

Molecular catalyst system enables recycling of polymeric consumer products.
Abstract
A transition metal catalyst system for the selective catalytic depolymerization of various polyester- and polycarbonate-based materials is presented. The use of a molecular ruthenium catalyst with selected triphos ligands enabled a selective hydrogenolysis of a large diversity of polymeric consumer products, paving the way to innovative and sustainable recycling strategies within a circular economy.
INTRODUCTION
A wide range of synthetic polymers are currently produced and designed to meet the various needs and requirements of industry and consumers. The raw materials for these durable products are predominantly centered on petroleum resources, and only recently is the possibility to use bio-based alternatives being industrially explored (1). Moreover, the steadily increasing production and use of these polymers are largely unsustainable, as efficient recycling strategies for these nonbiodegradable materials remain elusive (2). This results in a tremendous growth of waste plastics, and the environmental pollution with the respective material is becoming an enormous problem. Consequently, current research is trying to develop new strategies to either substitute traditional polymers by bio-based alternatives or effectively recycle postconsumer plastics. However, the former approach would not lead directly to sustainable polymers and a waste-free economy, as these new polymeric materials are often accompanied by comparable end-of-life problems. The direct reuse of polymeric matter is the straightforward approach, but rather limited to only selected applications. Regarding thermoplastic polymers, mechanical recycling can be accomplished by melding and remolding, representing a useful strategy for certain applications. However, despite the losses in polymer quality, political regulations on food packaging prevent a closed-loop recycling, exemplified on the difficult reuse of polyethylene terephthalate (PET) (3). Thus, almost 70% of postconsumed PET is currently used for the production of synthetic fibers (4). A more promising approach is the chemical recycling through depolymerization, but current chemical strategies such as hydrolysis (5–7), methanolysis (8, 9), and glycolysis (5, 10, 11) are not cost-competitive with the use of new petrol-based monomers (12).
Consequently, the European Environmental Agency is fostering research toward a “circular economy” with effective recycling strategies as a core task (13). The recycling strategies envisage a selective process in which polymer wastes are depolymerized back to their starting monomer. These compounds are then purified and reused to yield polymeric materials with comparable quality or to define educts/building blocks for new value-added polymeric products (14, 15).
The groups of D. Milstein and J. Robertson initiated research in this direction and used homogeneous catalysts with molecular hydrogen for a catalytic depolymerization of polyesters to alcohol monomers (16, 17). They presented a successful attempt of catalytic polymer breakdown via hydrogenolysis, using ruthenium-N,N,P-pincer complexes in the presence of base, and obtained high yields of the respective monomeric products. Later, T. Cantat extended this approach and used hydrosilanes as reduction equivalents to hydrogenate selected polymers (18). More recently, the group of M. Clarke enhanced the hydrogenolysis of PET by using innovative ruthenium-N,N,P-pincer complexes. However, high catalyst loadings, complex solvents, and long reaction times hamper so far the translation of this approach to consumer products and engineered polymers (19).
Herein, the selective catalytic hydrogenolysis using a molecular ruthenium complex of a variety of the most abundant polyester and polycarbonate polymers is presented, resulting in novel possibilities for efficient polymer recycling within a circular economy approach (Scheme 1).
Scheme 1. Reductive depolymerization of polymeric waste with a molecular ruthenium catalyst.

Recently, effective molecular catalysts for the reduction of challenging functionalities could be developed, exemplified by the highly versatile and stable ruthenium complex [Ru(triphos)tmm] [A, triphos = (1,1,1-tri(diphenylphosphinomethyl)ethane, tmm = trimethylenemethane] as privileged system. This important advancement moved this molecular catalyst, and especially recently optimized catalyst structure [Ru(triphos-xyl)tmm] [B, triphos-xyl=1,1,1-tri(bis(3,5-dimethylphenyl)phosphinomethyl)ethane], into the spotlight for novel transformations and in special cases closer to the harsher processing conditions of heterogeneous catalyst systems (20, 21). Consequently, the application of these systems in the challenging transformation of robust and stable polymeric materials was envisaged, using molecular hydrogen for the respective hydrogenolysis reaction.
RESULTS AND DISCUSSION
In a first set of experiments, the selected polyester and polycarbonate materials were used for the initial investigations (for details on polymer grade, see the Supplementary Materials). Referring to the repetition unit of the polymers, 1 mmol of each polymer granulate was transformed, using 1 mole percent (mol %) catalyst and bis(trifluoromethanesulfonyl)imide (HNTf2) as cocatalyst in 1,4-dioxane (Table 1).
Table 1. Hydrogenolysis of selected polyesters and polycarbonate material using [Ru(triphos)tmm] (A) or [Ru(triphos-xyl)tmm] (B) and HNTf2.*.
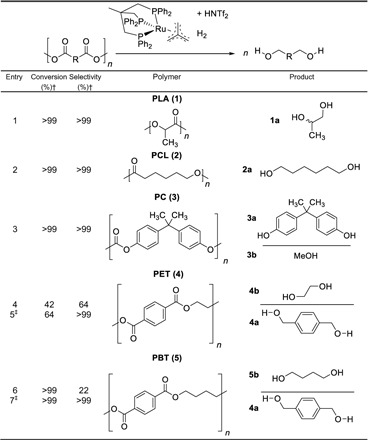
*Reaction conditions: H2 (100 bar), polymer (1 mmol; calculated on the repetition unit), 1 mol % [Ru(triphos)tmm] (A) and HNTf2, 3 ml of 1,4-dioxane, 140°C, 16-hour reaction time.
†Conversion and selectivity were determined by 1H NMR spectroscopy using mesitylene as internal standard.
‡[Ru(triphos-xyl)tmm] (B) (1 mol %) was used instead of A.
The first reaction was conducted in the presence of catalyst A with the aliphatic polyester polylactic acid (PLA, 1). In this transformation, full conversion could be achieved, and only 1,2-propanediol (1a) was obtained as product, confirming the suitability of this molecular catalyst for the hydrogenolysis of polyesters (Table 1, entry 1). Subsequently, polycaprolactone (PCL, 2) was converted in a high-pressure autoclave, and only 1,6-hexanediol (2a) could be detected in the product mixture (Table 1, entry 2). Exemplarily for polycarbonates, the most abundant representative, based on bisphenol A (PC, 3), was hydrogenated using [Ru(triphos)tmm] and HNTf2 as catalyst system. The application of former stated reaction conditions gave full conversion toward bisphenol A (3a) and methanol (3b; Table 1, entry 3). The hydrogenolysis of PET (4), the most common polyester, was more challenging, and only 42% conversion with a good selectivity of 64% toward 1,4-benzene dimethanol (4a) and ethylene glycol (4b) was achieved (Table 1, entry 4). Besides the formation of the respective diols, the acid-activated catalyst tends to catalyze the consecutive formation of ether products, strongly reducing the selectivity of the transformation. This effect was even more pronounced in the hydrogenolysis of polybutylene terephthalate (PBT, 5), where the polymeric material was fully converted, but only 22% of product could be assigned to the monomeric diols 4a and 1,4-butandiol (5b; Table 1, entry 6) (22). In these reactions, nuclear magnetic resonance (NMR) studies of the postreaction solution revealed the formation of a catalyst dimer and connected liberation of free acid as basis for the strongly enhanced etherification.
Consequently, a molecular catalyst suppressing these side reactions, should be established, enabling the selective formation of monomeric alcohols in the product solution. Recently, we introduced a ruthenium triphos complex showing higher activity and stability in many challenging hydrogenation reactions (20). The use of this modified catalyst B in the hydrogenolysis of PET (Table 1, entry 5) and PBT (Table 1, entry 7) confirmed the assumptions and excellent selectivity toward the respective diol species could be obtained in both cases.
In the subsequent development step, selected consumer products based on these polymers were investigated. As depicted in Fig. 1, various transparent or nontransparent (indicating partly crystalline domains in the respective material) PET sources were used. Thus, in the first set of experiments, a commercially available water bottle was cut into pieces and either shredded in a freeze mill or directly used without any further pretreatment. Subjecting those PET flakes (4) to the established reaction conditions gave full conversion with excellent selectivity (>99%) toward the diols 4a and 4b (Table 2, entry 1). Subsequently, the catalyst loading was decreased to 0.2 mol %, and again full conversion of the challenging substrate 4 could be obtained (Table 2, entry 2). Since remarkable results could be obtained with PET derived from a standard water bottle, PET based on other customary sources was tested.
Fig. 1. Overview of the commercially available PET sources used for the catalytic hydrogenolysis.

Table 2. Hydrogenolysis of PET of commercially available sources using [Ru(triphos-xyl)tmm] (B)/HNTf2 as catalyst.*.
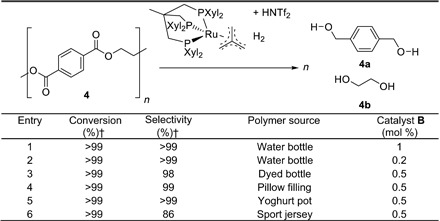
*Reaction conditions: H2 (100 bar), PET (2 mmol), 1 equivalent HNTf2 related on the catalyst [Ru(triphos-xyl)tmm] (B), 16 hours, 140°C, 4 ml of 1,4-dioxane.
†Conversion and selectivity were determined by 1H NMR spectroscopy using mesitylene as internal standard.
In detail, PET from a dyed soda bottle (Table 2, entry 3), a synthetic pillow filling (Table 2, entry 4), and a yoghurt pot (Table 2, entry 5) were used as complex starting materials. Before the hydrogenolysis reaction, the polymers were cut into pieces and used without any further treatment, as depicted in Fig. 1. As the substrates were only poorly soluble in the indicated solvents, the reaction had to be performed with the swollen polymers. Gratifyingly, all tested consumer products were fully converted to the diols 4a and 4b with excellent selectivity. In a challenging experiment, the depolymerization conditions were applied to a sport jersey, and again full conversion with a selectivity of 86% was obtained (Table 2, entry 6). These results indicate the broad applicability of the approach and, additionally, the negligible influence of polymer additives and color pigments on the catalyst activity.
After the successful demonstration of catalytic hydrogenolysis of polymers with catalyst structure B, the optimization of the system with respect to catalytic activity was targeted, paving the way to a broader applicability. In the first approach, PBT could be completely hydrogenated with a low catalyst loading of 0.2 mol % [Ru(triphos-xyl)tmm] B/HNTf2 (Table 3, entry 1). Second, PLA granulate was fully hydrogenolyzed using an even lower catalyst loading of 0.01 mol %, giving a maximum turnover number (TON) of 10,000 (Table 3, entry 2). Striving toward the depolymerization of consumer products, PLA obtained from a beverage cup, was used for further experiments. However, repeating the former experiment with customary PLA gave only 35% conversion (Table 3, entry 3) (for further information, see the Supplementary Materials). Thus, the reaction medium was altered, and excellent performance was observed with 1,2-propanediol as a solvent, yielding full conversion under standard hydrogenolysis conditions with 0.1 mol % [Ru(triphos)tmm] A/HNTf2 (Table 3, entry 4), facilitating the subsequent purification and downstream processing.
Table 3. Hydrogenolysis of polymer granules and commercially available products using [Ru(triphos)tmm] (A)/[Ru(triphos-xyl)tmm] (B) and HNTf2 as catalyst.*.
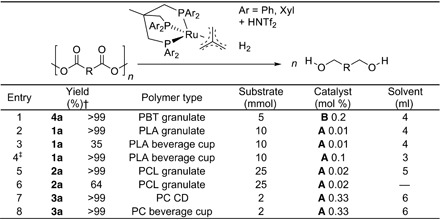
*Reaction conditions: H2 (100 bar), x mol % [Ru(triphos)tmm] (A)/[Ru(triphos-xyl)tmm] (B) and HNTf2, 16-hour reaction time, 140°C, x ml of 1,4-dioxane.
†Yield was determined via 1H NMR spectroscopy using mesitylene as internal standard.
‡Reaction was performed in 3 ml of 1,2-propanediol.
The second pure aliphatic polyester, PCL, could be hydrogenated to 2a with a low catalyst loading of only 0.02 mol % (Table 3, entry 5). Because of its rather low melting point (Tm, ~65°C) and high decomposition temperature (TD, ~350°C), the hydrogenolysis was performed at 140°C in the polymer melt and thus in the absence of external solvent (23). This resulted in 64% conversion (TON 3167) toward 2a (Table 3, entry 6; for further experiments, see the Supplementary Materials). Demonstrating the application of the polycarbonate hydrogenolysis on consumer products, a compact disc (CD) and a regular beverage cup were subjected to the hydrogenolysis conditions. Although commercially available PC contains impurities such as labeling dye, aluminum coating, or metallic glitter, it could be fully converted to 3a and 3b using only 0.33 mol % [Ru(triphos)tmm]/HNTf2 (Table 3, entries 7 and 8).
The properties of plastics are often tailored to the respective application by blending different polymers in the production procedure, either via a mechanical or a chemical process. This complexity in the material transforms the selective upcycling of those waste plastics into an even greater challenge. Thus, in the course of our investigations, the impact of complex polymer mixtures on the catalytic depolymerization reaction was tested. In the presence of polypropylene (PP), polyethylene (PE), nylon (PA 6), polyvinylchloride (PVC), or polystyrene (PS), the hydrogenolysis of PET was repeated under standard conditions. In these challenging experiments, full conversion of PET to 4a/4b was observed, and none of the described additional polymers hampered the selective depolymerization of the polyester. Moreover, most of the tested additional nonpolyester polymers were insoluble in 1,4-dioxane, opening a convenient pathway toward polymer separation.
On the basis of these findings, in the next development step, the separation of different polyesters via hydrogenolysis was investigated. 2 mmol PET and PLA from customary sources were depolymerized at different reaction temperatures (Fig. 2, top). Reaction temperatures of 140° and 120°C led to the full hydrogenolysis of both polymers, but lowering the reaction temperature resulted in a decreased conversion of PET, maintaining full conversion of PLA. Then, at 45°C, PET remained unconverted, while PLA got fully depolymerized (Fig. 2, top). Gratifyingly, this selectivity could also be achieved at 140°C if the solvent was changed to 1,2-propanediol 1a. Because of its insolubility in 1,2-propanediol and 1,4-dioxane, PET can be easily filtered off the reaction solution and depolymerized in a subsequent reaction. Consequently, this approach enables catalytic hydrogenolysis as a method to transform complex polymer mixtures by taking into consideration the respective physicochemical properties of the starting materials (Fig. 2, bottom).
Fig. 2. Selective separation of two different polyesters via the catalytic hydrogenolysis approach.
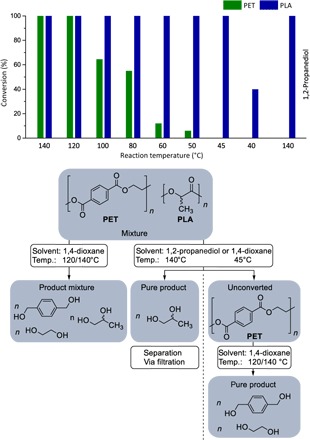
Top: Selective polymer hydrogenolysis of PLA and PET using [Ru(triphos-xyl)methylallyl]NTf2; reaction conditions: PLA (2 mmol), PET (2 mmol), [Ru(triphos-xyl)methyllaly]NTf2 (10 μmol), 1,4-dioxane (4 ml), H2 (100 bar), 16 hours. Conversion and selectivity were determined by 1H NMR spectroscopy using mesitylene as internal standard. Bottom: Flow diagram, illustrating the envisioned processing of polymer mixtures.
In the final experiments, the possibility to translate this development to a larger scale was investigated. Thus, a complete coated PLA beverage cup was cut into pieces and subjected to the established hydrogenolysis conditions in a 500-ml autoclave (Fig. 3 and Table 4). Gratifyingly, at a low catalyst loading of 0.05 mol % [Ru(triphos)tmm] A/HNTf2, full conversion to 1,2-propanediol 1a could be achieved, highlighting the feasibility of the novel approach (Fig. 3, top row, and Table 4, entry 1). Solids originating from the labeling of the cup could be easily filtered off the postreaction solution, as they are insoluble in the solvent. Subsequently, the most abundant polyester, PET, was hydrogenated on a gram scale using an entire 0.5-liter water bottle with screw cap (PP) and label (PE). Using a rather low catalyst loading of 0.2 mol %, 4 was selectively depolymerized to diols 4a and 4b, while PP and PE remained molten but unconverted (Fig. 3, middle row, and Table 4, entry 2). The unconverted polymers were simply filtered off the solution and could be easily transferred to a subsequent recycling process. Performing the reaction in the absence of screw cap and label with only 0.1 mol % catalyst (TON 1000) resulted in full conversion and a homogeneous solution without any residues (Table 4, entry 3). Remarkably, the scale-up experiment allowed a lowering of the minimum catalyst loading, thus highlighting the possibility for future optimization strategies. In a final reaction, a whole CD was cut into pieces and could be fully converted to 3a and 3b using 0.5 mol % [Ru(triphos)tmm]/HNTf2 (Fig. 3, bottom row, and Table 4, entry 4). Again, the solid residues from the aluminum layer and the coating remained unconverted and could be easily separated by filtration.
Fig. 3. Hydrogenolysis up-scaling for selected polyesters and polycarbonate consumer products.

Table 4. Hydrogenolysis scale-up of consumer polymer products using [Ru(triphos)tmm] (A)/[Ru(triphos-xyl)tmm] (B) and HNTf2.*.
| Entry | Conversion (%)† | Selectivity (%)† | Polymer source (g) | Catalyst (mol %) |
| 1 | >99 | >99 | Drinking cup, PLA (11.4) | A 0.05 |
| 2‡ | >99 | 98 | 0.5-Liter water bottle, PET (13.2) + screw cap, PP and labeling, PE (2.9) | B 0.2 |
| 3‡ | >99 | >99 | 0.5-Liter water bottle, PET (13.2) | B 0.1 |
| 4 | >99 | >99 | CD, PC (16.1) | A 0.5 |
*Reaction conditions: The reactions were performed in a 500-ml steel autoclave using 16-hour reaction time, x mol % [Ru(triphos)tmm] (A) and HNTf2, 140°C, 120 ml of 1,4-dioxane, and 90-bar constant hydrogen pressure.
†Conversion and selectivity were determined by 1H NMR spectroscopy using mesitylene as internal standard.
‡[Ru(triphos-xyl)tmm] (B) and HNTf2 were used.
CONCLUSION
In conclusion, the effective and selective catalytic depolymerization of waste polyesters/polycarbonates could be established, and the key enabler for this tailored recycling concept was the development of adapted molecular catalyst systems. On the basis of this approach, the polymers from these waste plastics are transformed to the respective value-added diols, enabling a recycling concept within the circular economy vision. Moreover, the detailed studies on a larger scale revealed tolerance of the catalyst toward various polymer additives and even allowed the consecutive transformation and separation of polymer mixtures. In addition, the effectiveness and impact of this method can be enlarged, envisioning a catalytic process with “green hydrogen” from water electrolysis based on renewable energy. Moreover, the catalytic combination of recycled diols with carbon dioxide allows the sustainable synthesis of linear and cyclic acetals only from waste carbon resources (24, 25), and thus, this strategy may pave the way to innovative open-loop recycling methods.
MATERIALS AND METHODS
General procedure for the hydrogenative depolymerization
Polymer (1 mmol; referring to its repetition unit) was filled in a glass insert, equipped with a stir bar, and subsequently placed in a high-pressure stainless steel autoclave. [Ru(triphos)(tmm)] (7.8 mg, 0.01 mmol) or [Ru(triphos-xyl)tmm] (9.5 mg, 0.01 mmol), HNTf2 (2.8 mg, 0.01 mmol), and 1,4-dioxane (3 ml) were added through a cannula under argon. Once the autoclave was sealed and pressurized with hydrogen (100 bar), the reaction mixture was stirred for 16 hours at 140°C. Afterward, the reaction was cooled in an ice bath, and the pressure was carefully released. The crude reaction mixture was analyzed by 1H NMR spectroscopy, using mesitylene as internal standard.
Supplementary Material
Acknowledgments
Funding: This work was supported in part by the Cluster of Excellence “Tailor-Made Fuels from Biomass,” which is funded by the Excellence Initiative by the German Federal and State Governments to promote science and research at German universities, and by the German Federal Ministry of Education and Research within the Kopernikus Project P2X: Flexible use of renewable resources—Exploration, validation and implementation of “Power-to-X” concepts. Further, we are grateful for a gift of ruthenium precursor from Umicore AG. Author contributions: S.W. and J.K. conceived the idea. S.W. and J.I. performed the experiments. S.W., J.I., and J.K. designed the experiments, analyzed the results, and wrote the manuscript. J.K. guided the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat9669/DC1
Section S1. General procedure
Section S2. Synthesis of the catalysts
Section S3. General procedure for autoclave reactions
Section S4. Additional Information
Section S5. NMR spectroscopy data of the products
Section S6. Crude NMR spectra of the products
Section S7. NMR spectra of the isolated products
Table S1. Hydrogenolysis of PLA derived from S-PLA granulate and a beverage cup using [Ru(triphos-derivative)tmm] complexes and HNTf2.
Table S2. Hydrogenolysis of PET with [Ru(triphos-xyl)tmm] and HNTf2 in the presence of a polymer “additive/impurity.”
Table S3. Hydrogenolysis of PCL in the polymer melt using [Ru(triphos)tmm] and HNTf2.
Table S4. Separation of PLA and PET via selective hydrogenolysis at low temperatures using [Ru(triphos-xyl)tmm] and HNTf2.
Fig. S1. Pressure drop curves of the hydrogenolysis of PCL with different molecular weights using [Ru(triphos)tmm] and HNTf2 as catalyst.
Fig. S2. Pressure drop curve of the hydrogenolysis of polyesters and polycarbonates.
Fig. S3. 1H NMR spectrum (600 MHz) [0 to 7.5 parts per million (ppm)] of the crude 1,4-dioxane reaction mixture of the PLA (1) hydrogenolysis to 1,2-propanediol (1a).
Fig. S4. 13C NMR spectrum (150 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the PLA (1) hydrogenolysis to 1,2-propanediol (1a).
Fig. S5. 1H NMR spectrum (600 MHz) (0 to 7.5 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PCL (2) to 1,6-hexanediol (2a).
Fig. S6. 13C NMR spectrum (150 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PCL (2) to 1,6-hexanediol (2a).
Fig. S7. 1H NMR spectrum (400 MHz) (0 to 7.5 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PET (4) obtained from a water bottle to benzene dimethanol (4a) and ethylene glycol (4b).
Fig. S8. 13C NMR spectrum (100 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PET (4) obtained from a water bottle to benzene dimethanol (4a) and ethylene glycol (4b).
Fig. S9. 1H NMR spectrum (400 MHz) (0 to 7.5 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PBT (5) to benzene dimethanol (4a) and 1,4-butanediol (5b).
Fig. S10. 13C NMR spectrum (100 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PBT (5) to benzene dimethanol (4a) and 1,4-butanediol (5b).
Fig. S11. 1H NMR spectrum (400 MHz) (0 to 7.5 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of polycarbonate (bisphenol A) (3), obtained from a CD, to 4,4′(propane-2,2-diyl)diphenol (bisphenol A, 3a) and methanol (3b).
Fig. S12. 13C NMR spectrum (75 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of polycarbonate (bisphenol A) (3), obtained from a CD, to 4,4′(propane-2,2-diyl)diphenol (bisphenol A, 3a) and methanol (3b).
Fig. S13. 1H NMR spectrum (400 MHz) (0 to 7.5 ppm) of the isolated 1,2-propanediol (1a) obtained from a postconsumed beverage cup in CDCl3.
Fig. S14. 13C NMR spectrum (100 MHz) (10 to 230 ppm) of the isolated 1,2-propanediol (1a) obtained from a postconsumed beverage cup in CDCl3.
Fig. S15. 1H NMR spectrum (400 MHz) (0 to 12.5 ppm) of the isolated bisphenol A (3a) obtained from a postconsumed CD in dimethyl sulfoxide (DMSO)–d6.
Fig. S16. 13C NMR spectrum (100 MHz) (0 to 230 ppm) of the isolated bisphenol A (3a) obtained from a postconsumed CD in DMSO-d6.
Fig. S17. 1H NMR spectrum (400 MHz) (0 to 8 ppm) of the isolated 1,4-benzene dimethanol (4a) obtained from a postconsumed PET bottle in a mixture of CDCl3 and DMSO-d6.
Fig. S18. 13C NMR spectrum (100 MHz) (0 to 210 ppm) of the isolated 1,4-benzene dimethanol (4a) obtained from a postconsumed PET bottle in a mixture of CDCl3 and DMSO-d6.
REFERENCES AND NOTES
- 1.Hong M., Chen E. Y.-X., Chemically recyclable polymers: A circular economy approach to sustainability. Green Chem. 19, 3692–3706 (2017). [Google Scholar]
- 2.Geyer R., Jambeck J. R., Law K. L., Production, use, and fate of all plastics ever made. Sci. Adv. 3, e1700782 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.V. Komolprasert, P. Turowski, Food Law Compliance of Poly(ethylene Terephthalate) (PET) Food Packaging Materials (ACS Symposium Series; American Chemical Society, 2014), vol. 1162. [Google Scholar]
- 4.Karayannidis G. P., Achilias D. S., Chemical recycling of poly(ethylene terephthalate). Macromol. Mater. Eng. 292, 128–146 (2007). [Google Scholar]
- 5.Carta D., Cao G., D’Angeli C., Chemical recycling of poly(ethylene terephthalate) (pet) by hydrolysis and glycolysis. Environ. Sci. Pollut. Res. 10, 390–394 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Yoshioka T., Sato T., Okuwaki A., Hydrolysis of waste PET by sulfuric acid at 150°C for a chemical recycling. J. Appl. Polym. Sci. 52, 1353–1355 (1994). [Google Scholar]
- 7.Austin H. P., Allen M. D., Donohoe B. S., Rorrer N. A., Kearns F. L., Silveira R. L., Pollard B. C., Dominick G., Duman R., El Omari K., Mykhaylyk V., Wagner A., Michener W. E., Amore A., Skaf M. S., Crowley M. F., Thorne A. W., Johnson C. W., Woodcock H. L., McGeehan J. E., Beckham G. T., Characterization and engineering of a plastic-degrading aromatic polyesterase. Proc. Natl. Acad. Sci. U.S.A. 115, E4350–E4357 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genta M., Yano F., Kondo Y., Matsubara W., Oomoto S., Development of chemical recycling process for post-consumer PET bottles by methanolysis in supercritical methanol. Tech. Rev. 40, 1–4 (2003). [Google Scholar]
- 9.Mishra S., Goje A. S., Kinetic and thermodynamic study of methanolysis of poly(ethylene terephthalate) waste powder. Polym. Int. 52, 337–342 (2003). [Google Scholar]
- 10.Güçlü G., Kaşgöz A., Özbudak S., Özgümüş S., Orbay M., Glycolysis of poly (ethylene terephthalate) wastes in xylene. J. Appl. Polym. Sci. 69, 2311–2319 (1998). [Google Scholar]
- 11.Troev K., Grancharov G., Tsevi R., Gitsov I., A novel catalyst for the glycolysis of poly(ethylene terephthalate). J. Appl. Polym. Sci. 90, 1148–1152 (2003). [Google Scholar]
- 12.Zhang X., Fevre M., Jones G. O., Waymouth R. M., Catalysis as an enabling science for sustainable polymers. Chem. Rev. 118, 839–885 (2018). [DOI] [PubMed] [Google Scholar]
- 13.EEA, “Circular economy to have considerable benefits, but challenges remain” (European Environment Agency, 2016).
- 14.Stahel W. R., The circular economy. Nature 531, 435–438 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Kiser B., Circular economy: Getting the circulation going. Nature 531, 443–446 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Krall E. M., Klein T. W., Andersen R. J., Nett A. J., Glasgow R. W., Reader D. S., Dauphinais B. C., Mc Ilrath Sean P., Fischer A. A., Carney M. J., Hudson D. J., Robertson N. J., Controlled hydrogenative depolymerization of polyesters and polycarbonates catalyzed by ruthenium(II) PNN pincer complexes. Chem. Commun. 50, 4884–4887 (2014). [DOI] [PubMed] [Google Scholar]
- 17.E. Balaraman, B. Gnanaprakasam, C. Gunanathan, D. Milstein, J. Zhang, Novel ruthenium complexes and their uses in processes for formation and/or hydrogenation of esters, amides and derivatives thereof, U.S. Patent WO2012052996A2 (2013).
- 18.Feghali E., Cantat T., Room temperature organocatalyzed reductive depolymerization of waste polyethers, polyesters, and polycarbonates. ChemSusChem 8, 980–984 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Fuentes J. A., Smith S. M., Scharbert M. T., Carpenter I., Cordes D. B., Slawin A. M. Z., Clarke M. L., On the functional group tolerance of ester hydrogenation and polyester depolymerisation catalysed by ruthenium complexes of tridentate aminophosphine ligands. Chemistry 21, 10851–10860 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Meuresch M., Westhues S., Leitner W., Klankermayer J., Tailor-made ruthenium-triphos catalysts for the selective homogeneous hydrogenation of lactams. Angew. Chem. Int. Ed. Engl. 55, 1392–1395 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Westhues S., Meuresch M., Klankermayer J., Ruthenium-catalyzed modular synthesis of cyclic tertiary amines from lactams. Angew. Chem. Int. Ed. Engl. 55, 12841–12844 (2016). [DOI] [PubMed] [Google Scholar]
- 22.vom Stein T., Meuresch M., Limper D., Schmitz M., Hölscher M., Coetzee J., Cole-Hamilton D. J., Klankermayer J., Leitner W., Highly versatile catalytic hydrogenation of carboxylic and carbonic acid derivatives using a Ru-triphos complex: Molecular control over selectivity and substrate scope. J. Am. Chem. Soc. 136, 13217–13225 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Li S., Pignol M., Gasc F., Vert M., Synthesis, characterization, and enzymatic degradation of copolymers prepared from ε-caprolactone and β-butyrolactone. Macromolecules 37, 9798–9803 (2004). [Google Scholar]
- 24.Thenert K., Beydoun K., Wiesenthal J., Leitner W., Klankermayer J., Ruthenium-catalyzed synthesis of dialkoxymethane ethers utilizing carbon dioxide and molecular hydrogen. Angew. Chem. 128, 12454–12457 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Schieweck B. G., Klankermayer J., Tailor-made molecular cobalt catalyst system for the selective transformation of carbon dioxide to dialkoxymethane ethers. Angew. Chem. Int. Ed. Engl. 56, 10854–10857 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat9669/DC1
Section S1. General procedure
Section S2. Synthesis of the catalysts
Section S3. General procedure for autoclave reactions
Section S4. Additional Information
Section S5. NMR spectroscopy data of the products
Section S6. Crude NMR spectra of the products
Section S7. NMR spectra of the isolated products
Table S1. Hydrogenolysis of PLA derived from S-PLA granulate and a beverage cup using [Ru(triphos-derivative)tmm] complexes and HNTf2.
Table S2. Hydrogenolysis of PET with [Ru(triphos-xyl)tmm] and HNTf2 in the presence of a polymer “additive/impurity.”
Table S3. Hydrogenolysis of PCL in the polymer melt using [Ru(triphos)tmm] and HNTf2.
Table S4. Separation of PLA and PET via selective hydrogenolysis at low temperatures using [Ru(triphos-xyl)tmm] and HNTf2.
Fig. S1. Pressure drop curves of the hydrogenolysis of PCL with different molecular weights using [Ru(triphos)tmm] and HNTf2 as catalyst.
Fig. S2. Pressure drop curve of the hydrogenolysis of polyesters and polycarbonates.
Fig. S3. 1H NMR spectrum (600 MHz) [0 to 7.5 parts per million (ppm)] of the crude 1,4-dioxane reaction mixture of the PLA (1) hydrogenolysis to 1,2-propanediol (1a).
Fig. S4. 13C NMR spectrum (150 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the PLA (1) hydrogenolysis to 1,2-propanediol (1a).
Fig. S5. 1H NMR spectrum (600 MHz) (0 to 7.5 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PCL (2) to 1,6-hexanediol (2a).
Fig. S6. 13C NMR spectrum (150 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PCL (2) to 1,6-hexanediol (2a).
Fig. S7. 1H NMR spectrum (400 MHz) (0 to 7.5 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PET (4) obtained from a water bottle to benzene dimethanol (4a) and ethylene glycol (4b).
Fig. S8. 13C NMR spectrum (100 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PET (4) obtained from a water bottle to benzene dimethanol (4a) and ethylene glycol (4b).
Fig. S9. 1H NMR spectrum (400 MHz) (0 to 7.5 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PBT (5) to benzene dimethanol (4a) and 1,4-butanediol (5b).
Fig. S10. 13C NMR spectrum (100 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of PBT (5) to benzene dimethanol (4a) and 1,4-butanediol (5b).
Fig. S11. 1H NMR spectrum (400 MHz) (0 to 7.5 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of polycarbonate (bisphenol A) (3), obtained from a CD, to 4,4′(propane-2,2-diyl)diphenol (bisphenol A, 3a) and methanol (3b).
Fig. S12. 13C NMR spectrum (75 MHz) (0 to 160 ppm) of the crude 1,4-dioxane reaction mixture of the hydrogenolysis of polycarbonate (bisphenol A) (3), obtained from a CD, to 4,4′(propane-2,2-diyl)diphenol (bisphenol A, 3a) and methanol (3b).
Fig. S13. 1H NMR spectrum (400 MHz) (0 to 7.5 ppm) of the isolated 1,2-propanediol (1a) obtained from a postconsumed beverage cup in CDCl3.
Fig. S14. 13C NMR spectrum (100 MHz) (10 to 230 ppm) of the isolated 1,2-propanediol (1a) obtained from a postconsumed beverage cup in CDCl3.
Fig. S15. 1H NMR spectrum (400 MHz) (0 to 12.5 ppm) of the isolated bisphenol A (3a) obtained from a postconsumed CD in dimethyl sulfoxide (DMSO)–d6.
Fig. S16. 13C NMR spectrum (100 MHz) (0 to 230 ppm) of the isolated bisphenol A (3a) obtained from a postconsumed CD in DMSO-d6.
Fig. S17. 1H NMR spectrum (400 MHz) (0 to 8 ppm) of the isolated 1,4-benzene dimethanol (4a) obtained from a postconsumed PET bottle in a mixture of CDCl3 and DMSO-d6.
Fig. S18. 13C NMR spectrum (100 MHz) (0 to 210 ppm) of the isolated 1,4-benzene dimethanol (4a) obtained from a postconsumed PET bottle in a mixture of CDCl3 and DMSO-d6.