

Abstract
Background
Although methylene blue (MB) had long been proposed to counteract the effects of cyanide (CN) intoxication, research on its mechanisms of action and efficacy has been abandoned for decades. Recent studies on the benefits of MB in post-anoxic injuries have prompted us to reexamine the relevance of this historical observation.
Methods
Our study was performed in adult male Sprague–Dawley rats and on HEK293T epithelial cells. First, the effects and toxicity of MB (0–80 mg/kg) on circulation and metabolism were established in four urethane-anesthetized rats. Then nine rats received a lethal infusion of a solution of KCN (0.75 mg/kg/min) and were treated by either saline or MB, at 20 mg/kg, a dose that we found to be innocuous in rat and to correspond to a dose of about 4 mg/kg in humans. MB was also administered 5 min after the end of a sub-lethal exposure to CN in a separate group of 10 rats. In addition, ATP/ADP ratio, ROS production, mitochondrial membrane potential (Δψm) and cellular O2 consumption rate (OCR) were determined in HEK293T cells exposed to toxic levels of CN (200 μM for 10 min) before and after applying a solution containing MB (1–100 μM for 10 min).
Results
Methylene blue was found to be innocuous up to 50 mg/kg. KCN infusion (0.75 mg/kg/min) killed all animals within 7–8 min. MB (20 mg/kg) administered at the same time restored blood pressure, cardiac contractility and limited O2 deficit, allowing all the animals to survive, without any significant methemoglobinemia. When administered 5 min after a non-lethal CN intoxication, MB sped up the recovery of lactate and O2 deficit. Finally, MB was able to decrease the production of ROS and restore the ATP/ADP ratio, Δψm as well as OCR of epithelial cells intoxicated by CN.
Conclusions
The present observations should make us consider the potential interest of MB in the treatment of CN intoxication. The mechanisms of the antidotal properties of MB cannot be accounted for by the creation of a cyanomethemoglobinemia, rather its protective effects appears to be related to the unique properties of this redox dye, which, depending on the dose, could directly oppose some of the consequences of the metabolic depression produced by CN at the cellular level.
Keywords: Cyanide intoxication, methylene blue, mitochondrial poisons, circulatory failure, redox dyes
Introduction
Cyanide (CN), one of the most feared mitochondrial poisons, remains a significant source of intoxication in victims of smoke inhalation [1]. High intake of CN from the plant cassava is also linked to dreadful outbreaks of spastic paralysis with myoclonus in tropical regions of Africa [2]. The optimal strategy of treatment of CN intoxication is still debated, relying on symptomatic measures and antidotes [3–5]. These antidotes are aimed at (1) scavenging “free” CN, using cobalt containing molecules [6–8] and nitrite compounds [9–12] and/or (2) increasing CN elimination as thiocyanate, using sodium thiosulfate [10,13–15] or other sulfur donors, such as sulfanegen [16,17]. The present paper is revisiting the potential interest of a different therapeutic approach based on the redox cycle dye methylene blue (MB).
The first clinical report of the antidotal properties of MB against CN poisoning was published in 1932 [18]. A young man, who had ingested “15 grains of potassium cyanide in 4 oz. of water”, was admitted to the San Francisco Hospital [18]. While comatose, he received an aqueous solution of MB and regained consciousness within 5 min and “fully” recovered within fifteen. Following other experimental observations obtained in vitro and in vivo [19–21], also suggesting that MB could antagonize CN toxicity, a controversy rapidly grew on the mechanisms of MB antidotal properties [22,23]. The ensuing debate, which started between Dr. Matilda Moldenhauer Brooks, from the University of California [22] and Dr. William Wendel, from the University of Tennessee [23], was trying to resolve whether or not a methemglobinemia could have resulted from MB administration and have “trapped” free CN, accounting for the very intriguing protective properties of MB. Since MB was later found to treat methemoglobinemia [24–26] and novel molecules (nitrite or thiosulfate) were proposed, by Chen and Rose [27], to counteract the effects of CN intoxication, the interest in MB in treating CN intoxication fell into complete oblivion. For this reason, the questions related to the nature of MB–CN interactions and to the potential clinical relevance of these observations, have remained largely unanswered.
Methylene blue is a dye which possesses unique redox properties: as soon as it enters the blood, MB is readily reduced into its leuco-form, leucomethylene blue (LMB), by many of the reducing agents this molecule will encounter, e.g., NAD(P)H [28,29] or reduced glutathione [25,30,31]. MB is thus regarded as an oxidant, decreasing concentrations of NAD(P)H [32] or reduced glutathione and gaining electrons in the process. LMB, on the other hand, can then be re-oxidized into MB by molecules with a higher redox potential (such as O2 or most metallo-compounds), giving electrons in the process, and a new cycle of reduction can be initiated [32]. Diffusing rapidly into the cytoplasm and in the mitochondria of any cells, including neurons [33], the couple MB/LMB can exert different effects depending on the redox state of its immediate environment. The properties of MB under its oxidized and/or reduced forms are so multifarious that it is difficult to predict which mechanism(s) could be involved in the protection against CN intoxication. For example, in its oxidized form (high concentrations), MB acts as electron acceptor [25] and could indeed lead to a methemoglobiemia, a sink for CN present in the blood [23]. However, in its reduced form, LMB prevents such a phenomenon to occur but has been suggested to interact directly with the mitochondrial electron chain [34], providing electrons to the complexes I and III and/or allowing a partial restoration of the Krebs cycle [35], whenever NADH is oxidized by MB or even a resuscitation of the mitochondrial electron chain. The latter could, in turn, maintain some ATP production when the electron chain is in its reduced form by keeping the mitochondrial gradient of protons when complexes are inhibited [36–40] or ATP production through the TCA cycle [35]. There is no evidence, however, that such a recently proposed mechanism of action of MB is implicated in CN intoxication.
The present study was therefore undertaken to describe the metabolic, cardiovascular and respiratory responses to MB alone, at different doses, in order to determine what safe level(s) of MB could be used in sedated rats, in keeping to the dose used in humans. Then, the effects of MB were tested during a lethal CN intoxication [41] and following a non-lethal exposure to CN. In an attempt to pursue the debate initiated eight decades ago, we have tried to characterize the interactions between MB and CN in vivo, in solution and in the blood, and determine the effects produced by the couple LMB/MB on cellular metabolism.
Methods
Characterization of methylene blue solution
The commercial solution of MB (methylene blue, solution 10mg/ml, Akron) was analyzed by mass spectrometry (Waters Q-TOF Premier quadrupole/time-of-flight (TOF) mass spectrometer) and was found to contain almost exclusively MB (four methyl groups). The amount of demethylated compounds, Azure B or Azure A, was negligible (Supplementary Figure S1).
Animal preparation and measurements
Adult male Sprague–Dawley rats (402 ± 5 g) were studied. Number of animals used for each experiment is given in the result section. All experiments were approved by the Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee.
Anesthesia was initiated with a brief inhalation of isoflurane 3.5% in oxygen (O2) allowing an intra-peritoneal administration of urethane (1.2 g/kg). The rats were tracheostomized and mechanically ventilated, to maintain PaO2 around 80 mmHg, as previously described [41,42]. A catheter (PE-50 tubing) was inserted into the right femoral artery for continuous monitoring of systemic arterial blood pressure and arterial blood sampling. A similar catheter was inserted into the left ventricle via one of the carotid arteries for determination of left ventricular pressure and max dp/dt as previously described [41,42]. Venous catheters were inserted into the right femoral and right jugular veins for CN infusion and antidote administration, respectively. Adequate ventilation was monitored by periodic arterial blood gas measurements using i-STAT1 blood gas analyzer (Abaxis, Union City, CA, USA). The electrocardiogram was recorded by a bioelectric amplifier (Differential AC amplifier, Model 1700; A-M Systems, Sequim, WA, USA).
Expiratory flow was measured using a pneumotachograph (1100 Series; Hans Rudolph, Inc. Shawnee, KS, USA) [43] and minute ventilation was determined [44]. Mixed expired O2 and CO2 fractions were measured continuously using O2 (Oxystar-100; CWE Inc. Ardmore, PA, USA), CO2 (model 17630; VacuMed, Ventura, CA, USA) analyzers allowing the determination of pulmonary oxygen uptake (V̇O 2) and CO2 production (V̇O2) [44].
Experimental protocol
Effects of MB, dose response in the rat
Methylene blue (5 mg/ml in saline) was infused at a rate of 2 mg/kg/min until death occurred. Arterial blood was sampled for measurement of lactate and methemoglobin (GEM premier 4000 gas analyzer; Instrumentation Laboratory, Bedford, MA, USA).
In addition, another group of animals were followed over 1 h after administration of MB at a dose, i.e., about 20 mg/kg, that was deemed to be close to that in humans, (see discussion section). Lactate, MB and the spectrum of absorbance of hemoglobin were determined before and various times after the end of MB administration.
Effect of MB during intravenous potassium cyanide infusion
Cyanide solution was prepared as previously described [41] and infused at the rate of 0.75 mg/kg/min, which typically leads to death by Pulseless electrical activity (PEA) asystole within 10 minutes. PEA was defined as a systolic pressure below 20 mmHg associated with a pulse pressure of less than 5 mmHg [41]. A first group of animals received saline (used as control) throughout the period of CN exposure. Then, in a separate group of rat, CN was infused at the same rate during a period that corresponded to the longest delay required to produce a PEA in the saline group. In that group, MB was infused at the rate of 2 mg/kg/min during CN exposure.
Effect of MB after intravenous potassium cyanide infusion
KCN was infused at the same rate but for 5 min only. Therefore, CN exposure was interrupted 2–3 min before death. Arterial blood was sampled before and at the cessation of CN infusion, then at 5, 10, 15 and 30 min into the period of recovery from CN infusion. MB (20 mg/kg) or saline was administered 5 min into the period of recovery (infusion was maintained for 5 min).
In vitro interaction between MB and CN
Cyanide (KCN) was mixed with a solution containing MB in phosphate-buffered saline (PBS) or in blood. The samples were analyzed by spectrophotometry (Eppendorf BioSpectrometer® basic Eppendorf AG 22331 Hamburg, Germany) to determine MB concentration at 665 nm and the profile of absorbance of hemoglobin. CN was determined in the whole blood using the protocol of Lundquist and Sorbo [45] one minute after the reaction and an hour later. All data were obtained in triplicate.
Cell culture
HEK293T cells were obtained from American Type Culture Collection, maintained in Dulbecco’s modified Eagle’s medium, supplemented with 10% (v/v) fetal bovine serum (Lonza), 1 mM sodium pyruvate (Sigma-Aldrich USA), and 1% non-essential amino acids (Sigma-Aldrich, St. Louis, MO, USA). They were grown under a humidified atmosphere at 37°C (95 % air, 5 % CO2).
ATP/ADP assays
ADP/ATP Ratio Assay Kit (BioAssay Systems, Hayward, CA, USA) was used according to manufacturer’s instructions. The ATP/ADP assays were performed in triplicate and data are presented as the average of the ATP/ADP ratio. HEK293T cells were plated in 96 well plates at a density of 20,000 cells per well, 24 h prior to the experiment. Cells were treated with or without 200 μM CN for 10 min. MB (50 μM) was added for another 10 min, cells were then washed with PBS and ADP/ATP ratio was determined using the assay kit. In control experiments, MB was not added. The signal of fluorescence was also tested with MB versus PBS alone in the absence of cells to determine possible direct interaction between MB and the determination of ADP/ATP.
Flow cytometric analysis of mitochondrial ROS
HEK293T cells were seeded in cell culture flasks (25 cm2) at a density of 1 × 106 cells per condition. Cells were treated with and without CN 200 μM for 10 min, then were exposed to a solution of MB at various concentrations (1, 10, 50 100 μM) for 10 min. The control group was not treated. After three washes with PBS, mitochondrial superoxide indicator (MitoSOX Molecular Probes) was added and cells were incubated for 10 min at 37 °C (5% CO2). MitoSOX fluorescence level was determined using flow cytometry (Fortessa BD, Flow-cytometry Core Penn State Hershey).
Mitochondrial membrane potential (Δψm) determination
Δψm was assessed by a mitochondrial voltage-sensitive dye, 5,5′,6,6′-tetrachloro-1,1’,3,3′-tetraethylbenzimidazole carbocyanide iodide (JC-1, Molecular Probes). Mitochondria with intact membrane potential show red fluorescence, de-energized mitochondria cannot concentrate JC-1 and show green fluorescence. HEK293T cells were seeded at a density of 106 cells per condition. Cells were treated with or without CN (200 μM) for 10 min, and then various concentrations of MB (1, 10, 50, 100 μM) were added for another 10 min. The control group was not treated. HEK293T cells were incubated with 10 μM JC-1 in media for 30 min in the dark at 37 °C (5% CO2). After the cells were incubated with the dye, the different samples were fixed for 20 min in PAF 2%. After three washes with PBS, cells were subjected to FACS (fluorescence-activated cell sorting) analysis. The ratio of JC-1 aggregate (FL2, green) to monomer (FL1, red) intensity was calculated.
Oxygen consumption rate
HEK293T Cells were seeded in 96-well plates (1.5 ′ 105 cells/well; XF96 plates; Seahorse Bioscience, North Billerica, MA, USA) and incubated at 37 °C overnight. The following day, the medium was changed to XF Assay Medium (Seahorse Bioscience, Santa Clara, CA, USA) containing 2.5 mM glucose, 1 mM pyruvate, and 2 mM glutamine and the cells were incubated at 37 °C for 1 h. Steady-state (baseline) oxygen consumption rate was measured using an XF96 extracellular flux analyzer (Seahorse Bioscience, Santa Clara, CA, USA). The effect of adding MB or CN was investigated for various concentrations of MB (see result section). About 1 μM oligomycin was used to inhibit oxidative phosphorylation; while the addition of 1 μM Rotenone and Antimycin A were used to inhibit the mitochondrial complexes I and III, respectively.
Statistical analysis
All results are presented as mean ± SD (or ± SEM for cell culture data). All variables of interest over time were analyzed using either a Friedman test, a one-way repeated-measured ANOVA or a two-way repeated ANOVA, followed by Bonferroni’s post hoc comparisons (Graphpad Software, La Jolla, CA, USA) depending on the data. p < .05 was regarded as significant for any of these comparisons.
Results
Metabolic and circulatory effects of MB infusion
Methylene blue infusion at a rate of 2 mg/kg/min in four rats produced a rise in V̇O2 (p < .001, Figure 1), Until the cumulative of dose of 50 mg/kg was reached. This increase in metabolism was associated to a proportional increase in minute ventilation (p < .001). Mean arterial blood pressure and left ventricular dP/dt max also significantly increased (Figure 1). Lactate concentration decreased significantly up to 50 mg/kg with no evidence of methemoglobinemia (Figure 2). Above 50 mg/kg, V̇O2 leveled off then rapidly decreased below baseline followed by a rapid depression in breathing, which led to an apnea at a cumulative dose ranging between 70 and 80 mg/kg (Figure 1). Mechanical ventilation was initiated, but did not correct the drop in cardiac contractility, blood pressure or V̇O2, leading to a PEA at the dose of 80 mg/kg.
Figure 1.
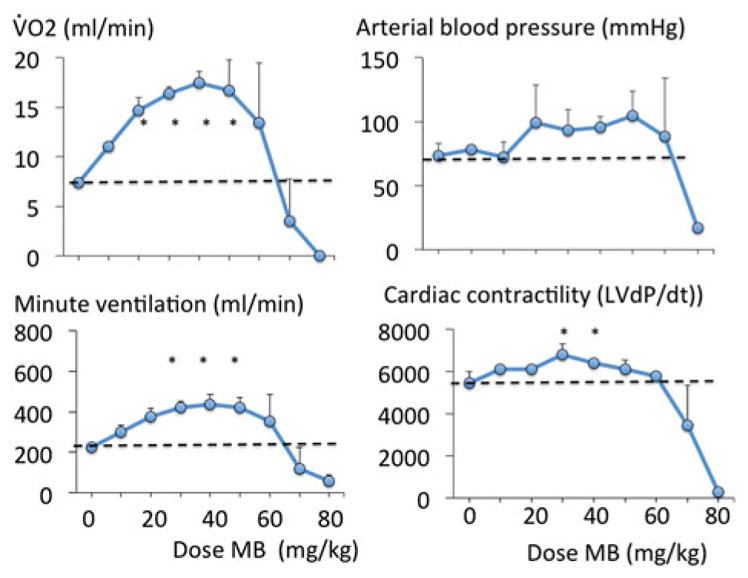
Effects of MB infusion during a ramp-like increase in MB cumulative dose (2 mg/kg/min) on V̇O2, arterial blood pressure, minute ventilation and cardiac contractility in four urethane-anesthetized rats. MB was very well-tolerated up to a cumulative dose of 50 mg/kg, producing an increase in metabolism, breathing and cardiac contractility. Above 50 mg/kg, a rapid decrease in blood pressure and cardiac contractility led to cardiogenic shock and death.
Figure 2.
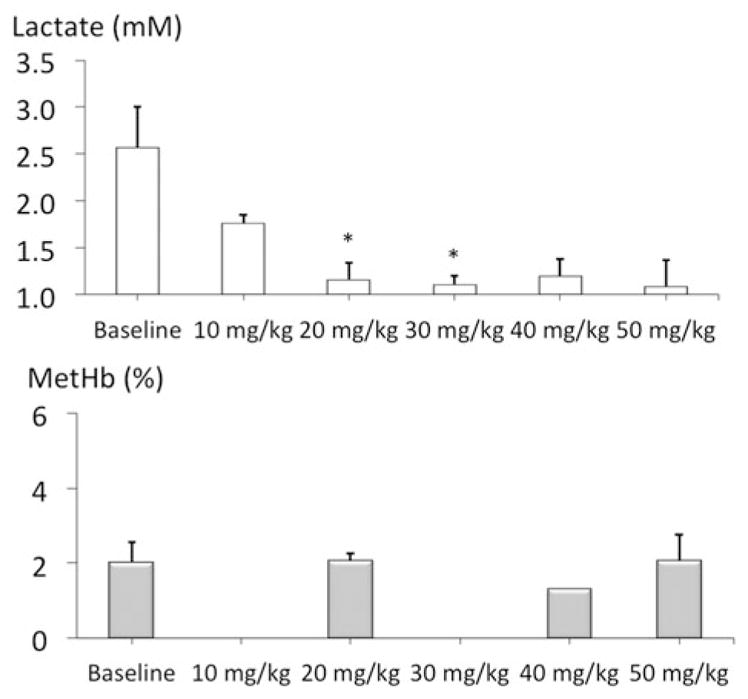
Lactate and methemoglobin concentrations during a ramp-like increase in MB cumulative dose (2 mg/kg/min). Note the decrease in blood lactate produced by MB and the lack of significant methemoglobinemia up to a cumulative dose of 50 mg/kg.
The dose of 20 mg/kg of MB was therefore selected for the rest of the study, as it was found to be innocuous and more importantly would be within the range recommended in humans (about 4 mg/kg), In keeping with the respective surface area of the two species (see discussion section for details). The effects of MB (20 mg/kg) were investigated in five rats over a 1-h recovery period. Five other rats received saline as control conditions. As expected from the ramp protocol, when compared to saline infusion, MB at the dose of 20 mg/kg increased V̇O2 and V̇CO2 (p < .001, Figure 3) but with a respiratory quotient ratio (QR = V̇CO2/V̇O2) that did not change from baseline (0.94 ± 0.05). Cardiac contractility, V̇O2 and body temperature, which increased during the infusion, returned to baseline within 1 h (Figure 4).
Figure 3.

Metabolic effects of MB at the dose of 20 mg/kg (2 mg/kg/min for 10 min) in five rats. This dose was the one used in the study (see also “Discussion”). Note that the increase in metabolism produced by MB consisted in a commensurate rise in V̇O2 and V̇CO2 (A) and no change in R (V̇CO2/V̇O2 ratio, B). (C) Averaged V̇O2 and arterial blood pressure before and at the end of MB infusion (10 min) along with O2 accumulation and body temperature changes (C) at the end of MB infusion (blue bars) compared to a group of five rats that received saline (open bars).
Figure 4.

Recovery (over 1 h) of the effects of MB infusion (20 mg/kg, 2 mg/kg/min for 10 min) in five rats. The changes in V̇O2, body temperature, arterial blood pressure, and cardiac contractility are displayed. Note that the increase in metabolism (V̇O2 and body temperature) or in cardiac contractility produced by MB spontaneously returned to baseline within 1 h.
Spectra of absorbance of the blood at the end of MB infusion
About 20 mg/kg MB did not change the profile of absorbance of hemoglobin in the blood of the rat that was sampled after the exposure, while the concentrations of MB averaged 99 ± 71 μM, 5 min after the end of MB infusion (Figure 5).
Figure 5.

Example of the spectrophotometry of blood samples from the same rat before and after receiving 20 mg/kg MB (left and middle panels), showing the lack of visible changes in the absorbance of oxyhemoglobin (two peaks), in the presence of MB reflecting the absence of significant methemoglobin. Note the peak of absorbance at 665 nm corresponding to MB (monomer).
Interactions between MB infusion and CN-induced lethal intoxication (n =9)
A rapid decrease in cardiac contractility, blood pressure, V̇O2 and heart rate was produced by KCN infusion (0.75 mg/kg/min) (Figure 6) leading to terminal asystole 7–9 min into CN exposure, as previously reported [41]. Infusion of MB (20 mg/kg), during a similar KCN exposure, maintained blood pressure and left ventricular contractility, preventing death in all rats (Figure 6 and 7). The O2 deficit was significantly reduced by MB when compared to the non-treated intoxicated animals. After cessation of CN exposure, the MB group displayed a rapid recovery of V̇O2 and circulation (Figure 8). All rats were alive at 30 min with blood lactate concentrations remained elevated as shown in Figure 8.
Figure 6.

Circulatory effects of an infusion of 0.75 mg/kg/min KCN with and without concomitant infusion of MB (20 mg/kg). The effects of MB alone are represented with black symbols (right panels). Upon infusion of CN, arterial blood pressure (BP), along with cardiac contractility, decreased leading to a rapid asystole. Despite receiving the same total dose of CN, the most significant effect of MB was the restoration of blood pressure and maintenance of left ventricular contractility preventing death in all rats exposed to a lethal intoxication by CN.
Figure 7.

Effects of an infusion of 0.75 mg/kg/min KCN with and without concomitant infusion of MB (20 mg/kg infused over 8 min) on V̇O2 and O2 deficit. The effects of MB alone are represented with black symbols (right panels). The O2 deficit was significantly lower in the group treated by MB exposed to CN than in the group intoxicated by CN without antidote.
Figure 8.

Recovery (over 1 h) of left ventricular pressure, blood pressure, V̇O2 and lactate concentrations in the nine animals exposed to lethal dose of CN. The group that received CN is displayed in red, while the group of rats that received 20 mg/kg MB and CN is shown in blue. All variables were back to baseline within 20 min in the group treated with MB except for lactate, which concentration remains elevated during all the recovery period. Note that one of the rats in the treated group had lower recovery values for VO2 and dP/dt than the rest of the group up to 15 minutes, accounting for a larger standard deviation during this period.
MB administered 5 min after the end of non-lethal CN intoxication (n =10)
All the 10 rats received KCN (0.75 mg/kg/min) for 5 min and presented a rapid decrease in blood pressure, but circulation returned toward baseline after CN infusion was stopped. All the animals survived. During the recovery period, the rats displayed a rapid increase in blood lactate concentration and developed a significant O2 deficit (Figure 9). MB infused 5 min into the recovery from CN exposure (n = 5), significantly decreased the concentration of lactate when compared to saline (n = 5) and allowed a much faster repayment of the O2 deficit produced by CN (Figure 9).
Figure 9.

(A) The left panel shows the lactate concentrations during the recovery period following the infusion of 0.75 mg/kg/min KCN for 5 min only. Blood was sampled after the end of CN exposure (time 0) then at 5, 10, 15 and 30 min. When CN infusion was stopped after 5 min, all animals survived and displayed a progressive rise in lactate reaching a maximum 10 min into recovery. The infusion of MB (20 mg/kg) or saline was started 5 min into recovery. Upon infusion of MB, lactate concentration dropped significantly (*p < .05). The administration of MB was associated (blue line) with a significantly faster recovery of O2 deficit (computed as the integral of V̇O2 over time, right panel) than when saline was infused (red line). (B) Concentrations of CN after 1 h of incubation in PBS alone (left panel) or in a solution of MB (65 μM/ml, middle panel) with an initial concentrations of CN of 200, 100, 50 and 25 μM, and in blood with an initial concentration of CN of 200 μM (right panel). CN concentrations were determined by spectrophotometry after reaction with sodium hypochlorite and barbituric acid-pyridine reagent (see method section for more details). The concentration of CN was not affected by the presence of MB in PBS but the it was lower in the blood in control condition than with MB, possibly reflecting a higher capacity of the blood to bind CN when MB was present (all tests were performed in triplicate).
In vitro interactions between MB and CN in solution
The concentrations of CN were identical in PBS whether or not incubated with MB (65 μM) at 1 or 60 min (Figure 9). In contrast, the concentration of CN incubated with blood and measured at 60 min, was higher in the presence of MB (Figure 9).
MB and mitochondrial function
Methylene blue restored ATP/ADP ratio depressed by CN (nine repetitions), as well as the level of mitochondrial membrane potential (Δψm, four repetitions) while limiting the production of ROS (six repetitions) (Figure 10). Of note, MB did not produce any artifact with ATP measurement and could not account for the modification of Δψm during CN (Figure 10).
Figure 10.

Effects of MB on ATP/ADP ratio, mitochondrial ROS and mitochondrial membrane potential (Δψm) during MB, CN and CN plus MB exposure in HEK293T cells. MB was able to restore most of the alterations of mitochondrial function produced by CN (see “Discussion” section for further details).
Exposing epithelial cells to MB resulted in an increase in O2 consumption (OR) (Figure 11, four repetitions) in a dose dependent manner. The blockade of any complex prevented the rise in OCR produced by MB, up to 50 μM (Figure 11). However, with larger doses of MB, the increase in OCR persisted after CN, rotenone or antimycin.
Figure 11.

Effects of MB on the O2 consumption rate (OCR) of HEK293T cells. MB produced an increase in OCR in a dose dependent manner. At the lowest concentrations, the effects of MB relied on the integrity of the mitochondrial electron chain, since the increase in O2 consumption rate was abolished after at blockade of the complexes I (by rotenone), III (by antimycin) or IV (by CN). At the highest doses of MB (above 50 μM), OCR remains elevated despite blockade of the mitochondrial complexes.
Discussion
We found that MB counteracted the effects of a lethal CN intoxication and prevented death by opposing the cardiovascular effects of CN. When injected after a non-lethal CN exposure, the recovery of lactate and the repayment of O2 deficit were dramatically sped up.
Effects of MB on metabolism and lactate
The most striking effect of MB in control animals was a two to threefold increase in metabolism (V̇O2 and V̇VO2) and body temperature. Similarly, in our cell cultures, MB increased V̇O2, echoing the historical observation that MB can increase O2 consumption and CO2 production in various tissues (46–48]. In vitro, the mechanisms involved appear to differ according to MB concentrations. At concentrations below 50 μM, MB-induced hyper-metabolism was abolished when the electron chain activity was partially or totally blocked (Figure 11), suggesting that MB increases O2 consumption through a mechanism that requires the integrity of the mitochondrial electron chain. At higher concentrations, similar to those produced in vivo (50–100 μM), the increase in O2 consumption was not affected by a partial or even a total inhibition of the mitochondrial electron chain activity. This “restoration” of O2 consumption after blockade of all complexes, including the complex IV, does not necessarily imply a recovery of the electron chain, as was previously proposed [36–40]. It may reflect the rate of re-oxidation of LMB by O2 (and consequently the rate of reduction of MB by NADH). In other words, whenever NADH [28,29] cannot be used/oxidized anymore by a blocked electron chain, it is still primarily oxidized by MB into LMB, while LMB will be re-oxidized by oxygen into MB, allowing a new redox cycle to take place. One can argue that, if true, this process should not be associated with any new ATP production, since the rate of O2 consumption is not anymore a marker of the electron chain activity, but only the reflection of the cyclic re-oxidation of MB in our cell culture preparation. This view is however difficult to reconcile with our in vivo data, wherein CO2 production always increases along with O2 consumption following MB administration, which implies that the Krebs (TCS) cycle must be involved. A possible explanation could be offered to resolve such a conundrum: whenever the electron chain activity is impeded, the ability of MB to maintain continuous oxidation of NADH at the mitochondrial level could have allowed some ATP production to be maintained by the glycolysis and the Krebs (TCA) cycle [35]. The accumulation of NADH (increased NADH/NAD ratio), when the electron chain activity is hindered, should have stopped the TCA cycle and produced an accumulation of lactate in the cytoplasm, as pyruvate cannot anymore be used in the mitochondria. The capacity of MB to oxidize NADH [28,29] could have allowed the TCA cycle to resume its activity, even in the absence of a functional electron chain. This could explain why in conditions of mitochondrial poisoning, molecules of ATP can still be produced by substrate-level phosphorylation in the mitochondria matrix via the TCA cycle, in the presence of MB, as has been recently suggested [35].
The observation that blood lactate concentrations decreased following MB administration is not a new finding [49]. This decrease could be accounted for by the consumption by MB of NADH, not available anymore for the transformation of pyruvate to lactate, which requires NADH as a co-enzyme [50]. In addition, the TCA cycle activity being “stimulated” by the consumption of NADH by MB in the mitochondria, allows pyruvate to be used in the mitochondria after transformation into acetyl-CoA, in turn decreasing the formation of lactate. In short, the decrease in lactate should be seen as a marker of (1) the oxidation of NADH by MB in the cytoplasm of the cells, limiting the formation of lactate from pyruvate, and (2) the utilization of pyruvate by a “stimulated” TCA cycle, as NADH/NAD ratio decreases in the mitochondria due the oxidative effects of MB [35].
Mechanisms of the antidotal effects of MB against CN intoxication
The antidotal action of MB against CN toxicity was initially assumed to be mediated via the production methemoglobinemia [23] (produced by the oxidative property of MB) causing ferric iron to “trap” free CN in the blood, akin to the effects of nitrite compounds [51]. Yet, we found that, in keeping with other older experiments [22], methemoglobinemia is not typically present following MB administration [52,53], at least not at the dose that was used in the present study and within the resolution of methemoglobinemia determination. It has been proposed that a cyclic conversion of hemoglobin into methemoglobin, and vice versa, could still be produced during the cyclic reduction MB into LMB then re-oxidation of LMB inside the red cells [23]. This phenomenon may indeed increase the chance for molecules of free CN to be trapped without any significant increase in mean concentration of methemoglobin. The larger pool of CN, that we found when MB and CN were incubated in blood (Figure 9), could reflect such a mechanism. This will make MB an intriguing molecule able to create, in contrast to nitrite-induced methemoglobinemia [54], a “virtual methemoglobinemia effect”, whenever CN is present leading to the formation of cyanomethemoglobin.
Clearly, the redox effects of LMB could have interacted with a large number of proteins which function could have been altered by an increased production of reactive oxygen species (ROS) during CN intoxication [55–58]. Among these proteins, numerous ion channels, including cardiac LCa channels [59], could see have affected the cardiac function, akin to the toxic effect of H2S in cardiomyocytes and its reversibility by MB [60].
As discussed in the previous section, another important site of interaction could be the redox effect of MB on substrate-level phosphorylation (depending on TCA cycle), which does not require a functional ATPase activity [35] as well as the mitochondrial respiration itself. Indeed, the couple MB/LMB has been suggested to be able to support the transfer of protons through the mitochondrial membrane, when the mitochondrial respiration is impeded [36–40,61,62]. For instance, Daudt et al. [38] found that MB, at the concentration of 1 and 10 μM can protect cells against the effects of rotenone (a blocker the mitochondrial complex I). In addition, reduced MB has also been suggested to increase the cytochrome c oxidase activity [63]. Zhang et al. [37] have shown that MB is able to reverse the oxidative stress and the depression oxygen consumption produced by rotenone in brain homogenates. Similarly, MB has been suggested to significantly increase glucose uptake and ATP production counteracting the effects of hypoxia [64]. One possible mechanism would be that the redox couple MB/LMB could provide electron to the complex IV by bypassing the complexes I and III, and maintaining some exchange of protons across the mitochondrial membrane. How CN, which inhibits the complex IV, could be counteracted by such a mechanism remains to be clarified.
It should be kept in mind that if the beneficial effect of MB requires the reduction of its oxidized form and/or the re-oxidation of LMB to be effective, one of the main limitations and possible toxicity of the couple MB/LMB will be dictated by the depletion in NAD(P)H or reduced glutathione, as well as by the increased production of H2O2 produced during the re-oxidation of LMB by O2. Of note, one can speculate that the latter is very likely to remains innocuous in vivo as long as large concentrations of catalase are present. The shift toward an oxidative intracellular and intra-mitochondrial milieu may well be one of the major factors limiting the use of high dosages of MB [30].
MB as a treatment of CN intoxication?
Methylene blue is a cationic tri-heterocyclic redox compound with a central aromatic thiazine ring system, which confers to this agent a high lipophilicity and the ability to concentrate in the central nervous system after systemic injection. MB diffuses extremely rapidly into most tissues including the brain, where it accumulates [33]. Its pharmacokinetics, both in the blood and tissues, have been established in various species [33,52]. MB has been shown to have a relatively long half-life (4–5 h in the rat) in the blood. Inside the cells, including neurons, MB concentrates in the mitochondria where it can interact with the mitochondrial complexes [37,38].
As mentioned in the introduction, MB is already used in various life-threatening conditions (methemoglobinemia or vasoplegic shock) [25,30,31]. Since a systemic injection must remain the standard of care during a life-threatening CN intoxication, due to circulatory and respiratory failure, intravenous or intra-osseous (i.o.) administrations of MB represent the best modalities to be considered in severe forms of CN intoxication. Intra-osseous MB has already been used during methemoglobinemia in infants and has been shown to be very effective [65]. Interestingly, there was no difference between the pharmacokinetics of MB using i.o. or i.v. routes in the rabbit [65]. Since an intra-osseous administration appears to be as effective as an intra-venous infusion, even during cardiopulmonary resuscitation, intra-osseous MB administration should be the method of choice whenever the placement of a venous line is unrealistic or impossible, like during a mass casualty situation, for instance.
Patients with moderate or no alteration in circulation could benefit from an intra-muscular (i.m.) administration of MB. However, fat or skin necrosis have been reported after local MB administration with the current formulation [66]. For this reason, a new formulation of MB that can be administered i.m. will need to be considered and developed. Suspension of MB in an oil (non-aqueous) carrier and subjected to bead milling may provide submicron suspension suitable for IM injection. The benefits of MB in moderate or very light forms of CN intoxication or more importantly when associated with current CN antidotes remains to be characterized.
Methylene blue can interact with selective serotonin reuptake inhibitors (SSRI) [67]. For obvious reasons, first responders are usually not aware of this prescription. The incidence of “serotonin syndrome” in patients treated by SSRI agents receiving MB is not known, but appears to be relatively low [67,68]. The risk of developing a serotonin syndrome, albeit rare, should be weighed against the consequences of CN intoxication, when a circulatory shock is present. Secondary/delayed mental status changes, neuro-muscular hyperactivity, and autonomic hyperactivity are characteristic of a serotonin syndrome and should indeed suggest the possibility of an adverse reaction of MB with SSRI drugs, which prognosis is favorable when identified early. MB has no serotonin “toxicity” by itself.
Finally, a deficit in G6PD is a classical contraindication, although partial, to the utilization of MB [30]. A G6PD deficit should, in theory, make red cells unable to produce enough NADPH, increasing the potential oxidative effects of MB. The clinical relevance of this question is well documented in the literature, as MB has been used as a potential treatment of malaria [69] in populations wherein the incidence of deficit in G6PD is high. MB appears to be very safe in children presenting a deficit in G6PD [70], as well as in adults with non-symptomatic G6PD deficit [71,72].
On the dose of MB
The doses of MB that are currently used in humans for the treatment of methemoglobinemia are about 1–2 mg/kg to be repeated, if needed [25,30]. How to translate such a dose to a 400 g animal, and vice versa, remains a difficult question to answer. This question is even more critical for a compound like MB, which modalities of action, efficacy and toxicity are dictated by many factors that are not simply related to purely allometric considerations. We have established that the dose of 20 mg/kg was innocuous and all the physiological changes were reversible within 30 min following the administration of MB (Figure 4). In addition, according to the recommendations for conversion of doses between animals and humans provided by the Department of Health and Human Services [73], the coefficient of conversion from mg/kg to mg/m2 is be about 40 in humans versus 8 in a 400 g mammal. In keeping with the surface area, a dose of 4 mg/kg in humans, corresponding to about 160 mg/m2 would therefore be equivalent to a dose of about 20 mg/kg in a rat. Using specific metabolic rate (V̇O2/kg) will lead to the same figure. In other words, the dose that was used in our rats would certainly correspond to a dose that would be well-tolerated in humans, and within a range recommended for the treatment of a methemoglobinemia for instance. Whether doses lower than 20 mg/kg in rat could be effective against CN intoxication remains to be determined.
Conclusions
Methylene blue increases immediate survival in a rat model of lethal CN intoxication. Beneficial effects were also observed when MB was administered after sublethal CN intoxication (faster O2 deficit recovery and lactate decline). Our study supports the historical proposal that MB can be used in a treatment of CN intoxication. The potential impact of MB on various metabolic pathways affected by CN, including the redox status of the mitochondrial complexes and the Krebs cycle inhibited by the accumulation of NADH in relevant organs (cardiomyocytes and medullary neurons) [35] remains to be characterized in intoxicated animals. The properties of MB represent a unique modality of action with no equivalent in the different families of drugs proposed in the treatment of CN intoxication. Due to its very fast effects, the benefits of associating MB to current CN antidotes, with well-demonstrated protective effects, should be considered.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Institutes of Health Office of the Director (NIH OD), and the National Institute of Neurological Disorders and Stroke (NINDS) [Grant Number R21 NS098991].
Footnotes
Supplemental data for this article can be accessed here.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Baud FJ, Barriot P, Toffis V, et al. Elevated blood cyanide concentrations in victims of smoke inhalation. N Engl J Med. 1991;325:1761–1766. doi: 10.1056/NEJM199112193252502. [DOI] [PubMed] [Google Scholar]
- 2.Tagwireyi D, Chingombe P, Khoza S, et al. Pattern and epidemiology of poisoning in the east african region: a literature review. J Toxicol. 2016;2016:1. doi: 10.1155/2016/8789624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baud FJ. Cyanide: critical issues in diagnosis and treatment. Hum Exp Toxicol. 2007;26:191–201. doi: 10.1177/0960327107070566. [DOI] [PubMed] [Google Scholar]
- 4.Borron SW, Baud FJ. Antidotes for acute cyanide poisoning. CPB. 2012;13:1940–1948. doi: 10.2174/138920112802273182. [DOI] [PubMed] [Google Scholar]
- 5.Bebarta VS. Antidotes for cyanide poisoning. Eur J Emerg Med. 2013;20:65–66. doi: 10.1097/MEJ.0b013e3283591730. [DOI] [PubMed] [Google Scholar]
- 6.Borron SW, Baud FJ, Megarbane B, et al. Hydroxocobalamin for severe acute cyanide poisoning by ingestion or inhalation. Am J Emerg Med. 2007;25:551–558. doi: 10.1016/j.ajem.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Bebarta VS, Pitotti RL, Dixon PS, et al. Hydroxocobalamin and epinephrine both improve survival in a swine model of cyanide-induced cardiac arrest. Ann Emerg Med. 2012;60:415–422. doi: 10.1016/j.annemergmed.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Lee J, Mahon SB, Mukai D, et al. The vitamin B12 analog cobinamide is an effective antidote for oral cyanide poisoning. J Med Toxicol. 2016;12:370–379. doi: 10.1007/s13181-016-0566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baskin SI, Horowitz AM, Nealley EW. The antidotal action of sodium nitrite and sodium thiosulfate against cyanide poisoning. J Clin Pharmacol. 1992;32:368–375. doi: 10.1002/j.1552-4604.1992.tb03849.x. [DOI] [PubMed] [Google Scholar]
- 10.Bebarta VS, Brittain M, Chan A, et al. Sodium nitrite and sodium thiosulfate are effective against acute cyanide poisoning when administered by intramuscular injection. Ann Emerg Med. 2017;69:718–725. e714. doi: 10.1016/j.annemergmed.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cambal LK, Swanson MR, Yuan Q, et al. Acute, sublethal cyanide poisoning in mice is ameliorated by nitrite alone: complications arising from concomitant administration of nitrite and thiosulfate as an antidotal combination. Chem Res Toxicol. 2011;24:1104–1112. doi: 10.1021/tx2001042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cambal LK, Weitz AC, Li HH, et al. Comparison of the relative propensities of isoamyl nitrite and sodium nitrite to ameliorate acute cyanide poisoning in mice and a novel antidotal effect arising from anesthetics. Chem Res Toxicol. 2013;26:828–836. doi: 10.1021/tx400103k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall AH, Dart R, Bogdan G. Sodium thiosulfate or hydroxocobalamin for the empiric treatment of cyanide poisoning? Ann Emerg Med. 2007;49:806–813. doi: 10.1016/j.annemergmed.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 14.Hall AH, Doutre WH, Ludden T, et al. Nitrite/thiosulfate treated acute cyanide poisoning: estimated kinetics after antidote. J Toxicol Clin Toxicol. 1987;25:121–133. doi: 10.3109/15563658708992618. [DOI] [PubMed] [Google Scholar]
- 15.Hall AH, Rumack BH. Hydroxycobalamin/sodium thiosulfate as a cyanide antidote. J Emerg Med. 1987;5:115–121. doi: 10.1016/0736-4679(87)90074-6. [DOI] [PubMed] [Google Scholar]
- 16.Patterson SE, Moeller B, Nagasawa HT, et al. Development of sulfanegen for mass cyanide casualties. Ann N Y Acad Sci. 2016;1374:202–209. doi: 10.1111/nyas.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patterson SE, Monteil AR, Cohen JF, et al. Cyanide antidotes for mass casualties: water-soluble salts of the dithiane (sulfanegen) from 3-mercaptopyruvate for intramuscular administration. J Med Chem. 2013;56:1346–1349. doi: 10.1021/jm301633x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geiger JC. Cyanide poisoning in San Francisco. JAMA. 1932;99:1944–1945. [Google Scholar]
- 19.Sahlin B. The antagonism between methylene blue and cyan potassium. Skandinavisches Archiv Fur Physiologie. 1926;47:284–291. [Google Scholar]
- 20.Eddy NB. Antagonism between methylene blue and sodium cyanide. J Pharmacol & Exper Therap. 1930;39:271. [Google Scholar]
- 21.Brooks MM. Methylene blue as antidote for cyanide and carbon monoxide poisoning. JAMA. 1933;100:59. [Google Scholar]
- 22.Brooks MM. The mechanism of methylene blue action on blood. Science. 1934;80:15–16. doi: 10.1126/science.80.2062.15-a. [DOI] [PubMed] [Google Scholar]
- 23.Wendel WB. The mechanism of antidotal action of methylene blue in cyanide poisoning. Science. 1934;80:381–382. [Google Scholar]
- 24.Wendel WB. The control of methemoglobinemia with methylene blue. J Clin Invest. 1939;18:179–185. doi: 10.1172/JCI101033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ginimuge PR, Jyothi SD. Methylene blue: revisited. J Anaesthesiol Clin Pharmacol. 2010;26:517–520. [PMC free article] [PubMed] [Google Scholar]
- 26.Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med. 1999;34:646–656. doi: 10.1016/s0196-0644(99)70167-8. [DOI] [PubMed] [Google Scholar]
- 27.Chen KK, Rose CL. Nitrite and thiosulfate therapy in cyanide poisoning. J Am Med Assoc. 1952;149:113–119. doi: 10.1001/jama.1952.02930190015004. [DOI] [PubMed] [Google Scholar]
- 28.Sevcik P, Dunford HB. Kinetics of the oxidation of NADH by methylene blue in a closed system. J Phys Chem. 1991;95:2411–2415. [Google Scholar]
- 29.Engbersen JFJ, Koudijs A, Van der Plas HC. Reaction of NADH models with methylene blue. Recl Trav Chim Pays-Bas. 2010;104:131–138. [Google Scholar]
- 30.Clifton J, 2nd, Leikin JB. Methylene blue. Am J Ther. 2003;10:289–291. doi: 10.1097/00045391-200307000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Schirmer RH, Adler H, Pickhardt M, et al. Lest we forget you–-methylene blue …. Neurobiol Aging. 2011;32:2325, 2325.e7–2316. doi: 10.1016/j.neurobiolaging.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Buchholz K, Schirmer RH, Eubel JK, et al. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob Agents Chemother. 2008;52:183–191. doi: 10.1128/AAC.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peter C, Hongwan D, Kupfer A, et al. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur J Clin Pharmacol. 2000;56:247–250. doi: 10.1007/s002280000124. [DOI] [PubMed] [Google Scholar]
- 34.Rojas JC, Bruchey AK, Gonzalez-Lima F. Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Prog Neurobiol. 2012;96:32–45. doi: 10.1016/j.pneurobio.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komlodi T, Tretter L. Methylene blue stimulates substrate-level phosphorylation catalysed by succinyl-CoA ligase in the citric acid cycle. Neuropharmacology. 2017;123:287–298. doi: 10.1016/j.neuropharm.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Wen Y, Li W, Poteet EC, et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem. 2011;286:16504–16515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Rojas JC, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox Res. 2006;9:47–57. doi: 10.1007/BF03033307. [DOI] [PubMed] [Google Scholar]
- 38.Daudt DR, 3rd, Mueller B, Park YH, et al. Methylene blue protects primary rat retinal ganglion cells from cellular senescence. Invest Ophthalmol Vis Sci. 2012;53:4657–4667. doi: 10.1167/iovs.12-9734. [DOI] [PubMed] [Google Scholar]
- 39.Atamna H, Nguyen A, Schultz C, et al. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. Faseb J. 2007;22:703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- 40.Poteet E, Winters A, Yan LJ, et al. Neuroprotective actions of methylene blue and its derivatives. PloS One. 2012;7:e48279. doi: 10.1371/journal.pone.0048279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haouzi P, Tubbs N, Rannals MD, et al. Circulatory failure during noninhaled forms of cyanide intoxication. Shock. 2017;47:352–362. doi: 10.1097/SHK.0000000000000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haouzi P, Van de Louw A. Uncoupling mitochondrial activity maintains body VO2 during hemorrhage-induced O2 deficit in the anesthetized rat. Respir Physiol Neurobiol. 2013;186:87–94. doi: 10.1016/j.resp.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Klingerman CM, Trushin N, Prokopczyk B, et al. H2S concentrations in the arterial blood during H2S administration in relation to its toxicity and effects on breathing. Am J Physiol Regul Integr Comp Physiol. 2013;305:R630–R638. doi: 10.1152/ajpregu.00218.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haouzi P, Van de Louw A. Persistent reduced oxygen requirement following blood transfusion during recovery from hemorrhagic shock. Respir Physiol Neurobiol. 2015;215:39–46. doi: 10.1016/j.resp.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 45.Lundquist P, Sorbo B. Rapid determination of toxic cyanide concentrations in blood. Clin Chem. 1989;35:617–619. [PubMed] [Google Scholar]
- 46.Harrop GA, Barron ES. Studies on blood cell metabolism: I. The effect of methylene blue and other dyes upon the oxygen consumption of mammalian and avian erythrocytes. J Exp Med. 1928;48:207–223. doi: 10.1084/jem.48.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barron ES. The catalytic effect of methylene blue on the oxygen consumption of tumors and normal tissues. J Exp Med. 1930;52:447–456. doi: 10.1084/jem.52.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bodine JH, Lu KH. Methylene blue, 2,4-dinitrophenol, and oxygen uptake of intact and homogenized embryos. Proc Soc Exp Biol Med. 1950;74:448–450. doi: 10.3181/00379727-74-17935. [DOI] [PubMed] [Google Scholar]
- 49.Levine S. Interaction between ethyl methylene blue and cyanide-induced increases in blood lactate. J Lab Clin Med. 1977;89:632–639. [PubMed] [Google Scholar]
- 50.Tranquada RE, Bernstein S, Grant WJ. Intravenous methylene blue in the therapy of lactic acidosis. Arch Intern Med. 1964;114:13–25. doi: 10.1001/archinte.1964.03860070059003. [DOI] [PubMed] [Google Scholar]
- 51.Hall AH, Saiers J, Baud F. Which cyanide antidote? Crit Rev Toxicol. 2009;39:541–552. doi: 10.1080/10408440802304944. [DOI] [PubMed] [Google Scholar]
- 52.Burrows GE. Methylene blue: effects and disposition in sheep. J Vet Pharmacol Ther. 1984;7:225–231. doi: 10.1111/j.1365-2885.1984.tb00904.x. [DOI] [PubMed] [Google Scholar]
- 53.Stossel TP, Jennings RB. Failure of methylene blue to produce methemoglobinemia in vivo. Am J Clin Pathol. 1966;45:600–604. doi: 10.1093/ajcp/45.5.600. [DOI] [PubMed] [Google Scholar]
- 54.Kohn MC, Melnick RL, Ye F, et al. Pharmacokinetics of sodium nitrite-induced methemoglobinemia in the rat. Drug Metab Dispos. 2002;30:676–683. doi: 10.1124/dmd.30.6.676. [DOI] [PubMed] [Google Scholar]
- 55.Shou Y, Gunasekar PG, Borowitz JL, et al. Cyanide-induced apoptosis involves oxidative-stress-activated NF-kappaB in cortical neurons. Toxicol Appl Pharmacol. 2000;164:196–205. doi: 10.1006/taap.2000.8900. [DOI] [PubMed] [Google Scholar]
- 56.Mills EM, Gunasekar PG, Pavlakovic G, et al. Cyanide-induced apoptosis and oxidative stress in differentiated PC12 cells. J Neurochem. 1996;67:1039–1046. doi: 10.1046/j.1471-4159.1996.67031039.x. [DOI] [PubMed] [Google Scholar]
- 57.Gunasekar PG, Sun PW, Kanthasamy AG, et al. Cyanide-induced neurotoxicity involves nitric oxide and reactive oxygen species generation after N-methyl-D-aspartate receptor activation. J Pharmacol Exp Ther. 1996;277:150–155. [PubMed] [Google Scholar]
- 58.Gunasekar PG, Borowitz JL, Isom GE. Cyanide-induced generation of oxidative species: involvement of nitric oxide synthase and cyclooxygenase-2. J Pharmacol Exp Ther. 1998;285:236–241. [PubMed] [Google Scholar]
- 59.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 60.Judenherc-Haouzi A, Zhang XQ, Sonobe T, et al. Methylene blue counteracts H2S toxicity-induced cardiac depression by restoring L-type Ca channel activity. Am J Physiol Regul Integr Comp Physiol. 2016;310:R1030–R1044. doi: 10.1152/ajpregu.00527.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scott A, Hunter FE. Support of thyroxine-induced swelling of liver mitochondria by generation of high energy intermediates at any one of three sites in electron transport. J Biol Chem. 1966;241:1060–1066. [PubMed] [Google Scholar]
- 62.Visarius TM, Stucki JW, Lauterburg BH. Stimulation of respiration by methylene blue in rat liver mitochondria. FEBS Lett. 1997;412:157–160. doi: 10.1016/s0014-5793(97)00767-9. [DOI] [PubMed] [Google Scholar]
- 63.Callaway NL, Riha PD, Wrubel KM, et al. Methylene blue restores spatial memory retention impaired by an inhibitor of cytochrome oxidase in rats. Neurosci Lett. 2002;332:83–86. doi: 10.1016/s0304-3940(02)00827-3. [DOI] [PubMed] [Google Scholar]
- 64.Ryou MG, Choudhury GR, Li W, et al. Methylene blue-induced neuronal protective mechanism against hypoxia-reoxygenation stress. Neuroscience. 2015;301:193–203. doi: 10.1016/j.neuroscience.2015.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herman MI, Chyka PA, Butler AY, et al. Methylene blue by intra-osseous infusion for methemoglobinemia. Ann Emerg Med. 1999;33:111–113. doi: 10.1016/s0196-0644(99)70427-0. [DOI] [PubMed] [Google Scholar]
- 66.Reyes F, Noelck M, Valentino C, et al. Complications of methylene blue dye in breast surgery: case reports and review of the literature. J Cancer. 2010;2:20–25. [PMC free article] [PubMed] [Google Scholar]
- 67.Ng BK, Cameron AJ. The role of methylene blue in serotonin syndrome: a systematic review. Psychosomatics. 2010;51:194–200. doi: 10.1176/appi.psy.51.3.194. [DOI] [PubMed] [Google Scholar]
- 68.Grubb KJ, Kennedy JL, Bergin JD, et al. The role of methylene blue in serotonin syndrome following cardiac transplantation: a case report and review of the literature. J Thorac Cardiovasc Surg. 2012;144:e113–e116. doi: 10.1016/j.jtcvs.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 69.Wainwright M, Amaral L. The phenothiazinium chromophore and the evolution of antimalarial drugs. Trop Med Int Health. 2005;10:501–511. doi: 10.1111/j.1365-3156.2005.01417.x. [DOI] [PubMed] [Google Scholar]
- 70.Meissner PE, Mandi G, Witte S, et al. Safety of the methylene blue plus chloroquine combination in the treatment of uncomplicated falciparum malaria in young children of Burkina Faso. Malar J. 2005;4:45. doi: 10.1186/1475-2875-4-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mandi G, Witte S, Meissner P, et al. Safety of the combination of chloroquine and methylene blue in healthy adult men with G6PD deficiency from rural Burkina Faso. Trop Med Int Health. 2005;10:32–38. doi: 10.1111/j.1365-3156.2004.01356.x. [DOI] [PubMed] [Google Scholar]
- 72.Muller O, Meissner P, Mansmann U. Glucose-6-phosphate dehydrogenase deficiency and safety of methylene blue. Drug Saf. 2012;35:85. doi: 10.2165/11597790-000000000-00000. Author reply 85–86. [DOI] [PubMed] [Google Scholar]
- 73.U.S. Department of Health and Human Services FaDA. Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers Edited by CDER. 2005 Available from: https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.