

SUMMARY
DNA interstrand crosslinks (ICLs) are extremely cytotoxic, but the mechanism of their repair remains incompletely understood. Using Xenopus egg extracts, we previously showed that repair of a cisplatin ICL is triggered when two replication forks converge on the lesion. After CDC45/MCM2-7/GINS (CMG) ubiquitylation and unloading by the p97 segregase, FANCI-FANCD2 promotes DNA incisions by XPF-ERCC1, leading to ICL unhooking. Here, we report that, during this cell-free ICL repair reaction, one of the two converged forks undergoes reversal. Fork reversal fails when CMG unloading is inhibited, but it does not require FANCI-FANCD2. After one fork has undergone reversal, the opposing fork that still abuts the ICL undergoes incisions. Our data show that replication fork reversal at an ICL requires replisome disassembly. We present a revised model of ICL repair that involves a reversed fork intermediate.
In Brief
DNA interstrand crosslinks (ICLs) are extremely cytotoxic lesions that are mainly repaired in a replication-coupled manner. Using a cell-free system, Amunugama et al. report that, during ICL repair, replication forks undergo reversal. Fork reversal requires replicative CMG helicase unloading.
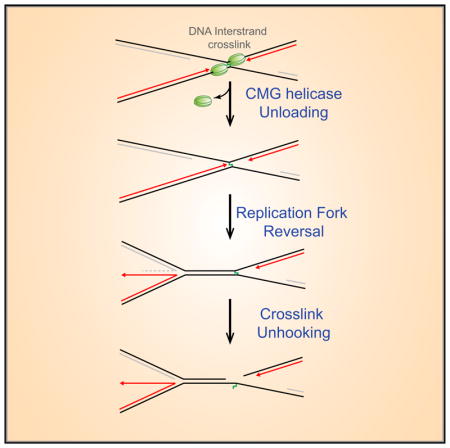
INTRODUCTION
DNA interstrand crosslinks (ICLs) impede DNA replication and transcription and are therefore extremely cytotoxic. In vertebrates, ICLs repair is coupled to replication and involves structure-specific endonucleases, translesion DNA polymerases, recombinases, and DNA helicases. Many of these proteins are mutated in Fanconi anemia (FA), a rare genetic disorder characterized by bone marrow failure, cancer predisposition, and hypersensitivity to ICLs (Clauson et al., 2013; D’Andrea, 2010; Deans and West, 2011; Kottemann and Smogorzewska, 2013). Although the FA pathway repairs ICLs induced by chemotherapeutic agents like cisplatin, the Fanconi anemia complementation group (FANC) proteins most likely evolved to counteract endogenous reactive aldehydes (Garaycoechea et al., 2012; Langevin et al., 2011; Rosado et al., 2011).
Using Xenopus egg extracts, we previously described a mechanism by which the FA pathway promotes cisplatin ICL repair in S phase (Figure 1A; Räschle et al., 2008). Repair is triggered when two DNA replication forks converge at the ICL, generating an “X-shaped” structure (Zhang et al., 2015). The leading strands of converging forks initially stall ~20–40 nt from the ICL due to the footprint of the CDC45/MCM2-7/GINS (CMG) helicase, which encircles and translocates along the leading strand template (Fu et al., 2011). The stalled CMGs then undergo ubiquitylation on their MCM7 subunits, and the p97 segregase removes the ubiquitylated CMG from chromatin, allowing approach of the leading strand to within 1 nt of the ICL (“−1” position; Fullbright et al., 2016; Long et al., 2014; Semlow et al., 2016). Concurrently, the FA core complex promotes ubiquitylation of FANCI-FANCD2, which binds near the lesion and recruits XPF(FANCQ)-ERCC1- and SLX4 (FANCP) (Klein Douwel et al., 2014; Knipscheer et al., 2009; Yamamoto et al., 2011). Unhooking allows a complex of the translesion DNA synthesis (TLS) polymerases REV1 and Pol ζ to mediate lesion bypass (Budzowska et al., 2015). Finally, the double-stranded break (DSB) generated by incisions is repaired by homologous recombination (HR) (Long et al., 2011).
Figure 1. Replication Fork Reversal Observed during ICL Repair.

(A) Current model of cell-free cisplatin ICL repair. See text for details.
(B) pICL was incubated in high-speed supernatant (HSS) of egg cytoplasm to license DNA and then supplemented with nucleoplasmic extract (NPE) to promote replication initiation (Walter et al., 1998; see Supplemental Experimental Procedures). 60 min after NPE addition, DNA was analyzed by EM, and representative images of late theta, figure 8, and reversed-fork intermediates, together with interpretive cartoons, are shown. Black arrowhead, reversed fork; red arrowhead, ssDNA on the lagging strand of the non-reversed fork.
(C) Quantification of late theta, figure 8, and reversed-fork intermediates. At the indicated times after NPE addition, samples were analyzed by EM as in (B). At least 100 interpretable molecules were analyzed for the quantification of repair intermediates at each time point. Error bars indicate the range in two independent experiments. A similar time-dependent decrease in figure 8 structures and increase in reversed forks was observed in Figures 2C and 2E.
The mechanism of ICL unhooking is unclear. On model fork substrates, XPF cuts the leading strand template within the duplex portion of the fork, 6 nt from the ICL (Abdullah et al., 2017; Figure S1A). This incised structure can be further processed by SNM1A, which digests past the ICL in the 5′ to 3′ direction, leading to ICL unhooking and formation of a one-ended double-stranded DNA (dsDNA) break (Figure S1A). This model is consistent with the epistatic relationship between XPF and SNM1A in ICL repair (Wang et al., 2011). In contrast, FAN1, which has a related activity to that of SNM1A (Wang et al., 2014), does not appear to operate in the FA pathway (Yoshikiyo et al., 2010; Zhou et al., 2012), and its depletion from egg extracts has no effect on unhooking (Klein Douwel et al., 2014). Importantly, the action of XPF-ERCC1 toward a converged fork intermediate is unlikely to be productive (Figure S1B). In summary, although strong evidence implicates XPF-ERCC1 and SNM1A in replication-dependent ICL unhooking, the structure they act on remains unclear.
Replication fork reversal, the reannealing of parental strands to create a four-way DNA structure (Holliday junction), was first postulated to occur in response to replication stress (Fujiwara and Tatsumi, 1976; Higgins et al., 1976). In bacterial and phage systems, partial replisome disassembly is a prerequisite for fork reversal (Manosas et al., 2012; Seigneur et al., 1998). Fork reversal has been observed in mammalian cells after exposure to a wide range of genotoxic agents (Neelsen and Lopes, 2015; Ray Chaudhuri et al., 2012; Zellweger et al., 2015). When forks stall in the absence of BRCA1 or BRCA2, fork reversal leads to the destruction of nascent strands (Kolinjivadi et al., 2017; Lemaçon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017). Based on these results, it has been proposed that fork reversal stabilizes and/or restarts stalled replication forks in wild-type cells, whereas it can cause fork degradation in cells lacking protective mechanisms (Neelsen and Lopes, 2015). However, the specific function of fork reversal in the damage response remains speculative, especially in eukaryotic cells.
Here, we show that ICL repair in Xenopus egg extracts is accompanied by fork reversal and that reversal requires unloading of the CMG helicase. We present a model of ICL repair in which fork reversal generates the substrate for ICL unhooking.
RESULTS
Replication Forks Undergo Reversal during ICL Repair in Xenopus Egg Extracts
To examine whether forks undergo reversal during ICL repair, we used egg extracts to replicate a plasmid containing a cisplatin ICL (pICL) (typically contaminated with ~7% undamaged plasmid; Knipscheer et al., 2009). Repair intermediates were cross-linked with psoralen and UV light to prevent branch migration (Neelsen et al., 2014), extracted, treated with E. coli single-strand binding protein (SSB) to decorate single-stranded regions, and examined by electron microscopy (EM). We grouped the observed DNA species into four categories (Figures S1C–S1E): (1) molecules that have not yet undergone incision (“pre-incision” intermediates), which include molecules where the replication forks have not reached the ICL (“late theta”), molecules where forks have converged on the ICL (“figure 8”), and a novel reversed fork intermediate (see below); (2) molecules that have undergone incisions (sigma and linear); (3) monomeric circular molecules that represent the ~7% of undamaged plasmids, or pICL plasmids that completed repair; and (4) uninterpretable structures containing multiple plasmids. 20 min after starting the reaction, pre-incision intermediates comprised primarily figure 8 and late theta structures (Figures 1B and 1C). However, later in the reaction, plasmids containing reversed forks (Figure 1B, black arrowhead) eventually became the dominant species (Figure 1C). At 45 min, reversed forks comprised 40% of all DNA on the grid (Figure S1D). Excluding circular molecules (most of which represent undamaged plasmids at this early time point; Räschle et al., 2008; Knipscheer et al., 2009) and sigma structures (which represent broken or incised figure 8 molecules), ~58% of pICL replication intermediates underwent reversal (Figure S1E). We conclude that replication fork reversal is a high-frequency event during pICL repair.
More detailed examination of reversed forks revealed that, in ~94% of cases, only one of the two forks underwent reversal (Figure 1B; data not shown), and these were usually fully coated with SSB (Figure 1B; see below). Given that both forks initially converged on the ICL, we infer that, after reversal of one fork, the ICL resides next to the un-reversed fork (Figure 1B, bottom cartoon). Because the nascent lagging strand undergoes active resection (Räschle et al., 2008), the reversed single-stranded DNA (ssDNA) represents nascent leading strands. In ~77% of the plasmids containing reversed forks, some SSB also bound to the non-reversed fork (e.g., Figure 1B, red arrowhead), probably due to ssDNA on the lagging strand template. To quantify fork reversal, we determined the length of the reannealed parental duplex (Figure S1F), which can be unambiguously measured, unlike the reversed leading strand that is compacted by SSB (Chrysogelos and Griffith, 1982). By 60 min, the median reversal reached ~1.5 kb (Figure S1F). Re-examination of our previous EM images of ICL repair (Räschle et al., 2008) also revealed indirect evidence of reversed forks (see Supplemental Experimental Procedures). Collectively, our data show that cell-free cisplatin ICL repair is accompanied by efficient reversal of one of the two forks that converge on the lesion.
CMG Unloading Is Required for Fork Reversal and DNA Incisions
We previously showed that fork convergence is essential for the first known step of ICL repair, CMG unloading (Zhang et al., 2015). To determine whether fork convergence is required for fork reversal, we prevented arrival of one fork at the ICL with a 1.5-kb array of 48 Lac repressors (LacR) (Figure 2A, middle arrow; Zhang et al., 2015). In this setting, nearly 100% of the plasmids appeared as late theta structures of the expected dimensions, and they lacked any evidence of fork reversal (Figures 2Bi, 2C, and S2A). In the absence of LacR (Figures 2Bii and 2C), reversal occurred as usual. To examine the role of CMG unloading more directly, we added NMS873 (“p97i”), an allosteric inhibitor of p97 that allows fork convergence but prevents CMG unloading (Figures 2A, right arrow, and S2A; Semlow et al., 2016; Fullbright et al., 2016). In the presence of p97i, ~90% of replicated pICL molecules appeared as catenated dimers, which were difficult to interpret (Figures 2Dii and 2E). We therefore digested the DNA with HincII (Figure 2A) to remove catenanes and performed EM. In the absence of p97i, ~80% of molecules contained two forks with a reversed tail (Figure 2Diii). In the presence of p97i, we observed simple X-shaped structures lacking reversed forks (Figure 2Div). We conclude that fork reversal during cell-free ICL repair requires CMG unloading.
Figure 2. Fork Convergence and CMG Unloading Are Required for Fork Reversal during ICL Repair.

(A) Model depicting replication fork convergence, CMG unloading, and fork reversal in an unperturbed reaction (left), in the presence of lacR (middle), and in the presence of NMS 873 (“p97i”; right).
(B) EM image of late theta structures (black arrows) in a LacR-treated reaction (i) or in a mock (buffer)-treated reaction (ii) at 90 min.
(C) At the indicated times, late theta, figure 8, and reversed-fork structures from the experiment shown in (B) were quantified and graphed. Error bars indicate the range in two independent experiments.
(D) EM images of reversed fork structures or catenated structures in the presence of mock- (i and iii) or p97i-treated (ii and iv) conditions, respectively, before and after HincII digestion. See text for details.
(E) Quantification of late theta, catenated molecules, figure 8s, and reversed forks by EM in a mock (DMSO)-treated or p97i-treated reaction. Error bars indicate the range in two independent experiments.
(F) pICL incision assay in a mock-treated (buffer) or p97i-treated reaction. pICL and an undamaged, internal control plasmid (pQnt) were nick translated with [α-32P] dATP before addition to extracts to induce replication and repair. Repair intermediates were recovered from extract and digested with HincII, separated by denaturing agarose gel electrophoresis, and visualized by autoradiography. Incisions result in loss of the large parental X-shaped structure (red strands in schematic; quantified in graph) and accumulation of a linear species. A similar result was seen in a second, independent experiment.
To directly test whether CMG unloading and fork reversal is required for incisions, we examined the effect of p97i on incisions. As shown previously (Knipscheer et al., 2009), the majority of pICL molecules normally underwent incisions, as measured by loss of the parental X-shaped structure generated after cutting by HincII (Figure 2F; average of 73% in two experiments). In contrast, in the presence of p97i, no significant incision was observed (Figure 2F). Collectively, these data show that CMG unloading is required for fork reversal and ICL unhooking.
Detection of Fork Reversal by Gel Electrophoresis
We next asked whether reversed forks can be detected via gel electrophoresis, as previously reported in bacteria (Fierro-Fernández et al., 2007). As shown in Figure 3A, fork convergence generated a discrete, “slow figure 8” intermediate (Figure 3A, red arrowhead; Räschle et al., 2008). Its slow mobility on gels after protein extraction is likely caused by the reannealing of ssDNA that was protected by CMG in the extract and subsequent catenation of the linked sisters (Figures S2Bi, S2Bii, and S2C). Next, a “fast figure 8” species appeared, which results from CMG unloading in the extract (Figures 3A, blue arrowhead, S2B, and S2C). The fast figure 8 structure subsequently underwent progressive retardation, generating species of intermediate mobility (Figure 3A, green arrowhead). Based on a diagnostic digest with the Holiday junction resolvase RuvC (Figures S2D and S2E) and EM of DNA extracted from agarose gels (Figures S2F and S2G), the intermediate mobility species corresponds to reversed forks. Quantification of these reversed forks at 45 min (Figure 3B, lane 3, green bar) shows that they comprise ~33% ± 1.2% (SEM; n = 5) of total replicated DNA and ~57% ± 0.8% (SEM; n = 5) of pre-incision intermediates, in reasonable agreement with our EM results (Figures S1D and S1E). We conclude that reversed forks are readily detected via gel electrophoresis, even without cross-linking.
Figure 3. Efficient Resection of the Nascent Lagging Strand Is Impaired when CMG Unloading Is Blocked.

(A) pICL plasmid was replicated in NPE containing [α-32P] dATP, and repair intermediates were separated on a native agarose gel and visualized by autoradiography. OC, open circular; SC, supercoiled.
(B) pICL was replicated in extract with [α-32P]dATP. 15 min after initiation of replication, extract was supplemented with cytosine arabinoside triphosphate (araCTP) (2 mM) or aphidicolin (50 μM). Replication intermediates were separated on a native agarose gel after deproteinization and visualized by autoradiography. Red arrowhead, slow figure 8; Blue arrowhead, fast figure 8; green arrowhead, reversed forks. To determine the absolute fraction of reversed forks, the radioactivity adjacent to the green bar in lane 3 was divided by the total radioactivity in the lane. To determine the fraction of pre-incision intermediates comprising reversed forks, the radioactivity in the green bar was divided by the radioactivity adjacent to the pink bar. The gel is representative of three independent experiments.
(C) Model depicting the rightward stalled fork in the presence of LacR, together with the BsaI site and primer used to generate the sequencing ladder.
(D) Same as (C) but in the presence of p97i.
(E) pICLLacO was replicated with [32P-α]dATP in the presence of LacR or p97i, and nascent strand products were analyzed by denaturing PAGE after digestion with BsaI. Red arrow, stalled leading strand; orange line, lagging strands of the rightward fork. The sequencing ladder was generated with primer R (C) and (D). Similar results were obtained in a second, independent experiment.
Approach Is Not Required for Fork Reversal
To address whether leading strand approach is required for fork reversal, we allowed forks to converge on the ICL and then added cytosine arabinoside triphosphate (araCTP), which terminates growing strands wherever cytosine is incorporated (Figure S2H, compare lane 6 with 13–18). AraCTP did not prevent the formation of fast figure 8 molecules, consistent with normal CMG unloading, or the accumulation of reversed fork structures (Figure 3B, lanes 11–14, green arrowhead). Interestingly, araCTP blocked generation of the well products, suggesting that the formation of stable joint molecules requires DNA synthesis. These conclusions were confirmed using EM (data not shown). Unlike araCTP, aphidicolin inhibited CMG unloading, as indicated by the persistence of the slow figure 8 (Figure 3B, lanes 15–21; its gradual decay is due to resection of nascent strands) and chromatin immunoprecipitation (ChIP) (Long et al., 2014). The greater effect of aphidicolin versus araCTP on CMG unloading may reflect aphidicolin’s greater inhibition of the leading strand polymerase (Figure S2H), whose activity might help evict CMG from the stalled forks (Long et al., 2014). Altogether, our data indicate that replication fork reversal does not require approach of leading strands to the ICL, indicating that the removal of CMG per se is essential for reversal.
Efficient Resection of the Nascent Lagging Strand Requires CMG Unloading
When forks converge on an ICL, the nascent lagging strand is resected (Räschle et al., 2008). To address how resection is affected by the inhibition of fork reversal, we blocked fork reversal with LacR (Figure 3C) or p97i (Figure 3D). We cut pICL repair intermediates 1.1 kb to the left of the ICL with BsaI to visualize nascent leading (Figure 3E, red arrow) and lagging strands (Figure 3E, orange bracket). Compared to the control, inhibition of fork convergence or CMG unloading, both of which prevent reversal, led to a dramatic stabilization of lagging strands (Figure 3E). These results suggest that efficient resection of nascent lagging strands is impaired when fork reversal is inhibited due to defective CMG unloading.
The Non-reversed Fork Abutting the ICL Is Subject to DNA Incisions
We envisioned three possibilities how fork reversal interfaces with ICL repair. First, after lagging strand resection, the reversed leading strand invades near the ICL, re-generating a figure 8 structure with a D-loop at the crosslink (Seigneur et al., 1998), followed by incisions (Figure 4A, i and ii). Second, the reversed fork is restored and incisions occur on a simple figure 8 structure (Figure 4A, iii and iv). Third, the fork is not restored before incisions (Figure 4A, v). The first two scenarios predict that, if incisions are prevented, reversed forks should no longer be visible (Figure 4A, i and iii). In contrast, we found that depletion of FANCD2, which is required for incisions, led to accumulation of reversed forks (Figures S3A and S3B, green arrowhead). In addition, we saw no evidence of the D-loop structures that would be predicted if the regressed tail reinvaded the plasmid near the ICL (Figure 4A, i; data not shown). The third scenario predicts that, after unhooking, a “sigma” structure with a four-way junction containing the reversed arm will form (Figure 4A, v, gray box). 76% of observed sigma structures contained the expected structure (Figure 4B, black arrowheads). These data suggest that the non-reversed fork abutting the ICL is incised.
Figure 4. The Non-reversed Fork Abutting the ICL Is Subject to DNA Incisions.

(A) Model depicting three possible pathways of incision, after one fork has undergone reversal, and one pathway for breakage. The sigma structure expected if the non-reversed fork on the reversed intermediate undergoes incisions is highlighted in gray. See text for other details.
(B) EM image of pICL repair intermediates at 90 min in a mock-treated condition. Black arrowheads, sigma structures containing a reversed fork; red arrowheads, linear species. Similar results were seen in two other independent experiments.
(C) Quantification of linear structures during ICL repair in a mock-treated or p97i-treated reaction. Error bars indicate the range in two independent experiments. A time-dependent increase in linear species of similar magnitude was observed in two other experiments, but the data were not included in the quantification due to different time points or slightly different conditions.
(D) A series of 3′-radiolabeled (red asterisks) splayed arm and X-shaped substrates containing or lacking nascent strands (dotted arrows) were incubated with XPF-ERCC1 in the presence or absence of RPA for 60 min and the DNA analyzed by denaturing PAGE. M, radiolabeled marker oligonucleotides of indicated structures and sizes. Blue arrow, approximate position of incision. See Supplemental Experimental Procedures and Figure S3D for details of model substrate preparation.
In FANCD2-depleted extracts, we detected a large population of sigma structures (Figure S3C, brown arrowheads), which were also readily observed via agarose gel electrophoresis (Figure S3B). This indicates that, in the absence of FANCI-FANCD2, a significant fraction of reversed forks undergo either incision or breakage. Notably, only ~15% of such molecules contained a single-stranded tail at the four-way junction (Figure S3C; data not shown). These data suggest that, in the absence of FANCI-FANCD2, figure 8 structures undergo de-regulated breakage (Figure 4A, vii), consistent with a replication fork protection function of FANCD2 (Lossaint et al., 2013; Schlacher et al., 2012).
We previously showed that the nascent leading strand is extended past the unhooked ICL during TLS (Räschle et al., 2008). Such a mechanism implies that the reversed leading strand is reannealed to the parental strand before TLS (“restoration”), a process that would release the incised sister chromatid as a linear species (Figure 4A, vi). Indeed, there was a time-dependent increase in linear species in unperturbed reactions, but not in the p97i-treated condition (Figures 4B, red arrowheads, and 4C). As predicted (Figure 4A, vi), many of the linear molecules contained ssDNA at one or both ends (Figure 4B). Thus, whereas incisions take place on figure 8 molecules with reversed forks, TLS occurs after restoration of the leading strand to its original position.
XPF Cannot Incise an X-Shaped Structure
We asked whether XPF could also incise an X-shaped structure, such as might exist prior to fork reversal (Figure 4A, top). As shown previously (Abdullah et al., 2017), purified XPF-ERCC1 incised the leading strand template of ICL-containing DNA replication forks six nucleotides internal to the duplex DNA (Figure 4D, lanes 3, 4, and 7), and replication protein A (RPA) suppressed the inhibitory effect of a leading strand on this reaction (Figure 4D, lanes 6 and 7). However, XPF exhibited no activity toward X-shaped structures whether or not they contained one or two leading strands and regardless of whether RPA was present (Figure 4D, lanes 8–19). Failure to incise was probably due to the absence of duplex DNA adjacent to the ICL. This observation is consistent with the idea that reversal of one fork promotes ICL unhooking by XPF, because reversal re-establishes a stretch of duplex DNA adjacent to the lesion.
AP-ICL Repair Does Not Involve Fork Reversal
We recently identified a second replication-coupled ICL repair pathway in egg extracts that operates on AP (abasic)-ICLs and psoralen ICLs (Semlow et al., 2016). In this pathway, the ICL is unhooked by the DNA glycosylase NEIL3 without formation of a DNA double-strand break intermediate. As shown in Figures S3E and S3F, plasmids undergoing AP-ICL repair showed no evidence of fork reversal. The small number of reversed forks that may have been detected during psoralen ICL repair (Le Breton et al., 2011) probably reflects the small fraction of these lesions that are unhooked by the incision pathway (Semlow et al., 2016). Therefore, fork reversal is specific to the incision pathway of ICL repair.
DISCUSSION
We report that, after two forks converge on a cisplatin ICL, one fork undergoes reversal, leaving the ICL next to the non-reversed fork (Figure S4, i–iii). Fork reversal requires fork convergence and the p97 ATPase, strongly indicating it depends on dissociation of CMG from chromatin. In the bacteriophage T4, fork reversal also requires disassembly of the replicative helicase from the fork (Manosas et al., 2012). Reversal does not depend on FANCI-FANCD2, suggesting that it occurs independently of incisions. We propose that incision of the non-reversed fork by XPF on the 5′ side of the ICL creates an entry point for SNM1A to digest 5′ to 3′ past the ICL (Figure S4, v), effectively unhooking the crosslink. Our model explains why neither incision is detected in XPF-depleted extracts (Klein Douwel et al., 2014). After unhooking, the fork is restored by an unknown mechanism to allow TLS (Figure S4, vi and vii; see vi’–ix’ for a possible alternative pathway). Whereas fork convergence is essential to unload CMG and allow reversal, our evidence suggests that, after reversal of one fork, the incision step is exerted on a “single fork” (Figure S4, iii–v, gray box), as envisioned in earlier models (Deans and West, 2011; Niedernhofer et al., 2005; Thompson and Hinz, 2009). Whether the first fork to lose CMG undergoes reversal or whether reversal of one fork occurs stochastically after removal of both CMGs remains to be determined. Either way, we postulate that CMG blocks reversal by preventing access of DNA-remodeling enzymes to the fork. Our data provide a concrete model of how replication fork reversal promotes DNA repair in a eukaryotic organism.
The following considerations are consistent with the idea that fork reversal is required for ICL unhooking. First, given the high efficiency of reversal (at least 58%) and incisions (73%), these two events are likely part of the same pathway. Second, two manipulations that blocked fork reversal (LacR addition and p97i) also inhibited ICL unhooking. Third, DNA incisions occurred preferentially in the context of figure 8 structures where one fork had undergone reversal (Figure 4B). Fourth, purified XPF-ERCC1 was not able to cut an X-shaped structure, implying a need for fork remodeling. Finally, we did not observe fork reversal during AP-ICL repair (Figures S3E and S3F), which does not involve incisions. Nevertheless, we cannot exclude that fork reversal in our system is a non-productive intermediate. To provide more definitive evidence for the role of reversal in ICL repair, it will be important to identify and neutralize the motor protein that promotes reversal and examine the effect on repair.
In contrast to our observation that fork reversal at chemically defined ICLs requires the convergence of two replisomes, other studies show that single forks can undergo reversal (Neelsen and Lopes, 2015; Ray Chaudhuri et al., 2012; Zellweger et al., 2015; Kolinjivadi et al., 2017). Importantly, the agents used in these studies lead to the formation of ssDNA at the fork, indicative of helicase uncoupling. We therefore propose that, when CMG uncouples from the polymerase, fork reversal may not depend on CMG unloading. In this case, reversal would “park” the CMG in double-stranded DNA for possible re-activation when the reversed fork is restored.
In mammalian cells, when a single fork encounters an ICL, it can pass over the ICL and keep moving without unhooking the lesion (Figure S4, “traverse”; Huang et al., 2013). Traverse gives rise to a structure that closely resembles the one generated after fork convergence and CMG unloading (Figure S4, ii). We therefore propose that, after traverse, one fork undergoes the same reversal process we observed after replication fork convergence. Our data suggest a unified model in which fork reversal is a general prerequisite for unhooking of ICLs that cannot be processed by the NEIL3 glycosylase pathway, whether they are encountered by one or two forks.
In our EM data, the majority of regressed tails were largely single stranded (Figure 1B), suggesting that fork reversal promotes resection. Furthermore, inhibition of CMG unloading caused a defect in resection of nascent lagging strands (Figure 3E). An attractive hypothesis is that fork reversal promotes resection of the regressed tail during ICL repair. Fork-reversal-mediated resection has been proposed in phage T4 and vertebrate systems in response to fork stalling events with hydroxyurea or aphidicolin (Long and Kreuzer, 2008; Thangavel et al., 2015; Kolinjivadi et al., 2017; Taglialatela et al., 2017; Mijic et al., 2017; Lemaçon et al., 2017). How reversal promotes resection at an ICL is an interesting question for future investigation.
In summary, we propose a revised mechanism of ICL repair that unifies previous single- and dual-fork models. In this view, after fork convergence or traverse, reversal of one fork places the ICL into the context of a single fork, which is the ideal substrate for unhooking.
EXPERIMENTAL PROCEDURES
For details, see Supplemental Experimental Procedures.
Xenopus Egg Extracts and DNA Replication
All experiments involving Xenopus laevis have been approved by the Harvard Medical Area Institutional Animal Care and Use Committee. Xenopus egg extracts were prepared essentially as described (Lebofsky et al., 2009; Walter et al., 1998). Replication of a plasmid containing a site-specific cisplatin inter-strand (pICL) (Räschle et al., 2008; Knipscheer et al., 2009) and pICL with an array of 48 Lac operator sequences (pICLLacO) (Zhang et al., 2015) have been described previously.
Crosslinking of DNA and Sample Preparation for EM Analysis
pICL was licensed in high-speed supernatant (HSS), and replication was initiated after addition of nucleoplasmic extract (NPE). At the indicated times, reactions were terminated in replication stop buffer III (see Supplemental Experimental Procedures), and DNA was crosslinked with trimethylpsoralen (TMP) (Sigma) and irradiation with UV light at 365 nM. Purified DNA was incubated with E. coli SSB protein followed by fixation of DNA-protein complexes with 0.3% glutaraldehyde and further purified by size-exclusion chromatography using agarose beads. Eluted fractions enriched for DNA-protein complexes were collected and mounted on grids for EM analysis.
Quantification of DNA Intermediates
Grids were examined either directly in the electron microscope and molecules were counted in order, as encountered, going across from top to bottom and from one side to the other and back so as to include all DNA in the viewed area, or they were imaged and subsequently analyzed. For quantification of figure 8, late theta, and reversed forks shown in Figures 1C, 2C, and 2E, only pre-incision intermediates were considered. In all cases, at least 100 pre-incision intermediates were counted. In Figures S1D and S1E, all DNA structures were accounted for. For quantification of how many sigma structures resembled the structure depicted in Figure 4Av, the 90-min mock time points of both repeats of the experiment shown in Figure 4C were examined.
Supplementary Material
Highlights.
Replication forks undergo reversal during repair of a defined DNA interstrand crosslink
Fork reversal requires replicative CMG helicase unloading
DNA incisions occur in the context of a single fork
Acknowledgments
We thank Kyle Vrtis, Gheorghe Chistol, and Denis Ptchelkine for preparation of reagents; Maria Ericson at the Harvard Medical School EM facility for help with EM; and Massimo Lopes, Ralph Scully, James C. Wang, David Long, Markus Räschle, and members of the Walter laboratory for helpful discussions. This work was supported by NIH grants HL098316 to J.C.W. and GM031819 and ES013773 to J.D.G. R.A.W. was supported by postdoctoral fellowship 131415-PF-17-168-01-DMC from the American Cancer Society. U.B.A. was supported by the Malaysia’s King Scholarship (Biasiswa Yang Di-Pertuan Agong). J.C.W. is an investigator of the Howard Hughes Medical Institute.
Footnotes
DATA AND SOFTWARE AVAILABILITY
Imaging data have been deposited in Mendeley Data and are available at https://doi.org/10.17632/h39pnpc265.1.
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.061.
AUTHOR CONTRIBUTIONS
R.A. and J.C.W. designed the experiments, interpreted the results, and prepared the manuscript. R.A. performed most experiments. R.A.W. prepared Figures 3B and S2H. S.W. and J.D.G. consulted on, performed, and analyzed the EM experiments. U.B.A. and P.J.M. designed the nuclease substrates and analyzed data. U.B.A. performed the nuclease assays. A.H.E.-S. and T.B. designed the DNA substrates for the nuclease assay, and A.H.E.-S. synthesized the substrates.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Abdullah UB, McGouran JF, Brolih S, Ptchelkine D, El-Sagheer AH, Brown T, McHugh PJ. RPA activates the XPF-ERCC1 to initiate the processing of DNA interstrand crosslinks. EMBO J. 2017;36:2047–2060. doi: 10.15252/embj.201796664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzowska M, Graham TG, Sobeck A, Waga S, Walter JC. Regulation of the Rev1-pol ζ complex during bypass of a DNA interstrand cross-link. EMBO J. 2015;34:1971–1985. doi: 10.15252/embj.201490878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrysogelos S, Griffith J. Escherichia coli single-strand binding protein organizes single-stranded DNA in nucleosome-like units. Proc Natl Acad Sci USA. 1982;79:5803–5807. doi: 10.1073/pnas.79.19.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauson C, Schärer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol. 2013;5:a012732. doi: 10.1101/cshperspect.a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362:1909–1919. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro-Fernández M, Hernández P, Krimer DB, Schvartzman JB. Replication fork reversal occurs spontaneously after digestion but is constrained in supercoiled domains. J Biol Chem. 2007;282:18190–18196. doi: 10.1074/jbc.M701559200. [DOI] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Schärer OD, Walter JC. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146:931–941. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y, Tatsumi M. Replicative bypass repair of ultraviolet damage to DNA of mammalian cells: caffeine sensitive and caffeine resistant mechanisms. Mutat Res. 1976;37:91–110. doi: 10.1016/0027-5107(76)90058-0. [DOI] [PubMed] [Google Scholar]
- Fullbright G, Rycenga HB, Gruber JD, Long DT. p97 promotes a conserved mechanism of helicase unloading during DNA cross-link repair. Mol Cell Biol. 2016;36:2983–2994. doi: 10.1128/MCB.00434-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, Patel KJ. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–575. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J Mol Biol. 1976;101:417–425. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell. 2013;52:434–446. doi: 10.1016/j.molcel.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D, Boonen RACM, Long DT, Szypowska AA, Räschle M, Walter JC, Knipscheer P. XPF-ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell. 2014;54:460–471. doi: 10.1016/j.molcel.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. Smar-cal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol Cell. 2017;67:867–881e7. doi: 10.1016/j.molcel.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- Le Breton C, Hennion M, Arimondo PB, Hyrien O. Replication-fork stalling and processing at a single psoralen interstrand crosslink in Xenopus egg extracts. PLoS ONE. 2011;6:e18554. doi: 10.1371/journal.pone.0018554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebofsky R, Takahashi T, Walter JC. DNA replication in nucleus-free Xenopus egg extracts. Methods Mol Biol. 2009;521:229–252. doi: 10.1007/978-1-60327-815-7_13. [DOI] [PubMed] [Google Scholar]
- Lemaçon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, Vindigni A. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun. 2017;8:860. doi: 10.1038/s41467-017-01180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Kreuzer KN. Regression supports two mechanisms of fork processing in phage T4. Proc Natl Acad Sci USA. 2008;105:6852–6857. doi: 10.1073/pnas.0711999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Räschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84–87. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Joukov V, Budzowska M, Walter JC. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol Cell. 2014;56:174–185. doi: 10.1016/j.molcel.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Décaillet C, Gari K, Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell. 2013;51:678–690. doi: 10.1016/j.molcel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- Manosas M, Perumal SK, Croquette V, Benkovic SJ. Direct observation of stalled fork restart via fork regression in the T4 replication system. Science. 2012;338:1217–1220. doi: 10.1126/science.1225437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, Lopes M. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun. 2017;8:859. doi: 10.1038/s41467-017-01164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol. 2015;16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, Chaudhuri AR, Follonier C, Herrador R, Lopes M. Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol Biol. 2014;1094:177–208. doi: 10.1007/978-1-62703-706-8_15. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Lalai AS, Hoeijmakers JHJ. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat Struct Mol Biol. 2011;18:1432–1434. doi: 10.1038/nsmb.2173. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC. Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. Cell. 2016;167:498–511e14. doi: 10.1016/j.cell.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol Cell. 2017;68:414–430e8. doi: 10.1016/j.molcel.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol. 2015;208:545–562. doi: 10.1083/jcb.201406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutat Res. 2009;668:54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Sun L, Newport J. Regulated chromosomal DNA replication in the absence of a nucleus. Mol Cell. 1998;1:519–529. doi: 10.1016/s1097-2765(00)80052-0. [DOI] [PubMed] [Google Scholar]
- Wang AT, Sengerová B, Cattell E, Inagawa T, Hartley JM, Kiakos K, Burgess-Brown NA, Swift LP, Enzlin JH, Schofield CJ, et al. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011;25:1859–1870. doi: 10.1101/gad.15699211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Persky NS, Yoo B, Ouerfelli O, Smogorzewska A, Elledge SJ, Pavletich NP. Mechanism of DNA interstrand cross-link processing by repair nuclease FAN1. Science. 2014;346:1127–1130. doi: 10.1126/science.1258973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K, Jiricny J, Takeda S, Hirota K. Involvement of SLX4 in inter-strand cross-link repair is regulated by the Fanconi anemia pathway. Proc Natl Acad Sci USA. 2011;108:6492–6496. doi: 10.1073/pnas.1018487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikiyo K, Kratz K, Hirota K, Nishihara K, Takata M, Kurumizaka H, Horimoto S, Takeda S, Jiricny J. KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc Natl Acad Sci USA. 2010;107:21553–21557. doi: 10.1073/pnas.1011081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, Walter JC. DNA Interstrand Cross-Link Repair Requires Replication-Fork Convergence. Nature Publishing Group; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, Diaz K, Lach FP, Bennett GR, Gee HY, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. 2012;44:910–915. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.