhet Oct4-GFPhet embryos. DR4 mice were bred with Balb/c to generate DR4 embryos. Sox2-GFP mice were mated with R26-rtTA mice to generate R26-rtTAhet Sox2-GFPhet embryos. Bex2-GFPhet (a Bex2 knockout reporter allele) females were mated with wild-type males to generate Bex2GFP/Y and littermate wild-type control embryos (Ito et al., 2014). Tfap2cfl/fl embryos were also generated (Schemmer et al., 2013). Embryos were harvested at E13.5–15.5, the head and internal organs were removed, and the remaining tissue was chopped and dissociated with trypsin to isolate MEFs. Tail tips of neonatal het/het mice were chopping and dissociating with trypsin to isolate TTFs.
METHOD DETAILS
Cell Culture and Reprogramming
MEFs were maintained in MEF medium [DMEM (Invitrogen) supplemented with L-glutamine, penicillin/streptomycin, nonessential amino acids, β-mercaptoethanol, and 10% FBS (Invitrogen)] and expanded to p3 or p4 prior to reprogramming. TTFs were expanded in MEF medium to p1 prior to reprogramming. DR4 MEFs were expanded to p3 or p4 and then irradiated (3,000 rads) to generate feeders. Fibroblasts were reprogrammed at low density on gelatin-coated cell culture plates, whereas sorted cells were plated on gelatin with irradiated DR4 feeder MEFs. Reprogramming experiments were performed in ESC medium [KO-DMEM (Invitrogen) with L-glutamine, penicillin/streptomycin, nonessential amino acids, β-mercaptoethanol, 1,000 U/mL LIF, and 15% FBS (Invitrogen)] supplemented with 1 ug/mL of doxycycline (dox) and 50 ug/mL of ascorbic acid (AA), unless indicated otherwise. For specific experiments, 3 uM GSKi (CHIR-99021, Tocris) or 1 uM Alk5i (EMD-616452, Calbiochem) were added to the ESC medium. Reprogramming intermediates derived from het/het MEF were used for the characterization of surface marker expression and the corresponding RNA-seq analysis. All adjusted reprogramming efficiency assays, RNA-seq of Eff and Ineff cells, ATAC-seq, and WGBS were done using reprogramming intermediates derived from ¾ MEFs. Established ¾ iPSCs were cultured in ESC medium and analyzed at p10 for molecular studies.
Flow Cytometry and Cell Sorting
MEFs, reprogramming intermediates, or iPSCs were dissociated with trypsin. For analysis of trypsin sensitive antigens (CD44, E-cadherin, and PECAM1) EDTA was used instead. Cells were then stained with combinations of the following antibodies: anti-mouse Thy1.2 (53-2.1), SSEA-1 (MC-480), PDGFRβ (APB5), VCAM1 (429), CD44 (IM7), CD73 (eBioTY/11.8), CD49d (R1-2), Sca-1 (D7), CD71 (R17217), EpCAM (G8.8), ICAM1 (eBioKAT-1), CD24 (M1/69), Prom1 (13A4), Podxl (FAB1556P), CEACAM1 (CC1), E-Cadherin (DECMA-1), c-Kit (2B8), PECAM1 (390), or an isotype control (eBR2a), all directly conjugated to phycoerythrin (PE), PE-Cy7, eFluor 450, or eFluor 660. DAPI was used for dead cell exclusion. For molecular analyses, ¾ iPSCs were sorted for Oct4-GFP, to eliminate contamination with differentiating cells. Flow cytometry was performed on a LSR-II (BD) and cell sorting was performed on a FACSAria-II (BD). Analysis was done with FlowJo software.
Assay for Adjusted Reprogramming Efficiency
To determine reprogramming efficiency, 10,000 sorted cells were plated in individual wells of 6-well dishes and exposed to dox for 0, 3, 6, 9, or 12 additional days. Dox was then withdrawn for at least 3 days prior to alkaline phosphatase (AP) staining (Vector Laboratories). AP+ dox-independent iPSCs were counted manually. Reprogramming efficiency was calculated as the number of iPSC colonies divided by the number of cells plated. To determine plating efficiency, cells from the same sort were sorted directly into 96-well plates with 24 wells each of 5 cells, 10 cells, 20 cells, and 40 cells. These wells were then exposed to dox for 4 additional weeks. Each well was assessed for Oct4-GFP+ colonies by inverted fluorescent microscopy. Cells were then dissociated with trypsin and plates were analyzed by flow cytometry on a MACSQuant (Miltenyi). Each well was scored individually as Oct4-GFP+, mCherry+, or negative for both fluorescent markers. A limiting dilution analysis was used to determine plating efficiency. Briefly, the log of % negative wells was determined for each input cell number and plotted to determine a best-fit line. Based on a Poisson distribution, the number of cells required for 37% of wells to be negative is the limiting dilution (LD). Plating efficiency was calculated as the inverse of the LD. Adjusted reprogramming efficiency was determined by dividing the reprogramming efficiency by the plating efficiency.
qRT-PCR
RNA purified from cells using the RNeasy Micro Kit (Qiagen) was converted to cDNA using the High-Capacity RNA-to-cDNA kit (Applied Biosystems). qRT-PCR reactions were set up in triplicate using the Brilliant III SYBR Master Mix (Agilent Genomics) and KiCqStart SYBR Green Primers (Sigma-Aldrich) to Oct4 (M_Pou5f1_2), Klf4 (M_Klf4_1), and Hprt (M_Hprt_1). Reactions were run on the LightCycler 480 PCR machine (Roche) with 40 cycles of 30s at 95C, 30s at 60C and 30s at 72C.
RNA-seq
Three replicates each of het/het MEFs, d3 SSEA-1−, d3 SSEA-1+, d6 SSEA-1−, d6 SSEA-1+, d9 SSEA-1−, d9 SSEA-1+, d12 SSEA-1−, d12 SSEA-1+, SSEA-1− after 12d of dox followed by at least 3d of withdrawal, and iPS (SSEA-1+ cells after 12d of dox followed by at least 3d of withdrawal) were isolated by FACS. Additionally, 2 replicates each of ¾ MEFs, d3 Ineff, d3 Eff, d6 Ineff, d6 Eff, and Oct4-GFP+ iPSCs were isolated by FACS. RNA was extracted from sorted cells using the RNeasy Micro Kit (Qiagen). cDNA libraries were generated using the NEBNext Ultra Directional RNA Library Prep Kit (NEB) based on poly-A selection. RNA and libraries were validated using a bioanalyzer (Agilent). Sequencing (50 cycles, paired-end) was performed using the HiSeq 2500 platform (Illumina), resulting in ~30 million reads per sample.
ATAC-seq
Two replicates each of ¾ MEFs, d3 Ineff, d3 Eff, d6 Ineff, d6 Eff, and Oct4-GFP+ iPSCs were isolated by FACS. ATAC-seq libraries were generated as previously described (Buenrostro et al., 2013). Briefly, 50,000 sorted cells were resuspended in nuclear isolation buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL). Nuclei were then treated with Tn5 transposase (Illumina). DNA was isolate using the MinElute Kit (Qiagen) and PCR-amplified using barcoded Nextera primers (Illumina). The DNA libraries were validated using a bioanalyzer (Agilent). Sequencing (50 cycles, paired-end) was performed using the HiSeq 2500 platform (Illumina), resulting in ~45 million reads per sample.
Whole Genome Bisulfite Sequencing (WGBS)
¾ MEFs, d3 Ineff, d3 Eff, d6 Ineff, d6 Eff, and Oct4-GFP+ iPSCs were isolated by FACS. Genomic DNA was purified from sorted cells, and WGBS library construction was performed as previously described (Gifford et al., 2013). Genomic fragments were sequenced using the HiSeq 2500 platform (Illumina).
siRNA Transfection and Analysis
Cells were transfected using Lipofectamine 2000 (Thermo Fisher) with pooled siRNA at a final concentration of 15–20 nM. All siRNA pools were esiRNA (Sigma-Aldrich) except for the siRNA targeting Dmrtc2 (GE-Dharmacon). Cells were treated with siRNA at d0, d3, d6, and d9 of reprogramming, after which dox was withdrawn. After 3–5 additional days, dox-independent iPSCs were stained for AP and reprogramming efficiency was determined (see above). Alternatively, d3 and d6 intermediates were analyzed by flow cytometry.
Lentivirus Production and Infections
293T cells were transfected with plasmids for lentiviral (LV) packaging (VSV-G and Δ8.9) and LV plasmids for either Stemcca, rtTA, tetO-Oct4, tetO-Sox2, tetO-Klf4, tetO-Myc, Bex2, tetO-Tfap2c, Cre-IRES-Puromycin, or Puromycin, using TransIT-293 Transfection Reagent (Mirus) to generate individual lentiviruses. MEFs were infected with virus combinations using Polybrene (Sigma). R26-rtTAhet Sox2-GFPhet were treated with LV-Oct4, LV-Sox2, LV-Klf4, and LV-Myc for individual vector reprogramming. Bex2GFP/Y and littermate control MEFs were infected with LV-Stemcca and LV-rtTA. Tfap2cflox/flox MEFs were infected with LV-Stemcca, LV-rtTA, and either LV-Cre-IRES-Puro or a LV-Puro control followed by puromycin selection prior to reprogramming. Het/het MEFs were infected with either LV-Bex2, LV-Tfap2c, or a LV-Puro control.
Generation of Utf1 Reporter Cells
Utf1-GFP reporter ESC lines were generated through CRISPR/Cas mediated gene targeting. A targeting construct was designed to integrate an E2A-GFP cassette in-frame with the final exon of Utf1. The construct was generated via PCR amplification and Gibson assembly (New England Biolabs) of E2A-GFP flanked on either side by 500bp of homology to the Utf1 locus. The targeting construct was cotransfected using Lipofectamine 2000 (Thermo) with a Utf1-targeting sgRNA (5′(GACTGATAACAAAGCTTTAT-3′) and a Cas9 expression construct into V6.5 ESCs that had previously been targeted with doxycycline inducible Col1a1-OKSM and Rosa26-rtTA. One week following the transfection, GFP+ cells were isolated by FACS and clonally expanded for analysis by Southern blot. Positive clones were injected into blastocysts and the resultant embryos were harvested at E15.5 for the preparation of high-grade chimeric MEFs.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistics including the number of replicates, mean, and S.D. are reported in the Figures and the Figure Legends. Data is judged to be statistically significant by two-tailed Student’s t-test (p < 0.05), where appropriate.
RNA-seq
RNA-seq reads were aligned to the mouse (mm9) reference transcriptome using STAR, a splice-aware alignment program. Read counts over transcripts were calculated using HTSeq based on a current Ensembl annotation file for NCBI37/mm9 assembly. For differential expression analysis the EdgeR package was used. DEGs were determined by a 2-fold or greater difference between two samples and a false discovery rate (FDR) below 0.05.
ATAC-seq
ATAC-seq reads were aligned to the mouse (mm9) reference genome using the BWA package. Only fragments with both ends unambiguously mapped to the genome that were longer than 100 bp were used for further analysis. Hotspot was used to detect significant peaks with an FDR cutoff of 0.05. Since the detected peaks were highly consistent between individual biological replicates, we merged replicate peak sets to produce the sets representing each population (MEF, d3 Ineff, d3 Eff, d6 Ineff, d6 Eff, and iPSC). The resulting peak regions were analyzed for changes in read density between populations. For the analysis of overlap between peak regions, we used the cutoff of 30% overlap in at least one of the two compared regions. DARs were determined by RPKM values for peak regions differing by 2-fold or greater between samples and an FDR below 0.05. TF-binding motif analysis was done with AME (http://meme-suite.org/tools/ame).
WGBS
Reads were aligned to the mouse (mm9) reference genome using BSMap. Methylation levels of individual CpGs were determined by observing bisulfite conversion in the aligned read compared to the reference genome. Region methylation levels were computed using CpGs covered by at least 5x in at least 4 samples. Differential methylation analysis was performed by using Fisher’s exact test to measure the significance of differential methylation at each CpG in a region. P-values of CpGs in a region were combined using Fisher’s method to calculate a region p-value. Regions covered by at least 3 CpGs, with a p-value of less than 0.01, and showing a weighted methylation difference of at least 30% were called differentially methylated.
DATA AND SOFTWARE AVAILABILITY
All sequencing data reported in this study were deposited at the gene expression omnibus (GEO) with the accession number GEO: GSE106838.
Supplementary Material
HIGHLIGHTS.
Cell surface markers allow for isolation of rare intermediates poised to reprogram
Transcriptional analysis of poised cells uncovers early regulators of reprogramming
Chromatin accessibility changes rapidly in reprogramming and precedes transcription
An early wave of DNA demethylation occurs in poised reprogramming intermediates
Acknowledgments
We thank Susan Schwarz, Bruno Di Stefano, and members of the Hochedlinger laboratory; Maris Handley and Meredith Weglarz of the MGH CRM/HSCI Flow Core; and members of the MGH Next Generation Sequencing Core. B.A.S. was supported through an MGH Pathology grant (NIH T32 CA921633) and an MGH ECOR fellowship; R.I.S. by NIH P30 DK40561; and K.H. by funds from MGH, the NIH (R01 HD058013, P01 GM099134), and the Gerald and Darlene Jordan Chair in Regenerative Medicine.
Footnotes
AUTHOR CONTRIBUTIONS
B.A.S., A.M, and K.H. designed the study. B.A.S., R.M.W., S.C., and H.G. performed and analyzed the experiments. J.L., A.K., and H.S. provided specific MEF lines. M.C., K.C., and R.I.S. performed the bioinformatics analysis. B.A.S. and K.H. wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no conflicting interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou E, Ferrari F, Walsh RM, Bar-Nur O, Stadtfeld M, Cheloufi S, Stuart HT, Polo JM, Ohsumi TK, Borowsky ML, et al. Genome-wide chromatin interactions of the Nanog locus in pluripotency, differentiation, and reprogramming. Cell Stem Cell. 2013;12:699–712. doi: 10.1016/j.stem.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auman HJ, Nottoli T, Lakiza O, Winger Q, Donaldson S, Williams T. Transcription factor AP-2γ is essential in the extra-embryonic lineages for early postimplantation development. Development. 2002;129:2733–2747. doi: 10.1242/dev.129.11.2733. [DOI] [PubMed] [Google Scholar]
- Bar-Nur O, Brumbaugh J, Verheul C, Apostolou E, Pruteanu-Malinici I, Walsh RM, Ramaswamy S, Hochedlinger K. Small molecules facilitate rapid and synchronous iPSC generation. Nat Methods. 2014;11:1170–1176. doi: 10.1038/nmeth.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinka S, Reimer MH, Pulakanti K, Rao S. Super-enhancers at the Nanog locus differentially regulate neighboring pluripotency-associated genes. Cell Rep. 2016;17:19–28. doi: 10.1016/j.celrep.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H, Jaenisch R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Markoulaki S, Hanna JH, Faddah DA, Buganim Y, Kim J, Ganz K, Steine EJ, Cassady JP, Creyghton MP, et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell. 2011;9:588–598. doi: 10.1016/j.stem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, Plath K. Cooperative binding of transcription factors orchestrates reprogramming. Cell. 2017;168:442–459. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng JC, Feng B, Han J, Jiang J, Kraus P, Ng JH, Orlov YL, Huss M, Yang L, Lufkin T, et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell. 2010;6:167–174. doi: 10.1016/j.stem.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Ito K, Yamazaki S, Yamamoto R, Tajima Y, Yanagida A, Kobayashi T, Kato-Itoh M, Kakuta S, Iwakura Y, Nakauchi H, et al. Gene targeting study reveals unexpected expression of brain-expressed X-linked 2 in endocrine and tissue stem/progenitor cells in mice. J Biol Chem. 2014;289:29892–29911. doi: 10.1074/jbc.M114.580084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. Chromatin accessibility predetermines glucocorticoid receptor binding patterns. Nat Genet. 43:264–268. doi: 10.1038/ng.759. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SI, Oceguera-Yanez F, Hirohata R, Linker S, Okita K, Yamada Y, Yamamoto T, Yamanaka S, Woltjen K. KLF4 N-terminal variance modulates induced reprogramming to pluripotency. Stem Cell Reports. 2015;4:727–743. doi: 10.1016/j.stemcr.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaupp AS, Buckberry S, Pflueger J, Lim SM, Ford E, Larcombe MR, Rossello FJ, de Mendoza A, Alaei S, Firas J, et al. Transient and Permanent Reconfiguration of Chromatin and Transcription Factor Occupancy Drive Reprogramming. Cell Stem Cell. 2017;21:834–845. doi: 10.1016/j.stem.2017.11.007. [DOI] [PubMed] [Google Scholar]
- Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, Bernstein BE, Meissner A. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DS, Shin JY, Tonge PD, Puri MC, Lee S, Park H, Lee WC, Hussein SM, Bleazard T, Yun JY, et al. An epigenomic roadmap to induced pluripotency reveals DNA methylation as a reprogramming modulator. Nat Commun. 2014;5:5619. doi: 10.1038/ncomms6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, Qin Y, Guo L, He J, Yu S, et al. Chromatin Accessibility Dynamics during iPSC Reprogramming. Cell Stem Cell. 2017;21:819–833. doi: 10.1016/j.stem.2017.10.012. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan E, Zunder ER, Ng YH, Goronzy IN, Nolan GP, Wernig M. Early reprogramming regulators identified by prospective isolation and mass cytometry. Nature. 2015;521:352–356. doi: 10.1038/nature14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milagre I, Stubbs TM, King MR, Spindel J, Santos F, Krueger F, Bachman M, Segonds-Pichon A, Balasubramanian S, Andrews SR, et al. Gender differences in global but not targeted demethylation in iPSC reprogramming. Cell Rep. 2017;18:1079–1089. doi: 10.1016/j.celrep.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley J, Skylaki S, Iwabuchi KA, Chantzoura E, Ruetz T, Johnsson A, Tomlinson SR, Linnarsson S, Kaji K. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature. 2013;499:88–91. doi: 10.1038/nature12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Wu T, Cyr AR, Woodfield GW, De Andrade JP, Spanheimer PM, Li T, Sugg SL, Lal G, Domann FE, et al. The role of Tcfap2c in tumorigenesis and cancer growth in an activated Neu model of mammary carcinogenesis. Oncogene. 2015;34:6105–6114. doi: 10.1038/onc.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak M, Jaenisch R. De novo DNA methylation by Dnmt3a and Dnmt3b is dispensable for nuclear reprogramming of somatic cells to a pluripotent state. Genes Dev. 2011;25:1035–1040. doi: 10.1101/gad.2039011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Anderssen E, Walsh RW, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schemmer J, Araúzo-Bravo MJ, Haas N, Schäfer S, Weber SN, Becker A, Eckert D, Zimmer A, Nettersheim D, Schorle H. Transcription factor TFAP2C regulates major programs required for murine fetal germ cell maintenance and haploinsufficiency predisposes to teratomas in male mice. PLoS One. 2013;8:e71113. doi: 10.1371/journal.pone.0071113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiba N, White CA, Lipsitz YY, Yachie-Kinoshita A, Tonge PD, Hussein SM, Puri MC, Elbaz J, Morrissey-Scoot J, Li M, et al. CD24 tracks divergent pluripotent states in mouse and human cells. Nat Commun. 2015;6:7329. doi: 10.1038/ncomms8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Stadtfeld M, Murphy G, Hochedlinger K, Kotton D, Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Borkent M, Hochedlinger K. A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat Methods. 2010;7:53–55. doi: 10.1038/nmeth.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Breault D, Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016;17:183–193. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- Vidal SE, Amlani B, Chen T, Tsirigos A, Stadtfeld M. Combinatorial modulation of signaling pathways reveals cell-type-specific requirements for highly efficient and synchronous iPSC reprogramming. Stem Cell Reports. 2014;3:574–84. doi: 10.1016/j.stemcr.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.