

Abstract
The lung is a complex organ with anatomically distinct pools of T cells that play specific roles in combating infection. Our knowledge regarding the generation and/or maintenance of immunity by parenchymal or circulating T cells has been gathered from either persistent (>60 days) or rapidly cleared (<10 days) infections. However, the roles of these distinct T cell pools in infections that are cleared over the course of several weeks are not understood. Clearance of the highly virulent intracellular bacterium, Francisella tularensis (Ftt) following pulmonary infection of immune animals is a protracted, T cell dependent process requiring approximately 30-40 days and serves as a model for infections that are not acutely controlled. Using this model, we found that intranasal vaccination increased the number of tissue resident CD4+ Teff cells and subsequent challenge of immune mice with Ftt led to a significant expansion of polyfunctional, parenchymal CD4+ Teff cells compared to the circulating pool. Despite the dominant in vivo response by parenchymal CD4+ T cells after vaccination and challenge, circulating CD4+ T cells were superior at controlling intracellular Ftt replication in vitro. Further examination in vivo revealed temporal requirements for resident and circulating T cells during Ftt infection. These requirements were in direct contrast to other pulmonary infections that are cleared rapidly in immune animals. The data herein provide important insights into the role of specific T cell populations that will be essential for design of novel effective vaccines against tularemia and potentially other agents of pulmonary infection.
INTRODUCTION
The development and dynamics of the T cell response within the lung following vaccination and infection are complex. Unlike secondary lymphoid organs, i.e. the spleen, in which T cells exist primarily in demarcated zones within the tissues, T cells in the lung reside in three anatomically distinct compartments: airway, parenchyma, or circulation. Historically, lung T cells have been analyzed at the tissue level without considering the unique contributions of each subset to pulmonary immunity. However, in the last few years, the role of specific T cell pools for protective immunity or its maintenance have been described for a variety of models of pulmonary infection. These studies have shown that the contribution of individual subsets appears to vary depending on the source and persistence of infection and immune status of the host. For example, airway-resident CD4+ T cells are critical for protection against MERS- and SARS-CoV while parenchyma-resident CD4+ T cells exhibit superior control of Mycobacterium tuberculosis infection [1, 2]. In contrast, circulating T cells are required for the host to effectively clear primary infection with Bordetella pertussis, but are dispensable during secondary infection [3]. Influenza A elicits resident T cell responses and while these cells are sufficient to control infection, and it has been suggested that circulating CD8+ T cells serve as a reservoir of long-lived memory T cells that re-populate this resident pool to maintain immunity long after infection [4, 5]. Each of these T cell subsets can be specifically targeted for expansion depending on the vaccination strategy [5, 6]. Thus, when considering the protective pulmonary T cell response necessary for development of effective long-lasting immunity, it is important to understand the nuanced response by subsets of T cells within the lung.
Francisella tularensis ssp. tularensis (Ftt) is a highly virulent intracellular bacterial pathogen that can cause fatal disease after exposure to 10 or fewer inhaled organisms. We and others have established that T cells are required for immune mice to survive Ftt infection [7, 8]. Following intranasal vaccination with the Live Vaccine Strain (LVS) and Ftt challenge in the C57Bl/6 mouse, immune animals depleted of CD4+ T cells succumb within one day of naïve mice while those lacking CD8+ T cells survived significantly longer [7]. Thus, while both sets of T cells contribute to survival of Ftt, these data suggested there was a pool of CD4+ T cells spatially poised to respond rapidly to Ftt challenge within the pulmonary compartment. In those studies, it was not determined whether this immunity was dependent on parenchymal CD4+ T cells, circulating CD4+ T cells, or both. Furthermore, since immune animals do not clear virulent Ftt until several weeks after infection, this model provided a unique opportunity to study pulmonary T cells in a protracted, but resolving, infection as compared to previously described persistent (>60 days) or rapidly cleared (<10 days) infections.
In this study, we characterized and identified the contributions of specific pulmonary T cell populations during vaccine-induced immunity against tularemia. LVS vaccination and Ftt challenge elicited a strong parenchymal CD4+ response. However, unlike other models of pulmonary bacterial infection, these T cells were inadequate for complete control of Ftt. Rather, we observed a temporal requirement for circulating T cells in control and ultimate survival of Ftt infection. Our data provide important insight into the role of T cells residing in specific pulmonary compartments during extended bacterial infection in the lung and will contribute to development of novel, effective vaccines.
MATERIALS AND METHODS
Bacteria
The Francisella tularensis subsp. holarctica live vaccine strain (LVS) was originally acquired from Fran Nano (University of Victoria, Victoria, British Columbia, Canada) and subsequently provided by Jean Celli (Rocky Mountain Laboratories [RML], NIAID, NIH, Hamilton, MT). F. tularensis subsp. tularensis strain SchuS4 (Ftt) was provided by Jeannine Peterson (Centers for Disease Control and Prevention, Fort Collins, CO). All Francisella stocks were generated as previously described [9]. Bacteria were thawed just prior to use and inocula were confirmed by enumerating viable bacteria on MMH agar after serial dilution.
Mice
Five to six-week-old specific-pathogen free C57Bl/6J (B6) female mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were housed in sterile microisolator cages in ABSL-2 and ABSL-3 facilities at RML. Studies were approved by and conducted in accordance with RML’s Animal Care and Use Committee.
Inoculation of mice
Mice were inoculated with Francisella as previously described [10]. Briefly, bacteria were thawed and serially diluted in phosphate buffered saline (PBS) to achieve the desired inoculum dose. Mice were anesthetized with 120 μl of 13.9 mg/ml ketamine/0.56 mg/ml xylazine given intraperitoneally. Bacteria were then delivered in 25 μL to a single nare. Mice were intranasally vaccinated with 150 CFU RML LVS. Twenty-eight days later, mice were intranasally challenged with 25 CFU Ftt. As indicated, 3 days prior to Ftt challenge and continuing every other day, mice were intraperitoneally injected with 1 mg/kg FTY720 (Sigma) or 5% dextrose in water as the vehicle control. Lymphopenia resulting from FTY720 treatment was confirmed by flow cytometric analysis of peripheral blood.
Enumeration of bacterial burdens and collection of tissue homogenate
Bacterial burdens in the spleen and lung were determined as previously described [11]. Briefly, tissues were aseptically removed and placed in cold tissue lysis buffer (150 mM Tris-HCl, 5 mM EDTA, and 10 mM Trizma base) that was supplemented with phosphatase inhibitor I, phosphatase inhibitor II, and protease inhibitor III (AG Scientific, San Diego, CA). Tissues were homogenized by grinding through a wire mesh screen using a syringe plunger. A portion of the homogenate was serially diluted in PBS and plated on MMH agar to determine bacterial burdens. The remaining homogenate was clarified by centrifugation at 14,000 × g for 30 minutes at 4°C. Homogenates were stored at −80°C until further analysis.
Intravenous labeling of circulating cells and tissue harvest
Mice were retroorbitally injected with 2.5 μg of CD45.2 FITC (Biolegend, San Diego, CA) in 100 μl PBS. Three minutes later, mice were euthanized and spleens, lungs, and mediastinal lymph nodes (mLN) were aseptically isolated and processed as previously described [12]. Spleens and mLNs were passed through a 70 μM cell strainer to generate a single cell suspension. The lung was minced and then digested with Liberase (Roche, Indianapolis, IN). Red blood cells in the spleen and lung were lysed by ACK lysis buffer (Life Technologies, Carlsbad, CA). The total number of viable cells in each tissue was determined by trypan blue exclusion using a TC10 Automated Cell Counter (Bio-Rad, Hercules, CA).
Flow cytometry
Cells were stained with Fixable Viability Dye eF780 (eBioscience) to distinguish live and dead cells. Cells were stained with the following directly conjugated antibodies: CD3 BV510 (clone 145-2C11), CD4 AF700 (clone GK1.5), CD8a BV605 (clone 53-6.7), CD44 PE-Cy7 (clone IM7), CD62L Pacific Blue (clone MEL-14), Ki67 APC (clone 16A8), IFN-γ PE (clone XMG1.2), TNF-α APC (clone MP6-XT22), CD69 APC-Cy7 (clone H1.2F3), CD103 BV711 (clone 2E7), CXCR3 BV650 (clone CXCR3-173), CX3CR1 BV785 (clone SA011F11). All antibodies were purchased from Life Technologies or Biolegend and were titrated on normal B6 splenocytes prior to use. For intracellular cytokine staining, a single cell suspension was incubated with 5 μg/ml Brefeldin A (Sigma-Aldrich) to inhibit cytokine secretion. Cells were fixed with 2% paraformaldehyde (Life Technologies) and permeabilized with 0.25% saponin (Sigma-Aldrich). For Ki67 staining, cells were fixed and permeabilized using the Transcription Factor Staining Buffer Set according to the manufacturer’s instructions (Life Technologies). Cells were washed after each staining step to remove residual unbound antibody. Data acquisition was performed on an LSRII (BD Biosciences, San Jose, CA) and data analysis was completed using FlowJo 10 (TreeStar, Ashland, OR). The gating strategy for data analysis is shown in supplemental figure 1.
IV− and IV+ CD4+ T cell enrichment
Lung parenchymal (IV−) and circulating (IV+) CD4+ T cells were discriminated from each other using intravenous staining as described above. Lungs were then processed into a single cell suspension and IV+ and IV− cells were sorted using a BD Aria sorter (BD Biosciences). CD4+ T cells were then positively selected from the sorted cells using Miltenyi CD4 Microbeads and MS columns according to the manufacturer’s instructions (Miltenyi Biotech Inc, San Diego, CA). This enrichment process resulted in >95% purity of each cell population as determined by flow cytometry.
Bone marrow macrophage (BMMs): T cell co-culture
BMMs were generated as previously described [13] from B6 mice. BMMs were seeded at 5×104 cells/well in a 96 well plate. BMM were inoculated with Ftt at a multiplicity of infection (MOI) of 25 in cDMEM. Ninety minutes later, bacteria-containing media was removed and cDMEM containing 50 μg/mL gentamicin (Life Technologies) was added for 45 minutes. Then, BMMs were washed twice and enriched CD4+ T cells in cDMEM containing 10 ng/ml M-CSF were added to the BMM cultures. BMMs and T cells were incubated for 48 hours at 37°C and 7% CO2, then supernatants were removed. BMMs were washed twice and then lysed in H2O. The number of intracellular bacteria were enumerated by plating serial dilutions of the lysate on MMH agar.
Cytometric Bead Assay and ELISAs
The concentrations of CCL5 and CXCL9 in tissue homogenates were quantified using a cytometric bead array (BD Biosciences) according to the manufacturer’s instructions. CXCL10 (R&D Systems Minneapolis, MN) concentration was quantified using enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions.
Histopathology
Tissues were processed for histopathology as previously described [10]. Briefly, tissues were fixed in 10% formalin, pH 7.4 for a minimum of 24 hours. Tissues were embedded in paraffin, placed in cassettes, and processed with a Sakura VIP-6 Tissue Tek, on a 12 hour automated schedule, using a graded series of ethanol, xylene, and ParaPlast Extra. Embedded tissues are sectioned at 5 μm and dried overnight at 42°C prior to staining. Tissues sections were stained with hematoxylin and eosin (H&E) and examined on an Olympus BX51 light-microscope equipped with an Olympus DP72 camera and associated cellSens Dimension 1.4.1 software. Tissues were assessed by a board-certified pathologist.
Statistics
Statistically significant differences between two groups were determined using an unpaired two-tailed t test and where necessary the Holm-Sidak method was used to correct for multiple comparisons. An ANOVA with Tukey’s post-test was used to compare three groups. CFU data was log-transformed prior to statistical analysis. Statistical significance is set at p < 0.05 and is indicated in the figures by *. Error bars represent standard error of the mean (SEM).
RESULTS
Intranasal vaccination elicits parenchymal CD4+ T cells
We previously established that pulmonary CD4+ T cells from immune mice directly control Ftt replication in BMMs and immune mice depleted of CD4+ T cells do not survive Ftt challenge [7]. Given the critical role of pulmonary CD4+ T cells during tularemia, it is important to trigger sufficient expansion of immune CD4+ T cells for effective immunity. Based on other models of pulmonary infection, specific subsets of T cells in the lung may be more important than others for controlling infection. These pools can be differently targeted for expansion following specific vaccination regimes, thus identifying parenchymal versus circulating T cells and their relative contribution to survival of Ftt infection is critical for development of more effective vaccines [5, 6]. Since these populations have not been examined following Francisella infection, we first determined the kinetics and contributions of parenchymal and circulating T cells after LVS vaccination and Ftt challenge. Pulmonary T cell populations were distinguished utilizing a previously described intravenous labeling technique which stains circulating, but not parenchymal, T cells with fluorochrome-conjugated antibody [14]. We confirmed that this technique yielded distinct circulating (IV+) and parenchymal (IV−) CD4+ T cells populations in naïve and immune mice (figure 1A). Consistent with previous reports, >90% of CD4+ T cells in a naïve mouse within the vasculature were positive for FITC CD45.2 [1]. FITC negative parenchymal CD4+ T cells were further differentiated from FITC positive circulating CD4+ T cells by the presence of CD69, CD103, and CXCR3 and absence of CX3CR1 on the surface of parenchymal cells as previously reported (supplemental figure 2) [1, 14]. We then compared these distinct pulmonary T cell populations among naive and immune animals. Compared to naïve animals, LVS immune mice had significantly increased the number of IV− CD4+ T cells in the lung (figure 1B). There was also a significant increase in the number and frequency of IV− CD4+ T cells with an effector phenotype (CD44+ CD62L−; Teff) in immune mice compared to naïve (figure 1C, D). In contrast, the number of IV+ CD4+ T cells was significantly lower in immune animals (figure 1E) compared to naïve mice. However, the number of CD4+ effector T cells present in the IV+ pool was similar among naïve and immune animals (figure 1F). This resulted in a significant increase in the ratio of IV+ CD4+ effector T cells to total CD4+ T cells among immune mice compared to naïve animals (figure 1G). Therefore, LVS vaccination established expanded pools of both IV− and IV+ CD4+ T cells in the lung.
Figure 1. Intranasal vaccination increases the number of IV− effector CD4+ T cells.
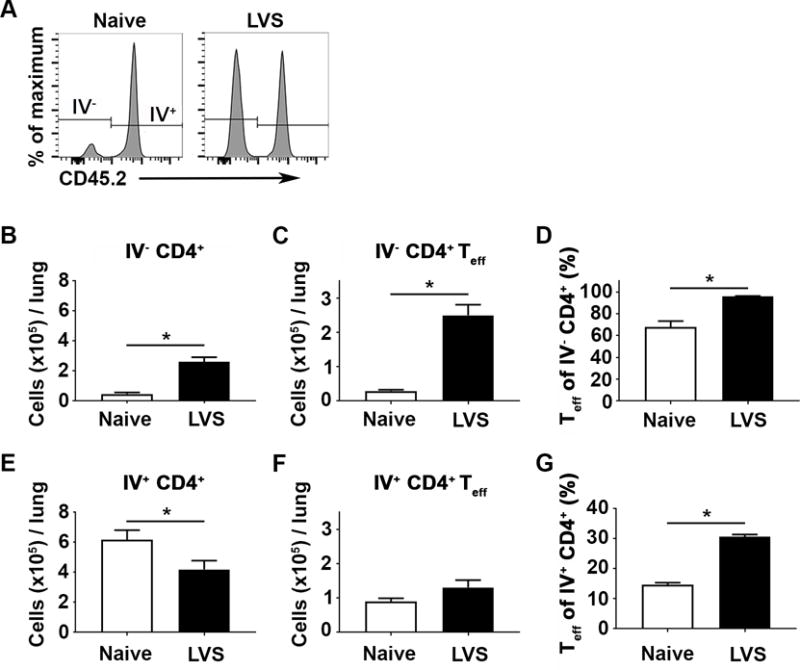
IV+ and IV− CD4+ T cell populations in naïve and immune mice were quantified 28 days after vaccination using intravascular staining. Mice were anesthetized, intravenously injected with anti-CD45.2 FITC and euthanized three minutes later. Lungs were harvested and cell populations were analyzed by flow cytometry. A) Representative histograms of lung CD4+ T cells from naïve and LVS vaccinated mice. Parenchymal T cells were defined as IV− and circulating cells were IV+. B) Number of IV− CD4+ T cells. C) Number of IV−CD4+ Teff (CD44+ CD62L−) cells. D) Percentage of IV− CD4+ with Teff phenotype. E) Number of IV+ CD4+ T cells. F) Number of IV+ CD4+ Teff cells. G) Percentage of IV+ CD4+ with Teff phenotype. Data are combined from 2 independent experiments; n=7-8 mice/group. Error bars represent standard error of the mean (SEM). Statistical significance was determined between naïve and vaccinated mice using a student’s t-test; * indicates p ≤ 0.05.
IV− CD4+ T cells are maintained and expand during the first week of Ftt challenge
As shown above, LVS vaccination successfully expanded the pools of the IV− and IV+ CD4+ Teff in the lung. However, it was unclear if these cell populations exhibit differential responsiveness, e.g. proliferation or cytokine production, following Ftt infection. Therefore, we quantified the number of IV− and IV+ CD4+ T cells and assessed their ability to produce cytokine directly ex vivo in mice challenged with Ftt 28 days after vaccination. On day 3 post-challenge, and continuing throughout the time points measured, the percentage of CD4+ T cells in the IV− pool was significantly greater than that in the IV+ population (figure 2A). The total number of IV− CD4+ was significantly greater than the number of IV+ CD4+ on day 7 after infection. However, at other time points after infection there were not statistical differences between the number of IV− and IV+ CD4+ T cells (figure 2B). Nearly all IV− CD4+ T cells maintained an effector phenotype throughout Ftt challenge (figure 2C). Between days 3 to 7 after Ftt infection, the percentage of IV+ CD4+ T cells with an effector phenotype increased (figure 2C) and there was a corresponding increase in the number of IV+ CD4+ Teff (figure 2D). However, there were significantly more IV− compared to IV+ CD4+ Teff cells at all time points measured (figure 2D).
Figure 2. The IV−, poly-functional CD4+ T cell pool expands early after SchuS4 challenge.
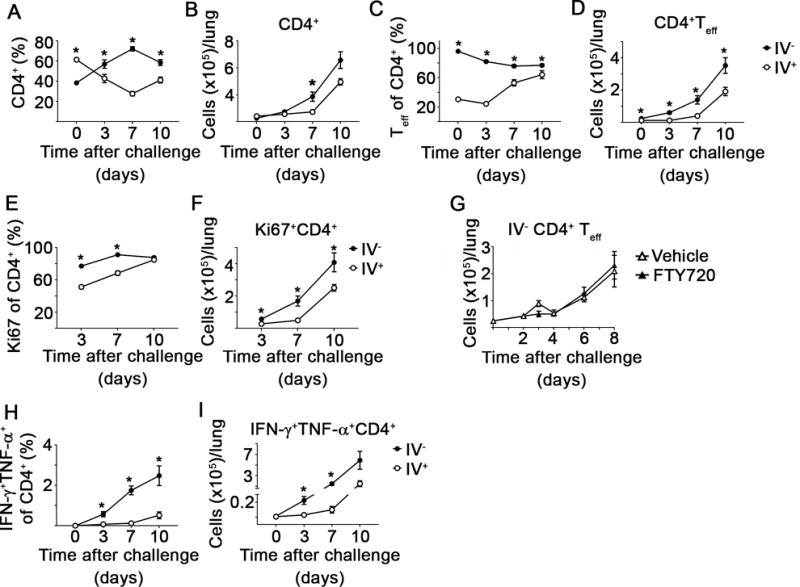
Naïve or vaccinated mice were challenged with 25 CFU Ftt 28 days after vaccination. At the indicated time point after Ftt challenge, IV+ and IV− CD4+ T cells populations were quantified. Additionally, cells harvested from lungs were assessed for proliferative capability using Ki67 or were immediately incubated with Brefeldin A followed by staining for intracellular IFN-γ and TNF-α. A) Percentage and B) number of IV− and IV+ CD4+ T cells. C) Percentage and D) number of CD4+ Teff. E) Percentage and F) number of Ki67+ CD4+. G) Number of CD4+ Teff after 5% dextrose in water (Vehicle) or FTY720 treatment. H) Percentage and I) number of IFN-γ+ TNF-α+ CD4+. Data are combined from two independent experiments; n=7-10 mice/group/time point. Error bars represent SEM. Statistical significance was determined using a student’s t-test using the Holm-Sidak method to correct for multiple comparisons; * indicates p ≤ 0.05.
The expansion of IV− CD4+ T cells could be a direct consequence of proliferation and/or recruitment of additional cells to the tissue. To determine the extent of T cell proliferation within each compartment, we stained for the proliferation marker, Ki67. Both IV− and IV+ CD4+ T cells proliferated after Ftt challenge, but a significantly higher proportion of IV− CD4+ T cells stained positive for Ki67 compared to IV+ CD4+ T cells (figure 2E). There was also a significant increase in the number of Ki67+ IV− CD4+ T cells compared to IV+ CD4+ T cells (figure 2F). Although IV− T cells are proliferating, this does not exclude the possibility that IV+ T cells have trafficked into the lung parenchyma thereby contributing to the IV− pool’s expansion. We therefore treated mice with FTY720 to inhibit extravasation of circulating T cells into the lung parenchyma and enumerated the number of CD4+ Teff during the first week of infection. Vehicle and FTY720-treated animals had similar numbers of IV− CD4+ Teff at all time points indicating local proliferation, and not T cell infiltration, is responsible for the expansion of the IV− CD4+ T cell pool (figure 2G).
During Ftt infection T cells that produce both IFN-γ and TNF-α are correlated with superior protective immunity in vaccinated animals [7]. Thus, we evaluated the ability of IV− and IV+ CD4+ T cells to produce the effector cytokines, IFN-γ and TNF-α directly ex vivo. There was a significant increase in the frequency and number of CD4+ T cells that produce both IFN-γ and TNF-α in the IV− pool compared to IV+ CD4+ T cells on days 3 and 7 after Ftt challenge (figure 2H, I). Together, these data indicated that within the first week of Ftt challenge, the IV− CD4+ Teff pool in vaccinated animals expanded via local proliferation and were the key producers of effector cytokines known to control Ftt intracellular replication.
IV+ CD4+ T cells exhibit superior control of intracellular Ftt replication
We previously demonstrated that bulk pulmonary CD4+ T cells from immune mice control Ftt replication in BMMs, but did not determine the relative contribution of IV− versus IV+ T cells in this process [7]. Given the increased proliferative capacity and heightened cytokine production in IV− CD4+ T cells following Ftt challenge of immune animals, we predicted this population would be superior at controlling intracellular Ftt replication. To test this hypothesis, IV− and IV+ cells from vaccinated mice were isolated and each population was individually assessed for its ability to control intracellular replication of Ftt in vitro. The number of IV− CD4+ T cells in naïve mice was prohibitively low for purifying and examining the activity of this population for the co-culture assay; therefore, we utilized total CD4+ T cells from the naïve lung as a negative control. Both IV− and IV+ CD4+ T cells from vaccinated mice controlled Ftt replication in BMMs compared to naïve CD4+ T cells (figure 3). Surprisingly, when the same number of IV− and IV+ CD4+ T cells were added to infected BMMs, IV+ CD4+ T cells controlled bacterial growth significantly better than IV− CD4+ T cells (figure 3). Due to the increased frequency of Teff in the IV− pool, we also overlaid the same number of IV− and IV+ CD4+ Teff and found the IV+ pool retained its superior ability to control Ftt replication (data not shown). Although purified T cells had >95% viability at the time of overlay, we were unable to conclusively demonstrate that differences in T cell viability did not contribute to the degree of bacterial control we observed. Overall, despite the dominant in vivo response by IV− CD4+ T cells, this subset was not as effective as IV+ CD4+ T cells at controlling Ftt intracellular replication on a per cell basis ex vivo.
Figure 3. IV+ CD4+ T cells exhibit superior control of intracellar Ftt replication in vitro.
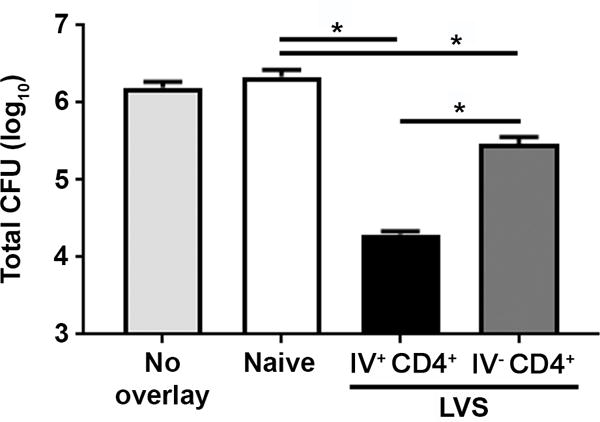
IV+ and IV− CD4+ T cell populations in immune mice were labeled 28 days after vaccination using intravascular staining. Lungs were processed into single cell suspension and sorted into FITC+ and FITC− populations. CD4+ T cells were enriched from these sorted pools or purified directly from naïve lungs and added to Ftt-infected bone marrow macrophages at a ratio of 1 T cell per 2 macrophages. Forty-eight hours later, the number of intracellular bacteria were enumerated. n=3 biological replicates per group. Data are representative of two independent experiments. Error bars represent (SEM). Statistical significance between naïve, IV− CD4+, and IV+ CD4+ was determined using an ANOVA with Tukey’s post-test; * indicates p ≤ 0.05.
Immune mice experience a protracted Ftt infection
The surprising result that IV+ CD4+ T cells exerted superior control of intracellular replication of Ftt compared to the IV− population suggested that there may be differences in the ability of T cells among immune animals to control intracellular replication of Ftt in vivo as compared to in vitro. Alternatively, it was possible that similar to our in vitro findings, IV+ T cells are more efficient at contributing to the control of Ftt replication in vivo. In other models of pulmonary bacterial infection, e.g. B. pertussis, resident T cells were sufficient to control secondary infection within a week of challenge of immune animals [3]. Therefore, we speculated that if IV− cells were sufficient for survival of in vivo infection, Ftt would be cleared within a week of challenge similar to other models of pulmonary bacterial infection. On the other hand, if clearance was protracted there may be specific temporal contributions of each CD4+ T cell population. Therefore, we determined the kinetics of bacterial dissemination and control in naïve and LVS vaccinated mice after intranasal Ftt challenge. There were no differences in the number of Ftt recovered from lungs of naïve or immune animals during the first 2 days of infection. As expected, within 4 days after Ftt infection naïve mice had high bacterial burdens in the lung and spleen and displayed signs of illness that met experimental endpoint criteria which required euthanasia in accordance with our ACUC protocol (figure 4A, B). However, among immune animals, mice had stabilized the number of bacteria in the lungs between days 2 and 14 after challenge (figure 4A). Ftt was eventually cleared from their lungs between day 14 and 21 with no detectable bacteria in most animals 42 days after Ftt challenge (figure 4A). Ftt infection in the peripheral compartment of vaccinated animals was similarly protracted. Bacteria were detected in the spleen 2 days after Ftt challenge and while immune mice control the infection, 40% of animals still had between 100-1000 bacteria on day 42 post-Ftt challenge (figure 4B). Overall, these data indicated the immune system must respond to Ftt infection for several weeks. Therefore, IV− and IV+ T cells may play important temporal roles in combatting this disseminated infection of intermediate duration, i.e. neither acute nor chronic.
Figure 4. Prolonged Ftt infection in immune animals.
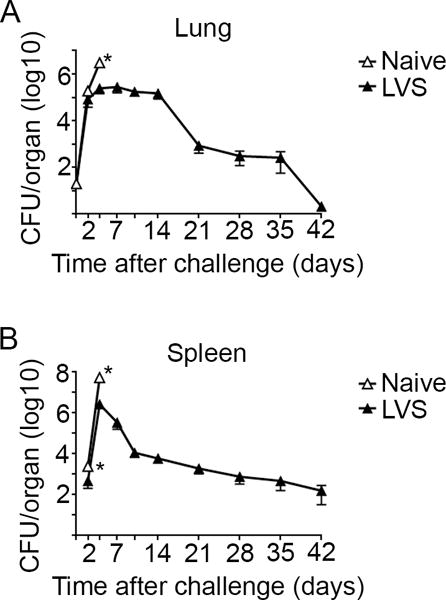
Naïve or mice intranasally vaccinated with 150 CFU LVS were intranasally challenged with 25 CFU Ftt 28 days after vaccination. At the indicated time point after challenge, mice were euthanized and bacterial burdens in the A) lung and B) spleen were determined. Data are combined from two independent experiments; n=6-10 mice/group/time point. Error bars represent SEM. Statistical significance was determined on log-transformed data using a student’s t-test with the Holm-Sidak method to correct for multiple comparisons; * indicates p ≤ 0.05.
Temporal roles for IV− and IV+ T cells after Ftt challenge
Given the prolonged nature of disseminated Ftt infection in vivo among immune animals and the difference in the ability of IV− and IV+ CD4+ T cells to control intracellular replication in vitro, we hypothesized that both IV− and IV+ T cells were necessary for protective immunity. To determine the requirements for control and survival of Ftt infection in immune hosts, we treated mice with FTY720 to prevent extravasation of circulating T cells into the lung parenchyma. At each time point up to 8 days post-challenge, we confirmed that FTY720 treated mice had significantly fewer IV+ T cells in the lung compared to vehicle treated controls (supplemental figure 2). While FTY720 treatment decreased the number of IV+ CD4+ and CD8+ T cells in the lung, it did not inhibit infiltration of innate immune cells, including neutrophils and monocytes, that could also contribute to bacterial control (supplemental figure 3). Therefore, the only detectable impairment in cellular recruitment among FTY720 treated animals was the inability of IV+ T cells to enter the tissue. It is difficult to clearly demarcate IV+ and IV− T cells in the spleen using intravenous staining. However, the systemic administration of FTY720 would impact the ability of IV+ T cells to traffic into other infected tissues, like the spleen. Consequently, IV+ T cells cannot aid in controlling the disseminated infection in FTY720 treated animals.
Following confirmation of the ability of FTY720 to restrict the presence of IV+ T cells in the lung of mice in our immune model, we first determined survival after Ftt challenge of mice treated with vehicle or FTY720. Consistent with our previous reports, 90% of vehicle treated immune mice survived Ftt infection (figure 5A). In contrast, FTY720 treated immune mice began to display endpoint criteria requiring euthanasia beginning on day 8 after infection and all animals succumbed to infection by day 13 after challenge (figure 5A). These data indicated that IV−T cells were sufficient to survive the first week of infection, but that IV+ T cells were required for immune animals to survive past this time point. To confirm that the lack of survival of Ftt infection in FTY720 treated immune mice was due to unrestricted bacterial replication, we assessed bacterial loads in these animals over time. There were no significant differences in bacterial loads among immune FTY720 treated mice compared to vehicle controls during the first 8 days of infection in the lung (figure 5B). Splenic bacterial burdens in vehicle and FTY720 treated immune mice were also not significantly different, except for day 2 post-challenge (figure 5C). However, bacterial loads in FTY720 treated animals at time points after day 8, e.g. day 10, 11, 12, had significantly higher bacterial burdens in their lungs and spleens compared to vehicle treated mice (figure 5B, C) indicating a global failure in restriction of bacterial growth. In addition to significantly higher bacterial burdens, we observed gross pathological and histological differences among FTY720 and vehicle control treated mice. Ftt challenged immune animals treated with vehicle displayed multifocal pulmonary lesions, characteristic of Ftt infection in animals that resolve infection (figure 5D) [10]. In contrast, FTY720 treated mice had coalescing lesions with necrotic debris, fibrin, and edema that filled and replaced alveoli and bronchioles (figure 5D). Naïve mice treated with vehicle or FTY720 had no evidence of histological changes, therefore drug treatment did not account for the observed events (figure 5D). Severe pathological changes were also evident in the spleens of FTY720 treated animals with red pulp expanded by increased deposition of fibrin, viable and degenerate neutrophils, and necrotic debris (figure 5E). The extensive inflammatory response evident in FTY720 treated animals suggested high levels of inflammatory chemokines. Indeed, FTY720 treated, Ftt challenged immune mice had significantly higher concentrations of CCL5, CXCL9, and CXCL10 (figure 5F) consistent with the dramatic inflammatory response observed in tissues of these animals. Together, these data indicate a temporal requirement for IV− and IV+ T cells in controlling Ftt infection among immune animals. Specifically, IV− cells were sufficient for mediating bacterial control within the first week after Ftt inoculation, however IV+ T cells were subsequently required for ultimate control of the infection and survival of immune animals.
Figure 5. Parenchymal T cells are adequate to control SchuS4 replication early after pulmonary infection in vivo.
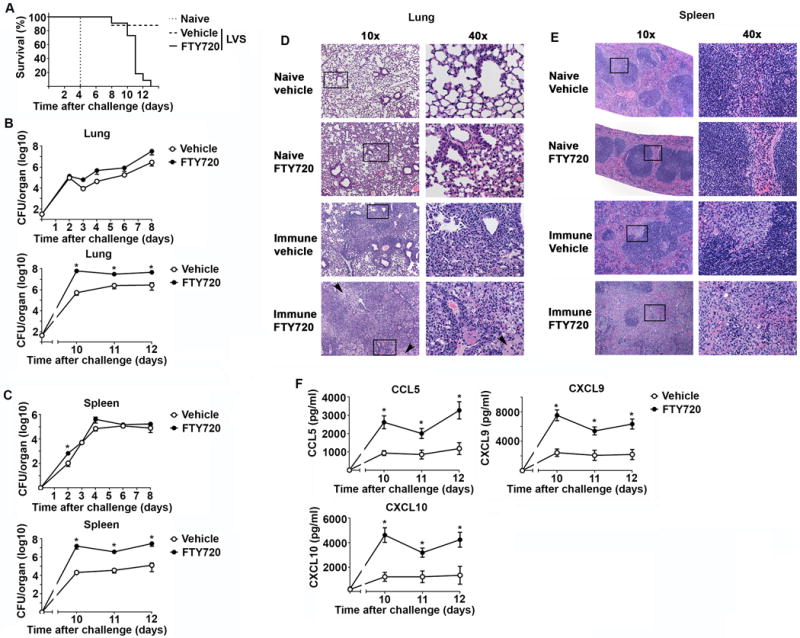
Naïve or vaccinated mice were challenged with 25 CFU Ftt 28 days after vaccination. At day -3 prior to Ftt challenge and continuing every other day, mice were intraperitoneally treated with 1 mg/kg FTY720 to retain T cells in peripheral lymphoid tissue. Mice treated with 5% dextrose in water served as vehicle controls (Vehicle). A) Survival after Ftt challenge. B) Bacterial burdens in lung homogenates were enumerated in animals that did not display endpoint criteria at the time of euthanasia. C) Bacterial burdens in spleen homogenates were enumerated in animals that did not display endpoint criteria at the time of euthanasia. D) Representative images of lung sections stained with Haemotoxylin and Eosin. Arrows indicate edema. E) Representative images of spleen sections stained with Haemotoxylin and Eosin. F) Concentrations of chemokines in lung homogenates. Data are combined from two independent experiments; n=7-10 mice/group/time point. Error bars represent SEM. Statistical significance was determined using a student’s t-test with the Holm-Sidak method to correct for multiple comparisons or Mantel-Cox test for survival (* vehicle vs FTY720); * indicates p ≤ 0.05.
DISCUSSION
Sublethal infections initiated in the pulmonary compartment typically expand pools of parenchymal CD4+ and/or CD8+ T cells ready to combat secondary challenge with the infectious agent. The importance of this population, as opposed to the circulating pool of effector memory T cells, is highlighted by the fact that parenchymal T cells are either completely sufficient or provide a greater contribution to the resolution of secondary infection with several pulmonary pathogens [1, 3, 5, 15-17]. Ftt infections initiated in the lung are typically considered the most dangerous type of tularemia. In part, this is due to the ability of the bacterium to undergo unrestricted replication over the first few days of infection prior to dissemination without triggering an inflammatory response in the lung [9]. Therefore, the presence of an effective, fast-acting immune response is essential for the host to restrict bacterial replication and survive pneumonic tularemia. Indeed, regardless of the route of vaccination or challenge, a strong T cell response driven by CD4+ T cells is required for resolution of Francisella infections [7, 8, 18]. However, prior to this report it was not known whether there were specific contributions by parenchymal and circulating CD4+ T cells in immune hosts.
Herein, we uncovered specific, temporal roles of parenchymal and circulating T cells among immune animals before and after Ftt challenge. We focused primarily on pulmonary T cells given the ease of identifying these populations using the well described intravascular staining technique [14]. Our initial experiments showed that several aspects of the pulmonary CD4+ T cell response after LVS vaccination and Ftt challenge were consistent with those observed in other models of chronic or acute bacterial or viral pathogens. First, as previously described for pulmonary infections of Bordetella pertussis, Influenza A, and Mycobacterium tuberculosis, intranasal vaccination with LVS significantly increased the number of IV− CD4+ T cells [3, 5, 6, 19, 20]. Second, consistent with data obtained from B. pertussis and Influenza A, IV− CD4+ T cells expand via local proliferation after Ftt infection [3, 19, 21]. These data suggested that IV− pulmonary T cells have generalizable features regardless of the infecting agent. In contrast to these similar features in CD4+ T cell responses directed against pulmonary infection, we identified differences in Ftt-driven parenchymal CD4+ T cells. Although IV− CD4+ T cells proliferated and produced cytokine in response to Ftt challenge, they were not sufficient to mediate bacterial clearance. Rather, IV+ T cells must gain access to the pulmonary tissue to limit Ftt replication and inflammation. This finding is unique compared to other pulmonary pathogens. For example, resident T cells are sufficient to control secondary infection with B. pertussis, Nippostrongylus brasiliensis, and Influenza A [3, 5, 19, 22]. Of note, these pathogens cause acute infections that are cleared within 7 days in immune hosts [3, 19, 22] whereas secondary Ftt infection takes 30-40 days to clear. Furthermore, B. pertussis and Influenza A infections are restricted to the lung whereas Ftt disseminates from this tissue and colonizes other organs within 48 hours after intranasal inoculation. Therefore, the difference in the requirements for protection likely reflects the duration and nature of secondary infection.
We propose the following model for the pulmonary T cell response to Ftt in immune animals. Initially, parenchymal T cells control Ftt replication as they are spatially poised to respond to local antigen. However, because these T cells are not as effective as IV+ T cells at controlling intracellular Ftt replication, the bacteria disseminate and infect peripheral organs such as the liver and spleen. Final resolution of infection requires recruitment of IV+T cells to the lung and likely other tissues infected with Ftt. Our approach could not distinguish the relative contributions of IV+ CD4+ and CD8+ T cells in restricting Ftt replication; it is possible, in fact likely, that both cell types cooperate to limit bacterial growth. However, in resting immune animals, lung CD4+ T cells exerted significantly greater control over intracellular Ftt replication as compared to CD8+ T cells in our co-culture assay (data not shown). Furthermore, unlike the CD4+ pool where nearly half of the total T cells are in the circulation, approximately 80% of CD8+ T cells are in the vasculature (data not shown). Together, these data suggest, at least at the onset of infection, that parenchymal CD4+ T cells are the predominant immune cell required to control bacterial replication but does not exclude the possibility that CD8+ T cells, particularly in the circulation, are not important for Ftt control and clearance.
There could be several mechanisms that underlie the difference between IV− and IV+ CD4+ T cells in their ability to control intracellular Ftt replication. One possibility is the up-regulation of the inhibitory receptor PD-1 on IV− CD4+ T cells after LVS vaccination compared to IV+ CD4+ T cells (data not shown). PD-1 is upregulated on T cells during chronic antigen exposure [23] and serves as a rheostat to modulate T cell function and control inflammation. After M. tuberculosis or Influenza A infection, IV− CD4+ T cells also express higher levels of PD-1 compared to their IV+ counterparts [1, 19]. The up-regulation of PD-1 by IV− T cells aids in constraining, but not eliminating, T cells responses and resulting inflammation, thereby maintaining lung homeostasis during an ongoing immune response. Despite increased PD-1 expression by IV− CD4+ T cells, adoptively transferred IV− T cells mediate significantly more control than transferred IV+ T cells in M. tuberculosis in the lung [1]. Similarly, IV− T cells are sufficient for mice to control secondary Influenza A infection [5]. These data suggest that resident T cells in these infection models can overcome the inhibitory effects of PD-1 expression. In contrast, IV− T cells present in Ftt immune animals may not properly modulate PD-1 expression to allow effective T cell responsiveness and concomitant control of exuberant inflammatory responses as observed in other infections. This inability to alter PD-1 expression, resulting in subpar T cell driven immunity among IV− T cells in Ftt immune mice, may also explain the dependence of IV+ T cells that are not under this restriction for resolution of infection.
An additional explanation for the dependence on both IV− and IV+ T cells is the nature of the infection. As opposed to infections mediated by other bacteria, the requirements for both IV− and IV+T cells for optimal control of Ftt infection is more reminiscent of the response to Leishmania major in the skin. Leishmaniasis, like Ftt, takes weeks to control suggesting that prolonged infections share the requirement for multiple T cell pools to effectively control infection, irrespective of the affected tissue. Furthermore, past exposure to L. major expands skin resident CD4+ T cells and while these cells control parasite burden, optimal protection occurs in the presence of resident and circulating T cells. Specifically, tissue resident T cells serve as immune sentinels and recruit circulating cells to the site of second exposure [24]. In both infection models, it remains unclear whether circulating T cell recruitment is an inherently slow process, taking approximately 1 week, or whether this delay is a feature of immunomodulation on the part of the pathogen. Similar to L. major, Ftt potently evades and suppresses inflammatory responses during infection [9, 13, 25]. Thus, it is possible that the delay in recruitment of circulating T cells may be a feature of the pathogen dampening appropriate chemokine/cytokine responses required for extravasation of effector T cells into the tissue. Alternatively, there may be a natural regulation of chemokine/cytokine production in the lung that limits potentially inflammation generating T cells into the tissue. Dissection of the relative contribution of pathogen versus host regulatory processes for controlling T cell recruitment to the lung will be essential for development of new vaccines. Irrespective of the timing of circulating T cell recruitment, the requirement for parenchymal T cells to help recruit circulating T cells in other models of infection could explain why adoptive transfer of immune T cells has not yet been shown to protect naïve animals from intranasal Ftt challenge [24, 26, 27]. Following adoptive transfer, a sufficient pool of parenchymal T cells may not be established. Alternatively, there may be insufficient numbers of circulating T cells to be recruited to the tissue. Our data suggest that either scenario would result in absence of protection in recipient mice. To overcome this hurdle, we plan to use mouse parabiosis to further define the contributions parenchymal and circulating T cells in protective immunity to tularemia.
Together, our data suggest distinct temporal roles for parenchymal and circulating immune T cells following infection with Ftt. Understanding this requirement is critical for development of more effective vaccines directed against Ftt as the route and nature of vaccination could influence the number, location, and effector function of memory T cells. For example, boosting BCG immunized mice intranasally with adenovirus expressing Ag85A resulted in significantly greater protection against secondary challenge compared to mice that received the adenovirus vaccine intramuscularly. This improved response was correlated with enhanced pulmonary T cell responses in intranasally boosted mice as compared to those vaccinated intramuscularly [28]. We have shown that prime/boost vaccination strategies significantly enhances the number of memory T cells in the lung and correlates with improved survival, however that study did not distinguish between parenchymal and circulating T cells [29]. Since both vaccinations were delivered intranasally, we predict that there was a significant expansion of parenchymal T cells. We are currently confirming this hypothesis and aim to further improve that pool of cells as a strategy to more quickly control Ftt infection.
Data presented herein suggests that identifying the contribution of specific T cell populations in the lung (and likely elsewhere) will aid in successful development of more effective vaccines against tularemia. More generally, our data also suggest that there is a critical requirement for expanding parenchymal and circulating T cells for infections that start in the lung but disseminate after colonization. Understanding the dynamics of the T cell response will be important for vaccine design against infections mediated by other virulent bacteria.
Supplementary Material
Acknowledgments
We thank Aaron Carmody of the RML Flow Cytometry Core for performing the cell sorting. We also thank Dan Long, Tina Thomas, and Rebecca Rosenke for preparing and staining tissues for histopathological analysis processing. Finally, we thank Robert Buntyn, Rio Hammond, and Drs. Forrest Jessop and Benjamin Schwarz for technical assistance.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Disease
References
- 1.Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, Barber DL. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol. 2014;192:2965–2969. doi: 10.4049/jimmunol.1400019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao J, Zhao J, Mangalam AK, Channappanavar R, Fett C, Meyerholz DK, Agnihothram S, Baric RS, David CS, Perlman S. Airway Memory CD4(+) T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity. 2016;44:1379–1391. doi: 10.1016/j.immuni.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilk MM, Misiak A, McManus RM, Allen AC, Lynch MA, Mills KHG. Lung CD4 Tissue-Resident Memory T Cells Mediate Adaptive Immunity Induced by Previous Infection of Mice with Bordetella pertussis. J Immunol. 2017;199:233–243. doi: 10.4049/jimmunol.1602051. [DOI] [PubMed] [Google Scholar]
- 4.Slutter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, Harty JT. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci Immunol. 2017;2 doi: 10.1126/sciimmunol.aag2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zens KD, Chen JK, Farber DL. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight. 2016;1 doi: 10.1172/jci.insight.85832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perdomo C, Zedler U, Kuhl AA, Lozza L, Saikali P, Sander LE, Vogelzang A, Kaufmann SH, Kupz A. Mucosal BCG Vaccination Induces Protective Lung-Resident Memory T Cell Populations against Tuberculosis. MBio. 2016;7 doi: 10.1128/mBio.01686-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts LM, Crane DD, Wehrly TD, Fletcher JR, Jones BD, Bosio CM. Inclusion of Epitopes That Expand High-Avidity CD4+ T Cells Transforms Subprotective Vaccines to Efficacious Immunogens against Virulent Francisella tularensis. J Immunol. 2016;197:2738–2747. doi: 10.4049/jimmunol.1600879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conlan WJ, Shen H, Kuolee R, Zhao X, Chen W. Aerosol-, but not intradermal-immunization with the live vaccine strain of Francisella tularensis protects mice against subsequent aerosol challenge with a highly virulent type A strain of the pathogen by an alphabeta T cell- and interferon gamma- dependent mechanism. Vaccine. 2005;23:2477–2485. doi: 10.1016/j.vaccine.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 9.Bosio CM, Bielefeldt-Ohmann H, Belisle JT. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J Immunol. 2007;178:4538–4547. doi: 10.4049/jimmunol.178.7.4538. [DOI] [PubMed] [Google Scholar]
- 10.Crane DD, Scott DP, Bosio CM. Generation of a convalescent model of virulent Francisella tularensis infection for assessment of host requirements for survival of tularemia. PLoS One. 2012;7:e33349. doi: 10.1371/journal.pone.0033349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chase JC, Bosio CM. The presence of CD14 overcomes evasion of innate immune responses by virulent Francisella tularensis in human dendritic cells in vitro and pulmonary cells in vivo. Infect Immun. 2010;78:154–167. doi: 10.1128/IAI.00750-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson RV, Crane DD, Bosio CM. Long lived protection against pneumonic tularemia is correlated with cellular immunity in peripheral, not pulmonary, organs. Vaccine. 2010;28:6562–6572. doi: 10.1016/j.vaccine.2010.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crane DD, Ireland R, Alinger JB, Small P, Bosio CM. Lipids derived from virulent Francisella tularensis broadly inhibit pulmonary inflammation via toll-like receptor 2 and peroxisome proliferator-activated receptor alpha. Clin Vaccine Immunol. 2013;20:1531–1540. doi: 10.1128/CVI.00319-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9:209–222. doi: 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, Barber DL. CD4 T Cell-Derived IFN-gamma Plays a Minimal Role in Control of Pulmonary Mycobacterium tuberculosis Infection and Must Be Actively Repressed by PD-1 to Prevent Lethal Disease. PLoS Pathog. 2016;12:e1005667. doi: 10.1371/journal.ppat.1005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connor LM, Harvie MC, Rich FJ, Quinn KM, Brinkmann V, Le Gros G, Kirman JR. A key role for lung-resident memory lymphocytes in protective immune responses after BCG vaccination. Eur J Immunol. 2010;40:2482–2492. doi: 10.1002/eji.200940279. [DOI] [PubMed] [Google Scholar]
- 17.Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187:5510–5514. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woolard MD, Hensley LL, Kawula TH, Frelinger JA. Respiratory Francisella tularensis live vaccine strain infection induces Th17 cells and prostaglandin E2, which inhibits generation of gamma interferon-positive T cells. Infect Immun. 2008;76:2651–2659. doi: 10.1128/IAI.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, Cauley LS. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. 2014;95:215–224. doi: 10.1189/jlb.0313180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Guan T, Hu Y, Cauley LS, Craft J, Kaech SM. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity. 2014;41:633–645. doi: 10.1016/j.immuni.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGill J, Legge KL. Cutting edge: contribution of lung-resident T cell proliferation to the overall magnitude of the antigen-specific CD8 T cell response in the lungs following murine influenza virus infection. J Immunol. 2009;183:4177–4181. doi: 10.4049/jimmunol.0901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thawer SG, Horsnell WG, Darby M, Hoving JC, Dewals B, Cutler AJ, Lang D, Brombacher F. Lung-resident CD4(+) T cells are sufficient for IL-4Ralpha-dependent recall immunity to Nippostrongylus brasiliensis infection. Mucosal Immunol. 2014;7:239–248. doi: 10.1038/mi.2013.40. [DOI] [PubMed] [Google Scholar]
- 23.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 24.Glennie ND, Yeramilli VA, Beiting DP, Volk SW, Weaver CT, Scott P. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med. 2015;212:1405–1414. doi: 10.1084/jem.20142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta G, Oghumu S, Satoskar AR. Mechanisms of immune evasion in leishmaniasis. Adv Appl Microbiol. 2013;82:155–184. doi: 10.1016/B978-0-12-407679-2.00005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol. 2013;14:509–513. doi: 10.1038/ni.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346:98–101. doi: 10.1126/science.1254536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santosuosso M, McCormick S, Zhang X, Zganiacz A, Xing Z. Intranasal boosting with an adenovirus-vectored vaccine markedly enhances protection by parenteral Mycobacterium bovis BCG immunization against pulmonary tuberculosis. Infect Immun. 2006;74:4634–4643. doi: 10.1128/IAI.00517-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts LM, Wehrly TD, Crane DD, Bosio CM. Expansion and retention of pulmonary CD4+ T cells after prime boost vaccination correlates with improved longevity and strength of immunity against tularemia. Vaccine. 2017;35:2575–2581. doi: 10.1016/j.vaccine.2017.03.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.