

SUMMARY
Influenza hemagglutinin (HA) is the canonical type-I viral envelope glycoprotein, and provides a template for the membrane fusion mechanisms of numerous viruses. The current model of HA-mediated membrane fusion describes a static “spring-loaded” fusion domain (HA2) at neutral pH. Acidic pH triggers a singular irreversible conformational rearrangement in HA2 that fuses viral and cellular membranes. Here, using single-molecule Förster resonance energy transfer (smFRET) imaging we directly visualized pH-triggered conformational changes of HA trimers on the viral surface. Our analyses reveal reversible exchange between the pre-fusion and two intermediate conformations of HA2. Acidification of pH and receptor binding shift the dynamic equilibrium of HA2 in favor of forward progression along the membrane fusion reaction coordinate. Interaction with the target membrane promotes irreversible transition of HA2 to the post-fusion state. The reversibility of HA2 conformation may protect against transition to the post-fusion state prior to arrival at the target membrane.
eTOC Blurb
Single molecule FRET is used to provide real-time visualization of influenza hemagglutinin conformational dynamics on the viral surface during triggering by low pH.
INTRODUCTION
Influenza virus entry into cells requires fusion of the viral membrane with an endosomal membrane. The trimeric viral envelope glycoprotein, HA, which resides on the surface of the influenza virion, promotes this fusion event. HA is translated as the polypeptide precursor, HA0. Each protomer in the HA0 trimer is cleaved by cellular proteases resulting in the formation of the two disulfide-linked subunits, HA1 and HA2, which comprise the receptor-binding domain, and the membrane fusion domain, respectively. During entry the influenza virion attaches to the cell surface by way of interactions between HA1 and sialic acid moieties in the plasma membrane. The virion is then endocytosed and trafficked to the late endosome. Acidification of the endosome triggers conformational changes in HA, which result in fusion of the viral and endosomal membranes, thereby releasing the viral genetic material into the host cell.
The current model of HA-mediated membrane fusion suggests an extensive conformational refolding of HA2 (Harrison, 2015). The structure of HA at neutral pH indicates that the fusion peptide on the N terminus of HA2 is sequestered in a hydrophobic pocket at the trimer interface (Wilson et al., 1981). According to this model, exposure to acidic pH in the late endosome leads to undocking of the HA1 domains from the trimer, allowing the B loop of HA2 to refold into a helix. This loop-to-helix transition moves the fusion peptide ~100 Å away from the virion surface where it inserts into the target membrane, forming the hypothesized extended intermediate conformation of HA2 (Bullough et al., 1994a; Carr and Kim, 1993). Collapse of the extended intermediate draws the opposing viral and endosomal membranes together and promotes fusion (Harrison, 2015). This transition results in HA2 forming a highly stable trimeric coiled-coil conformation (Bullough et al., 1994a; Chen et al., 1999). Direct observation of the conformational changes of HA2 has never been made, making the current model of HA-mediated membrane fusion unverified.
Numerous studies have hinted that the current model lacks essential features of HA conformational dynamics. First, additional HA conformations beyond those described by the current model may be adopted during the membrane fusion reaction (Bottcher et al., 1999; Fontana et al., 2012; Garcia et al., 2015; Gruenke et al., 2002; Ivanovic et al., 2013; Leikina et al., 2002; Stegmann et al., 1990; Xu and Wilson, 2011). Second, several studies provide evidence of reversible conformational changes of HA during refolding at acidic pH (Fontana et al., 2012; Korte et al., 1997; Krumbiegel et al., 1994; Leikina et al., 2002; Tatulian and Tamm, 1996; Weber et al., 1994). Of particular note, using an ensemble lipid-mixing assay, Leikina et al. proposed HA refolding involves an early reversible conformation with a lifetime on the order of minutes (Leikina et al., 2002). However, the dynamic and structural nature of these putative intermediate conformations, and how they integrate into the mechanism of membrane fusion remains unknown. This is due to a lack of methodology capable of directly observing the pH-triggered HA conformational trajectory. As a result, key questions remain unanswered. Is HA2 capable of undergoing reversible conformational changes? If so, what regulates those dynamics, and what advantage does that reversibility provide the virus? What is the order of events along the conformational trajectory of HA2 that leads from the pre-fusion conformation to the post-fusion coiled-coil conformation?
To address these questions, we developed a single-molecule Förster resonance energy transfer (smFRET) imaging assay to probe the conformational dynamics of the HA2 domain within the context of HA trimers on the surface of viral particles (Figure 1A). Our study suggests that HA is a dynamic machine, undergoing spontaneous and reversible transitions between multiple conformations. pH and receptor binding shifts the dynamic equilibrium in favor of forward progress along the reaction coordinate. Interaction with the target membrane promotes transition to the irreversible post-fusion coiled-coil conformation. Regulation of HA dynamics occurs through direct interactions with the receptor and the target membrane, as well as through a long-range allosteric connection between the receptor-binding site and the fusion peptide. This multi-step process may provide a strategy for the virus to limit premature inactivation, which would render it non-infectious.
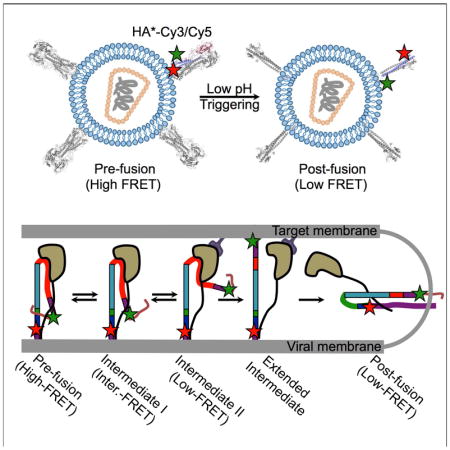
Figure 1. smFRET assay for direct visualization of HA conformational dynamics.

(A) Viral particles containing a single HA*-Cy3/Cy5 protomer, labeled at positions 17 and 127 in the HA2 domain, within an HA trimer were immobilized on a quartz microscope slide and imaged at room temperature with TIRF microscopy (STAR Methods). (B) Molecular models of HA*-Cy3/Cy5 predict that fluorophores placed at positions 17 and 127 in the HA2 domain will report on the transition from the pre-fusion to the coiled-coil conformation of HA2 via changes in FRET efficiency (models were based on PDB IDs 2FK0 and 1HTM, STAR Methods) (Bullough et al., 1994a; Stevens et al., 2006). See also Figure S4.
RESULTS
Site-specific attachment of fluorophores to non-canonical amino acids in HA2 on the surface of viral particles
We developed an smFRET-imaging assay that reports on the conformational changes of HA related to membrane fusion (Figure 1A). For this purpose, we attached one fluorophore at position 17 proximal to the fusion peptide of HA2 from the H5N1 strain A/Vietnam/1203/2004 (VN04) of influenza virus. We placed the second fluorophore at position 127 in the loop C-terminal to the D helix in HA2 (Figures 1B, S1A; see STAR Methods for details). We chose these sites to probe the position of the fusion peptide with respect to the base of the HA stalk since the fusion peptide undergoes dramatic displacements during the membrane fusion reaction as a result of HA2 refolding. Structural models of HA suggested a significant increase in the distance (ca. 65 Å) between the fluorophores after transition to the post-fusion coiled-coil conformation, which predicts a decrease in FRET efficiency (Figure 1B) (Bullough et al., 1994a; Stevens et al., 2006). To facilitate fluorophore attachment, we employed a genetic code expansion technique whereby amber stop codons (UAGs) at positions 17 and 127 were suppressed by an orthogonal tRNA/tRNA synthetase pair, and used to code for a non-canonical amino acid (ncAA) containing a trans-cyclooct-2-ene moiety (TCO*) (Figure S1) (Nikić et al., 2014). In this way we engineered HA to have an HA2 domain containing two TCO* residues at positions 17 and 127 (referred to as HA*). We subsequently formed virions with HA* and the HIV-1 core (Wang et al., 2008), followed by labeling of the TCO* residues by strain-promoted inverse electron-demand Diels-Alder cycloaddition (SPIEDAC) with Cy3- and Cy5-tetrazine (HA*-Cy3/Cy5; Figure S1, STAR Methods). Virions with either HA*-Cy3/Cy5 or mock-labeled HA were immobilized on quartz microscope slides and imaged using total internal reflection fluorescence (TIRF) microscopy (Figure S1, STAR Methods). This indicated specific labeling of HA* and no detectable labeling of particles with wild-type HA.
HA* and HA*-Cy3/Cy5 maintained approximately 80% and 76% functionality, respectively, as compared to wild-type HA in a virus entry assay (Cavrois et al., 2002), and were efficiently neutralized by antibodies recognizing conformational epitopes in the HA stalk (Figure S2A) (Dreyfus et al., 2012; Hai et al., 2012). We also evaluated the function of HA*-Cy3/Cy5 in a fluorescence-based virus-liposome fusion assay. The kinetics of fusion across a range of pHs were comparable for HA*-Cy3/Cy5 and wild-type HA, indicating maintenance of fusogenicity and pH dependence of the modified HA (Figure S2B, STAR Methods). Furthermore, to determine the extent to which HA*-Cy3/Cy5 maintains a native pre-fusion conformation, we performed ELISA assays with four different stalk-binding neutralizing antibodies (CR9114, KB2, FI6, and 6F12) (Corti et al., 2011; Dreyfus et al., 2012; Hai et al., 2012; Tan et al., 2012). We found identical binding affinities of the antibodies for virions with wild-type HA and HA*-Cy3/Cy5 (Figure S2C, STAR Methods). Taken together, these analyses indicate that HA*-Cy3/Cy5 maintains a high level of functionality as compared to wild-type HA, and a near-native global pre-fusion conformation. It remains possible that our modifications to HA result in localized changes in structure at the sites of fluorophore attachment, but these putative effects are of only minimal significance in the assays utilized here.
HA2 reversibly interconverts between three distinct conformations
For smFRET imaging, we formed virions with an excess of wild-type HA over HA*, such that each particle contained on average a single HA* protomer within an otherwise native HA trimer, among the native distribution of wild-type HA molecules, as done for a similar study of HIV-1 envelope glycoprotein dynamics (Munro et al., 2014). After labeling with Cy3- and Cy5-tetrazine, the virions were surface-immobilized and imaged using TIRF microscopy (Figure 1A). Visual inspection of the recorded fluorescence traces indicated that each visible virion was labeled with a single fluorophore pair on average (STAR Methods).
We first visualized the conformational landscape of HA2 at pH 7, a condition known to favor the pre-fusion conformation. smFRET trajectories displayed a predominant high-FRET state with short-lived transitions to intermediate- and low-FRET states (Figures 2A and S3A). We used hidden Markov modeling (HMM) to fit the trajectories to a three-state model with mean FRET values of approximately 0.95, 0.53, and 0.20. This procedure indicated 75% occupancy in the high-FRET state, as indicated in a histogram of the FRET trajectories (Figure 2A, Table S1). HMM analysis also enabled the formation of a transition density plot (TDP), which displays the relative frequencies of transitions between the observed FRET states. The TDP indicated transitions between the high- and intermediate-FRET states, and between the intermediate- and low-FRET states. Direct transition between the high- and low-FRET states was rarely observed. The high-FRET state is consistent with the 40-Å inter-fluorophore distance predicted by molecular dynamics (MD) simulation of the HA*-Cy3/Cy5 trimer, which is based on the structure of pre-fusion VN04 HA determined at neutral pH (Figures 1B and S4, STAR Methods) (Stevens et al., 2006). Also, fluorescence anisotropy and lifetime measurements were consistent with freely tumbling fluorophores and minimal interaction of the fluorophores with their surrounding microenvironment (Table S2). This indicates that changes in FRET efficiency principally report on changes in the distance between the fluorophores. The observed transitions out of the high-FRET state therefore suggest spontaneous and reversible fluctuations of HA2 out of the pre-fusion conformation. The short-lived intermediate-FRET state suggests a configuration in which the fusion peptide, and the proximal region containing the fluorophore attached to position 17, has moved away from the trimer axes and the base of the HA stalk, increasing the distance between the fluorophores. The low-FRET state suggests a conformation in which the fusion peptide has moved farther from the base of the stalk. We cannot rule out the possibility that movement of position 127 also contributes to the observed dynamics; but based on the existing structural data and the mechanistic model of HA-mediated membrane fusion we expect this contribution is modest.
Figure 2. Direct visualization of pH-triggered conformational changes in HA.
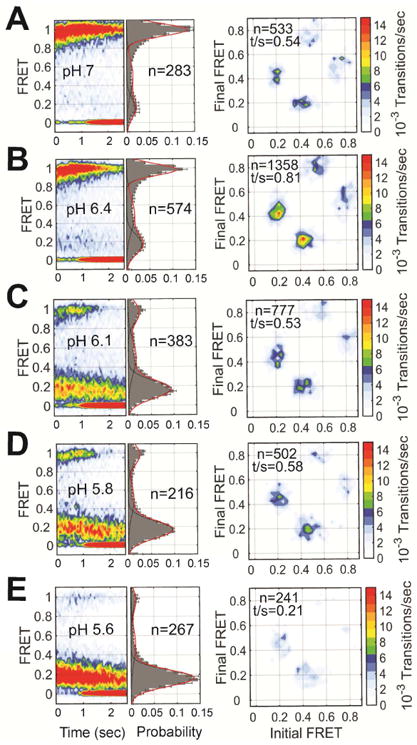
(A) (left) FRET contour plots constructed from the compilation of the population of smFRET trajectories. The contour plots were summed over time, generating FRET histograms, which are shown with three Gaussian distributions overlaid (red) with means ± standard deviations of 0.20 ± 0.08, 0.53 ± 0.09 and 0.95 ± 0.06 that represent the states identified through HMM analysis (black). The number of smFRET traces (n) compiled into each histogram is indicated. (right) TDPs displaying the distributions of initial and final FRET values for every observed transition in FRET. The number of observed transitions (n) and the total transitions per second (t/s) are indicated. (B–E) The same data acquired at the indicated pH. See also Figure S3 and Table S1.
Low pH amplifies the conformational dynamics of HA2 and promotes low-FRET states
We next determined how changes in pH affect the conformational equilibrium of HA2. Decreasing the pH from 7 to 6.4 led to a modest reduction in the occupancy of the high-FRET state (58%), and a corresponding increase in the occupancy of the low-FRET state (Figures 2A–B and S3A–B, Table S1). More dramatic was the increase in the level of dynamics reflected in the TDP. This suggests an increased propensity for the fusion peptide to move away from the base of the stalk (Bottcher et al., 1999; Fontana et al., 2012; Lin et al., 2014), which may be associated with localized changes in the B loop that precede triggering (Xu and Wilson, 2011). Further reduction to pH 6.1 led to a dramatic decrease in the occupancy of the high-FRET state (23%), and a corresponding increase in the occupancy of the low-FRET state (57%) (Figure 2C, Table S1). Despite the dramatic redistribution of the conformational equilibrium, fluctuations between the same three FRET states persisted. These data show that HA2 maintains the ability to spontaneously interconvert between minimally three conformations at neutral and moderately acidic pH. Further reduction to pH 5.8 had only modest effect on the equilibrium, slightly increasing the low-FRET state occupancy (Figure 2D, Table S1). At pH 5.6 the low-FRET state occupancy increased to 78% and minimal transitions were observed (Figures 2E and S3E, Table S1). Formation of a stable low-FRET state at acidic pH likely reflects, at least in part, transition to the coiled-coil conformation. Indeed, MD simulation of the labeled HA2 trimer based on the structure of HA determined at acidic pH predicts an inter-fluorophore distance of approximately 105 Å, in agreement with low FRET (Figures 1B and S4) (Bullough et al., 1994a). Here again, at pH 5.6 fluorescence anisotropy and lifetime measurements gave no indication of significant interactions between the fluorophores and their environment (Table S2), suggesting that FRET efficiency is still primarily reporting on the distance between the fluorophores. However, the coiled-coil conformation is highly thermodynamically stable (Lin et al., 2014; Melikyan et al., 2000). Therefore our observation of reversible sampling of low FRET at elevated and mildly acidic pH may indicate the existence of multiple conformations reflected by low FRET. Interestingly, the occupancy of the intermediate-FRET state was not sensitive to pH, remaining between 12% and 20% throughout the titration (Table S1).
We further validated our interpretation of the observed dynamics through mutagenesis of HA. First, we inhibited movement of the fusion peptide by mutating the furin cleavage site (RKKR to QKQQ). This prevents proteolytic processing of the non-fusogenic HA0, and maintains the covalent linkage of the fusion peptide in HA2 to HA1 (Chen et al., 1998). HA0 stably formed the high-FRET state at neutral pH (Figure S5A, Table S1). Surprisingly, HA0 also displayed reversible fluctuations to lower FRET states. This is consistent with the high-FRET state representing the pre-fusion conformation, and indicates that the fusion peptide has some mobility prior to proteolytic cleavage of HA. However, given the restrictions on the fusion peptide due to its linkage to HA1 prior to proteolytic cleavage, the lower FRET states seen here may reflect movements of the fusion peptide that are not comparable to those seen for cleaved HA. As expected, HA0 failed to form a stable low-FRET state at acidic pH (Figure S5B–C). This is consistent with the fusion peptide on HA0 being unable to stably adopt positions significantly distal to the base of the stalk. Second, the G4E mutation in the fusion peptide of HA2 led to stable low FRET and minimal dynamics at neutral and acidic pH (Figure S5D–E, Table S1). This is consistent with destabilization of the fusion peptide in the hydrophobic pocket and the resulting inability to maintain the pre-fusion conformation (Gething et al., 1986).
The conversion from the high-FRET pre-fusion conformation to low FRET occurs via the transient intermediate-FRET state
We next determined whether the intermediate-FRET state represents a conformation adopted during transition from the high-FRET pre-fusion conformation to the low-FRET state stabilized by acidic pH. We performed a pre-steady state smFRET experiment in which the conformation of HA2 was visualized during stopped-flow introduction of pH 5.6 buffer to surface-bound virions. The resulting trajectories were synchronized to the point of transition out of the high-FRET state, and compiled into a contour plot of time-resolved changes in FRET (Figure 3A–B). This treatment, along with the corresponding TDP, shows that the intermediate-FRET state represents a conformation adopted during pH-induced triggering of HA2 (Figure 3C). However, the TDP also indicates transitions directly from high to low FRET, likely reflecting the unstable nature of this intermediate conformation, and the limited time resolution of our measurements, which gives rise to missed dwells in the intermediate-FRET state. Thus, our data suggest that the intermediate-FRET state represents release of the fusion peptide from the hydrophobic pocket to a position that is adopted during transition from the pre-fusion conformation to the low-FRET configurations.
Figure 3. Pre-steady state smFRET imaging indicates an intermediate conformation adopted during triggering of HA by acidic pH.

(A) Representative fluorescence (donor, green; acceptor, red) and FRET trajectories (blue) obtained from a single HA*-Cy3/Cy5 protomer within an HA trimer on the surface of a viral particle. At each time point FRET efficiency was calculated as the ratio of acceptor fluorescence intensity to total fluorescence intensity. Overlaid on the FRET trajectory in red is an idealized trace generated through HMM analysis. (B) Contour plot of smFRET trajectories synchronized to the transition out of high FRET. Transition through the intermediate-FRET state is observed in route to low FRET. (C) As in Figure 2, the TDP displays the distribution in initial and final FRET values for every transition observed. The TDP indicates that trajectories predominantly transition from high to intermediate FRET, and from intermediate to low FRET. A minority of transitions occurs directly from high to low FRET with no observation of intermediate FRET. This can be explained by the instability of the intermediate-FRET state and the limited time resolution of our imaging.
HA2 stalk-targeting antibodies prevent stable adoption of low FRET conformations
We next asked if neutralizing antibodies that target the HA stalk domain in the pre-fusion conformation would inhibit the transition to low FRET configurations. We incubated the virions with antibody CR9114 prior to smFRET imaging at pH 6.1, which triggers transition out of the pre-fusion conformation in the absence of antibodies. CR9114 binds an epitope at the base of HA2 helix A, and blocks pH-induced conformational changes (Dreyfus et al., 2012). As expected, the CR9114 prevented stable formation of low FRET (37%), and maintained significant occupancy in the high-FRET state (37%) as compared to the absence of antibody at the same pH (Figure S5F, Table S1). We also observed substantial occupancy in the intermediate-FRET state (26%), indicating that the fusion peptide maintains mobility in the presence of CR9114, and that transition between pre-fusion high FRET and intermediate FRET is not inhibited. The TDP showed increased transitions between the intermediate- and low-FRET states. CR9114 therefore does not prevent transition to the low-FRET conformations, but rather limits the stability of those states. Given our observation that HA still transitions to low FRET in the presence of CR9114, and since CR9114 does not recognize the coiled-coil conformation (Dreyfus et al., 2012), this is further evidence that low FRET does not correspond uniquely to the coiled-coil conformation. We obtained the identical result for a second stalk-targeting antibody, KB2, which is also believed to inhibit conformational changes in HA2 related to membrane fusion (Figure S5G, Table S1) (Hai et al., 2012).
Return to neutral pH restores the high-FRET pre-fusion conformation
To further probe the origin of low FRET, we next asked if HA2 can return to the high-FRET pre-fusion conformation after transient exposure to acidic pH, followed by restoration of neutral pH. We incubated the virions at pH 5.6, 5.3, or 5.2 for varying time intervals. HA2 displayed low FRET when imaged immediately upon exposure to the acidic pHs, and the TDP showed transitions between low FRET and intermediate FRET (Figures 4A–C). In all cases, the high-FRET pre-fusion state was restored upon return to neutral pH after 5 minutes; the transitions between pre-fusion high FRET and intermediate FRET were also restored (Figures 4D–F). After exposure to pH 5.6 and 5.3 for 15 minutes, the high-FRET pre-fusion state was still largely restored, while 15 minutes at pH 5.2 displayed equal occupancies in pre-fusion high-FRET and low-FRET states after return to neutral pH (Figure 4G–I). After 30-minutes exposure to the acidic pHs, all cases were found in the low-FRET state after return to neutral pH (Figure 4J–L).
Figure 4. Restoration of the pre-fusion conformation after return to neutral pH.

FRET contour plot, FRET histogram and TDP acquired immediately after exposure of HA to (A) pH 5.6 (repeated from Figure 2), (B) pH 5.3, or (C) pH 5.2. The same data were acquired after returning to pH 7.5 following (D–F) 5 min, (G–I) 15 min, or (J–L) 30 min. Data are displayed as in Figure 2. See also Table S1.
These data suggest a model in which HA2 reversibly samples a low-FRET conformation. This state becomes increasingly stable with decreasing pH, but is fully reversible after limited exposure to acidic pH. Upon extended exposure to acidic pH HA2 transitions from the reversible low-FRET state to the coiled-coil conformation, which does not generate a change in FRET that is detectable in the current experimental setup. Consistent with estimations of the thermodynamic stability of the coiled-coil conformation (Lin et al., 2014; Melikyan et al., 2000), this state is irreversible even after restoration of neutral pH.
Binding to sialic acid allosterically regulates HA2 conformational dynamics
We next asked whether the presence of sialic acid and a target membrane might affect the observed dynamics, especially the transition from the reversible to the irreversible low-FRET state. Based on previous bulk studies of membrane fusion (Leikina et al., 2002), we hypothesized that the irreversible low-FRET conformation would be adopted more quickly by the presence of a sialic acid-containing target membrane at acidic pH. To test this idea, prior to smFRET imaging, we incubated the surface-bound virions with liposomes containing GD1a ganglioside, which presents the sialic acid receptor for HA (Figure 5A) (Floyd et al., 2008). At neutral pH, we observed interconversion between the same three FRET states as in the absence of liposomes (Figures 5B–C). However, notably, we observed increased dynamics and occupancy in the low-FRET states at neutral pH in the presence of receptor-containing liposomes (Figures 2A and 5C, Table S1). This suggests greater mobility of the fusion peptide in the presence of a receptor-containing target membrane. To determine whether interaction of HA with sialic acid, or with the liposome membrane induced the increased dynamics we imaged virions in the presence of LS-tetrasaccharide a (LSTa), which provides a soluble sialylated receptor analogue. We observed the same increased occupancy in the low-FRET states as seen in the presence of the GD1a-containing liposomes (Figure S6A–F). No such promotion of low FRET was seen in the presence of liposomes lacking GD1a, which were bound to the virions by way of a biotin-streptavidin linkage (Figure S6G–L). These data suggest that the presence of sialic acid bound to HA1 allosterically stimulates movement of the fusion peptide at neutral pH. In all cases, stepwise reduction in pH decreased the occupancy of the high-FRET pre-fusion conformation, and increased the occupancy in low FRET, as seen in the absence of receptor or liposomes (Figures 5C and S6, Table S1). But in the presence of receptor, this conversion occurred somewhat more rapidly, further evidence that receptor binding promotes refolding of HA2 (Figure 5D–E).
Figure 5. Interaction with a sialic acid-containing target membrane regulates HA conformational dynamics.

(A) Schematic of the smFRET-imaging assay in the presence of liposomes containing GD1a, which contains sialic acid (STAR Methods). (B) Representative fluorescence and FRET traces at pH 6.1 displayed as in Figure 2. (C) FRET contour plots (left), FRET histograms (middle) and TDPs (right) displayed as in Figure 2. Quantification of the occupancies in the (D) high-FRET pre-fusion and (E) low-FRET conformations in the absence (blue) and presence (red) of liposomes, which was determined through HMM analysis. See also Table S1.
Interaction with the target membrane promotes transition to the irreversible coiled-coil conformation
We next determined whether the receptor-containing target membrane affects the transition from the reversible to the irreversible low-FRET state. To this end, we incubated the virus bound to receptor-containing liposomes at pH 5.6 for 5 or 30 mins before restoration of neutral pH. Here again, HA2 displayed low FRET when imaged immediately upon exposure to acidic pH (Figures 6A). After only 5 mins exposure to acidic pH, the virions bound to receptor-containing liposomes had transitioned to the irreversible low-FRET conformation, failing to return to the high-FRET pre-fusion state after restoration of neutral pH (Figures 6B–C). This suggests that either receptor binding to HA1 or the presence of a target membrane promotes HA2 conversion from the reversible low-FRET state to the coiled-coil conformation. To distinguish between these two possibilities we incubated the virus at pH 5.6 for 5 or 30 mins in presence of either LSTa or liposomes lacking GD1a before restoration of neutral pH. We found that HA returned to the high-FRET pre-fusion conformation in the presence of LSTa. But this reversibility was inhibited by the presence of the liposome (Figure S7). This suggests that interaction with the target membrane promotes transition of HA2 to the irreversible coiled-coil conformation.
Figure 6. A sialic acid-containing target membrane promotes transition of HA to the irreversible coiled-coil conformation.

(A) FRET contour plot, histograms and TDPs acquired in the presence of GD1a-containing liposome immediately after exposure to pH 5.6. The same data were acquired after return to pH 7.5 following (B) 5 min, or (C) 30 min. Data are displayed as in Figure 2. See also Table S1.
DISCUSSION
The prevailing “spring-loaded” mechanism of HA-mediated membrane fusion presents a picture of a static pre-fusion HA undergoing a singular transition to the post-fusion coiled-coil conformation by way of the putative extended intermediate (Harrison, 2015). But direct observation of HA conformational changes in a viral membrane has gone unrealized due to the lack of methodology equipped to probe such dynamics on a relevant timescale. Our use of smFRET imaging, facilitated by the incorporation of ncAAs into HA, has provided the first real-time visualization of HA conformational dynamics. What emerges from our study is a view of HA as a dynamic machine, undergoing spontaneous, reversible fluctuations to multiple conformations on pathway to membrane fusion. Our results suggest that low pH, receptor binding, and interaction with the target membrane regulate the intrinsic conformational dynamics of HA, promoting forward progression along the membrane fusion reaction coordinate.
pH shifts the dynamic equilibrium of HA2 conformations
Our observations support a model in which HA2 spontaneously samples minimally three conformations, which correspond to the high-, intermediate-, and reversible low-FRET states. The high-FRET state reflects the pre-fusion conformation described by crystallography in which the fusion peptide is sequestered in a hydrophobic pocket (Wilson et al., 1981). The current smFRET data do not specify the nature of the global conformation of HA in the intermediate- and reversible low-FRET states, which we term intermediate states I and II, respectively. One possibility is that transition to intermediate state I involves release of the fusion peptide from the hydrophobic pocket. Transition to intermediate state II may involve further displacement of the fusion peptide either farther toward the top of the trimer, or laterally away from the trimer axis (Figure 7A). Models of HA conformations that could correspond to intermediate states I and II have been suggested by cryo-electron tomography experiments (Fontana et al., 2012) and MD simulations (Lin et al., 2014). This structural interpretation of the FRET states is consistent with a study of HA dynamics using hydrogen-deuterium coupled to mass-spectrometry (Garcia et al., 2015), as well as earlier studies of pH-dependent antibody binding (White and Wilson, 1987), and inter-subunit stabilization with disulfide crosslinking (Kemble et al., 1992). In all cases it was found that regions of HA associated with fusion peptide release became exposed prior to regions associated with head domain separation. But future structural investigations of HA are required to definitively describe the observed FRET states.
Figure 7. Kinetic model of HA2 conformational dynamics.

(A) smFRET analysis indicates a sequence of conformational changes in HA during conversion from the pre-fusion to the coiled-coil conformation. The HA2 domain reversibly interconverts between three conformations (pre-fusion, intermediate state I, and intermediate state II). Low pH stalls HA2 in intermediate state II in the absence of a receptor-containing membrane, from which it can return to the pre-fusion state by reneutralizing the pH. Interaction with the receptor-containing target membrane promotes formation of the coiled-coil conformation. (B) The rate constants k1, k−1, k2, are k−2 were determined by exponential fitting of the dwell time histograms obtained through HMM analysis (STAR Methods). Rates of transition between the observed states are shown as a function of pH in the (C–D) absence, or (E–F) presence of a receptor-containing target membrane. Error bars represent the 95% confidence intervals obtained during fitting of the dwell time histograms. The plotted lines represent linear fits. See also Table S3.
Reduction in pH shifts the dynamic equilibrium of HA2 conformations in favor of intermediate state II, ultimately leading to stable formation of this state. Kinetic analysis of the observed dynamics indicates that the rates of transition from the pre-fusion conformation to intermediate state I (k1), and from intermediate state I to II (k2) increase when the pH drops from 7 to 6.4, while the occupancies of the observed FRET states do not change appreciably (Figure 7C–D, Table S3, STAR Methods). Reduction in pH below 6.4 leads to further increase in k1 and k2, and concomitant increase in the occupancy of intermediate state II consistent with an extensive alteration of the global conformational equilibrium (Figure 7C–D, Table S3). In the same manner, the rate of return to the pre-fusion conformation decreases with decreasing pH (k−1) (Figure 7C, Table S3). Yet, notably, the rate of transition from intermediate state II to intermediate state I is insensitive to pH (Figure 7D, Table S3), suggesting that the stability of this state is not determined by the protonation state of HA.
Long-range allostery regulates fusion peptide dynamics
In the presence of a sialic acid-containing target membrane, the same general trend was observed: k1 and k2 increase with decreasing pH, while k−1 decreases with decreasing pH, and k−2 is insensitive to pH (Figure 7E–F, Table S3). The only rate constant that is significantly affected by the presence of sialic acid bound to HA1 is k2, which partially accounts for the greater occupancy in low FRET at neutral pH as compared to unliganded HA (Figure 7F). If formation of intermediate state II involves separation of the head domains, this would indicate that binding to sialic acid, present either in soluble LSTa or incorporated in a target membrane, may facilitate this motion. Whatever the precise mechanism, this long-range allosteric connection between the receptor-binding site in HA1, and the fusion peptide in HA2 has been suggested previously. A mutational study of residues surrounding the fusion peptide demonstrated that destabilization of the fusion peptide enhanced exposure of an epitope at the interface of adjacent head domains (Yewdell et al., 1993). This allosteric connection between receptor binding to the head domain and fusion peptide dynamics may serve to restrain the fusion peptide until a target membrane is present, thus enhancing efficiency of membrane fusion and limiting premature inactivation.
An active role for the target membrane in promoting fusion
In the absence of sialic acid and a target membrane, after exposure to acidic pH HA stalls in intermediate II, from which it returns to the pre-fusion conformation upon reneutralization of the pH (Figure 7A). Only after extended incubation in acidic pH does HA slowly transition to the irreversible coiled-coil conformation. This transition likely occurs by way of the hypothesized extended intermediate in which the fusion peptide protrudes outward perpendicular to the viral membrane. Although not explicitly defined, the extended intermediate would probably put the fluorophores more than 150 Å apart with the current labeling strategy, which would register as a state with nearly zero FRET. Probing formation of the extended intermediate will require alternative fluorophores or labeling strategies. The presence of a target membrane accelerates transition from intermediate state II to the irreversible coiled-coil conformation (Figure 7B). This indicates that interaction with the membrane promotes conformational changes in HA related to fusion. One possibility is that insertion of the fusion peptide into the target membrane, which likely occurs during formation of the extended intermediate, is irreversible and prevents transition back to intermediate state II, effectively increasing the probability of collapse into the coiled-coil conformation. Similarly, the target membrane was reported to promote an irreversible conformational change of HA reconstituted in supported phospholipid bilayers (Tatulian et al., 1995). Experiments that directly correlate membrane fusion with HA conformational changes are needed to test this hypothesis. Another likely possibility is that binding receptor-containing membranes locally concentrates HA trimers, promoting intermolecular interactions. This could give rise to cooperativity in HA transitioning to the irreversible coiled-coil conformation, as seen in bulk fusion assays (Markovic et al., 2001). The observation that the irreversible coiled-coil conformation can be slowly adopted in the absence of a target membrane is in agreement with the previous observation that the fusion peptide can insert into the viral membrane (Weber et al., 1994).
Biological roles for the reversibility in HA2 conformation
Prior to virion assembly and budding HA folds into the pre-fusion conformation in the endoplasmic reticulum and transports through the Golgi network in the cellular secretory pathway. During passage through the Golgi network HA0 is cleaved—at least partially, depending on the strain of influenza—by the cellular furin proteases into HA1 and HA2. This cleavage is nearly complete in the case of the H5 HA studied here owing to its highly basic cleavage site (Kawaoka and Webster, 1988). For HAs that are cleaved by furin, the acidic trans-Golgi network (TGN) presents a challenge to the virus (Llopis et al., 1998). How do these HAs pass through the TGN without being prematurely activated or inducing fusion of TGN membranes? At least two answers to this question have been identified. First, in many influenza serotypes, the extent of cleavage by furins in the TGN is minimal, and HA0 requires cleavage by trypsin-like proteases in the airway of the host in order to become fusogenic. But this cannot be a general strategy for preventing premature inactivation since some HAs are efficiently cleaved by furin and still maintain their fusogenicity. Second, the viral M2 proton channel may insert into the TGN membrane, raising the pH and preventing premature inactivation of cleaved HA molecules (Ciampor et al., 1992; Sakaguchi et al., 1996). However, M2-mediated increase in the TGN pH also would reduce the activity of furin (Takeuchi and Lamb, 1994), and some HAs exit the cell cleaved and fusogenic. Furthermore, the present study, as well as numerous others, show that cleaved and fusogenic HA is formed in the absence of M2. Our present observations suggest two additional benefits to the observed reversibility in HA conformation. First, HA may maintain reversibility in its conformation until an appropriate target membrane is present. In this case, the acidic TGN would stabilize HA in intermediate state II, but following budding of newly formed virions into the pH-neutral extracellular space, HA would refold into the pre-fusion conformation. Second, the reversibility of HA conformation might facilitate synchronization of the irreversible transition to the post-fusion coiled-coil conformation with adjacent HA trimers. This would be consistent with the positive cooperativity observed among neighboring HA molecules during transition to the coiled-coil conformation (Ivanovic et al., 2013; Markovic et al., 2001), which may enhance the efficiency of membrane fusion. These explanations provide biological rationales for why maintaining reversibility in HA conformation is advantageous to the virus.
Here we have shown that HA is a dynamic machine, intrinsically sampling multiple conformations in equilibrium. These intrinsic dynamics are mediated by pH, receptor binding, and the target membrane, which regulate the function of HA during membrane fusion. The mechanisms of this regulation are diverse, occurring through both long-range allosteric and direct interactions with the fusion domain. Given that HA is the canonical type-I glycoprotein, these observations are of critical importance to our understanding of the mechanisms of numerous other viral fusogens. The smFRET imaging methodology presented here provides a platform for probing the dynamics of these diverse molecules, and will surely facilitate future investigations.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, James B. Munro; james.munro@tufts.edu
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
HEK293T and A549 cells were maintained in DMEM (Gibco), supplemented with 10% fetal bovine serum (Gemini Bioscience), 100 U/ml penicillin/streptomycin (Gibco), and 2 mM L-glutamine (Gibco). HEK293T cells were derived from female human embryonic kidney cells, into which the simian virus 40 T-antigen was inserted. A549 cells were derived from lung carcinomatous tissue explanted from an adult male.
METHOD DETAILS
Pseudotyping VN04 HA onto the HIV-1 core
We formed pseudotyped virions consisting of the HIV-1 core and hemagglutinin (HA) from the A/Vietnam/1203/2004 (VN04) H5N1 strain of influenza A virus. This influenza strain was chosen for three reasons. First, VN04 HA efficiently forms a fusogenic pseudovirus on the HIV-1 core (Wang et al., 2008). Second, owing to a highly basic furin cleavage site (RKKR), HA0, the uncleaved non-fusogenic precursor, is efficiently cleaved by cellular furins during trafficking to the plasma membrane (Kawaoka and Webster, 1988; Stieneke-Gröber et al., 1992). This results in a heterodimeric protomer consisting of the HA1 receptor-binding domain and the HA2 membrane fusion domain, which maintain linkage by way of an inter-domain disulfide bond. In contrast, other influenza strains require extracellular proteolytic cleavage in order to become fusogenic. We reasoned that VN04 HA would be less heterogeneous in the extent of proteolytic cleavage. Finally, the structure of VN04 HA has been characterized in atomic detail (Ha et al., 2001; Stevens et al., 2006; Zhu et al., 2015).
VN04 HA pseudotyped virions were formed following published protocols (Wang et al., 2008). Briefly, HEK293T cells were maintained in DMEM (Gibco), supplemented with 10% FBS (Gemini Bioscience), 100 U/ml penicillin/streptomycin (Gibco), and 2 mM L-glutamine (Gibco). At 50–75% confluency the cells were co-transfected with plasmids encoding VN04 HA and HIV-1 Gag-Pol. At 24 hours post-transfection fresh medium containing 7mU/ml bacterial neuraminidase from Vibrio cholera (Roche) was added to the cells to allow the release of the pseudovirions (McKay et al., 2006; Sandrin et al., 2002). Virus was harvested from the supernatant at 48 hours post-transfection, passed through a 0.45 μm filter to remove cell debris, and concentrated 10-fold by centrifugation for 2 hours at 20,000 × g over a 10% sucrose cushion. The particles were then shown to be positive for HA and HIV-1 p24 by Western blot (Figure S1C). As a negative control for proteolytic cleavage, particles were also formed with HA0, the uncleaved precursor, generated by mutating the RKKR furin cleavage site to QKQQ (Chen et al., 1998). Importantly, wild-type VN04 HA was >90% proteolytically cleaved.
Incorporation of the non canonical amino acids (ncAA) TCO* into VN04 HA
We used an amber stop codon-suppression technique to incorporate two TCO* ncAAs into a single HA2 subunit of HA (referred to as HA*) from the VN04 H5N1 strain of influenza. TCO* can then be labeled with tetrazine-conjugated fluorophores by strain-promoted inverse electron-demand Diels-Alder cycloaddition (SPIEDAC) chemistry. To this end, TAG codons were introduced by site-directed mutagenesis at single-codon intervals at positions 15 to 21 in the region of HA2 immediately C-terminal to the fusion peptide. Likewise, TAG codons were introduced at positions 127 and 133 in the loop C-terminal to the D helix at the base of the HA2 stalk. In this method, translation proceeds through the UAG codons on the mRNA only in the presence of an orthogonal tRNA (tRNAPyl), which recognizes the UAG codon, and a corresponding aminoacyl-tRNA synthetase (NESPylRSAF) (Nikić et al., 2014; 2016; 2015). The engineered synthetase aminoacylates the suppressor tRNA with TCO*, facilitating its incorporation at specific locations in HA by the ribosome (Figure S1).
Readthrough of the UAG codons was evaluated in the context of the HA0 mutant so that detection of truncation at positions proximal to the fusion peptide were not complicated by the presence of HA1. The translation of full-length VN04 HA0 was first evaluated by transfecting HEK293T cells with plasmids encoding either TAG-mutated HA0 and NESPylRSAF/tRNAPyl, or wild-type HA0 with HIV-1 Gag-Pol. The growth medium was supplemented with 0.5mM TCO* ncAA (SiChem). As before, fresh medium containing neuraminidase was added at 24 hours post-transfection. At 48 hours post-transfection cells were lysed and evaluated by Western blot. Maximal UAG readthrough efficiency was achieved at positions 17 and 127. Therefore, TAG mutations at both positions were combined into a single construct (HA17TAG/127TAG; Figure S1A). In the presence of UAG codons at both positions 17 and 127, full-length HA0* was translated at approximately 5–10% efficiency as compared to HA and was dependent on the presence of NESPylRSAF/tRNAPyl and TCO* (Figure S1C). In addition, virions were harvested, concentrated, and evaluated by Western blot for the presence of HA0* and HIV-1 p24. Full-length HA0* was incorporated efficiently into the virion, and was dependent on NESPylRSAF/tRNAPyl and TCO* ncAA (Figure S1C).
Virus entry assay
The functionality of the modified VN04 HA was verified in a viral membrane fusion assay (Cavrois et al., 2002). HEK293T cells were co-transfected with HA17TAG/127TAG or wild-type HA plasmid, HIV-1 Gag-Pol plasmid and a reporter plasmid encoding β-lactamase fused to the HIV-1 protein, Vpr (BlaM-Vpr). Alternatively, as a positive control for fusion, VSV-G plasmid was transfected in place of HA. Translation of full-length HA* was facilitated through the additional presence of the NESPylRSAF/tRNAPyl plasmid, and the inclusion of 0.5 mM TCO* ncAA in the growth medium. Virions were harvested, and concentrated by centrifugation, as described above. Virus pellets were resuspended in phenol red-free DMEM medium (Gibco) (supplemented as for the growth medium) and used to infect A549 cells in a 96-well plate. The plate was centrifuged for 30 minutes at 3700 rpms at 4°C. The cells were washed with HBSS and the media was replaced. The cells were incubated at 37°C for 90 minutes. The cells were then loaded with the CCF4-AM fluorophore (LiveBlazer FRET B/G kit, ThermoFisher) in the presence of 250 mM probenecid at 11°C overnight. The infection was monitored by detecting cleavage of the CCF4-AM by BlaM using a Synergy HT plate reader (Biotek). Virus particles containing HA17TCO/127TCO demonstrated fusion at approximately 80% the efficiency of wild-type HA (Figure S2A). Alternatively, virus particles were incubated with 1.5 μM KB2 or CR9114 antibody prior to introduction to the A549 target cells. Under these conditions, the antibodies neutralized the virions containing HA* as efficiently as those containing wild-type HA (Figure S2A).
ELISA assay
ELISA assays were performed with a panel of four neutralizing antibodies (KB2, CR9114, FI6, and 6F12) that are specific to pre-fusion HA to ensure that incorporation of two TCO* residues and attachment of fluorophores did not disrupt the native conformation of HA. Pseudovirions containing HA or HA* were immobilized on high binding-capacity 96-well plates (Thermo). Antibodies were bound to virions for 1 hour at 37C at the concentrations indicated in Figure S2C, followed by detection using horseradish peroxidase-conjugated secondary antibodies and SuperSignal ELISA Pico chemiluminescent substrate (Thermo). Absorbance at 450 nm was read using a Synergy HT plate reader (Biotek).
Virus-liposome fusion assay
The membrane of virions containing either HA or HA*-Cy3/Cy5 was labeled with 40 μM DiO through incubation for 3 hours at room temperature, followed by purification on an OptiPrep gradient as described above. Liposomes were formed as described below, and combined to a final concentration of 30 nM with virions containing either HA or HA*-Cy3/Cy5, and DiO-labeled membrane. The liposome and virus mixture was allowed to incubate at room temperature for 5 minutes to enable virus-liposome binding. Buffer was then added by stopped-flow to adjust the pH to the desired level, followed by time-based measurement of DiO fluorescence. DiO was excited at 450 nm, and fluorescence was detected at 525 nm at 1 second intervals for 300 seconds at room temperature in a QuantaMaster 400 bulk fluorimeter (Horiba; Figure S2B).
Fluorescently labeling HA* for smFRET imaging
For smFRET imaging virus particles were formed that contained on average a single doubly labeled HA* protomer. HEK293T cells were transfected with a 1:1 ratio of HA17TAG/127TAG to wild-type HA plasmid, HIV-1 Gag-Pol plasmid and NESPylRSAF/tRNAPyl plasmid, and grown in the presence of 0.5 mM TCO* ncAA. This HA17TAG/127TAG to wild-type HA plasmid ratio equates to an approximately 10- to 20-fold excess of wild-type HA protein over HA* protein, given the limited efficiency of reading through two stop codons during translation of HA* (Figure S1). Virus was harvested and concentrated by centrifugation as described above. Virus pellets were resuspended in PBS buffer, and labeled by incubation with 2 mM 3-(p-Benzylamino)-1,2,4,5-tetrazine-Cy3 and 2 mM 3-(p-Benzylamino)-1,2,4,5-tetrazine-Cy5 for 10 minutes at 37°C (Nikić et al., 2015). Following the labeling reaction, DSPE-PEG2000-biotin (Avanti Polar Lipids) was added to the mixture at a final concentration of 6 μM (0.02 mg/ml). The mixture was then incubated for an additional 30 minutes at room temperature. The labeled virus was purified away from unbound dye and lipid by ultracentrifugation for 1 hour at 35,000 × g over a 6–30% Optiprep (Sigma) gradient in 50 mM Tris pH 7.4, 100 mM NaCl. The gradient was fractionated, and the fractions containing virus were identified by p24 Western blot. The labeled virus was stored at −80°C until use in smFRET imaging experiments. Fluorescent labeling of the virions was dependent on the presence of HA* (Figure S1D).
smFRET imaging with TIRF microscopy
smFRET imaging was performed using a custom-built prism-based TIRF microscope, which centers on a Rapid Automated Modular Mounting (RAMM) microscope frame (Applied Scientific Instrumentation). The fluorescently labeled virions were immobilized on polyethylene glycol (PEG)-passivated, streptavidin-coated quartz microscope slides, and mounted on an XY automated stage (Applied Scientific Instrumentation). Buffer and reagents were delivered to the surface-bound virions with a syringe pump (Harvard Apparatus). The surface-bound virions were illuminated by the evanescent field generated by total internal reflection of a 532-nm Genesis MX solid-state laser (Coherent) at an intensity of 0.2 kW/cm2. Fluorescence emission was collected through a 1.2-NA 60X water-immersion objective (Olympus), and passed through a T550LPXR dichroic filter (Chroma) to remove scattered laser light. Donor and acceptor emission were separated with a T635LPXR dichroic filter (Chroma) mounted within a MultiCam-LS (Cairn Research), and imaged on two parallel ORCA-Flash4.0-V2 sCMOS cameras (Hamamatsu) connected to a computer via the Camera Link acquisition board (Hamamatsu). Images were collected at 25 frames/second using custom-built LabView acquisition software (National Instruments).
Experiments were performed in Imaging Buffer containing either 50 mM Tris pH 6.4–7.5 or 50 mM citric acid pH 5.2–6.1, and 50 mM NaCl. Also contained in the imaging buffer was a cocktail of triplet-state quenchers (1mM trolox, 1mM cyclooctatetraene, 1 mM nitrobenzyl alcohol) (Dave et al., 2009), and an enzymatic system for removal of molecular oxygen, which included 2 mM protocatechuic acid and 8 nM protocatechuate 3,4-deoxygenase (Aitken et al., 2008). All smFRET experiments were conducted at room temperature.
Liposome preparation
Receptor-containing liposomes were formed according to published procedures (Floyd et al., 2008). Briefly, liposomes were composed of a 4:4:2:0.1 ratio of 1,2,dioleoyl-sn-glycero-3-phosphocholine (DOPC; Avanti Polar Lipids), 1-oleoyl-2-palmitoyl-snglycero-3-phosphocholine (POPC; Avanti Polar Lipids), cholesterol (Avanti Polar Lipids; Avanti Polar Lipids), and bovine brain disialoganglioside GD1a (Sigma). Lipids dissolved in chloroform were mixed in a glass vile. The solvent was removed by evaporation under a stream of argon gas. The dried lipid film was then suspended in HNE buffer (5 mM HEPES, 145 mM NaCl, 0.2 mM EDTA, pH 7.5) to a final concentration of 10 mg/ml. Liposomes were extruded through a polycarbonate membrane filter with pore sizes of 100 nm. Liposome preparations were stored at 4°C and used within 24 hours.
smFRET imaging in the presence of liposomes
Virions were immobilized on passivated streptavidin-coated quartz microscope slides as described above. GD1a-containing liposomes were prepared as described above to a concentration of approximately 30 nM, introduced to the surface-bound virions, and incubated at room temperature for five minutes. Given the exceedingly low effective concentration of surface-bound virions, during incubation liposomes were present at a saturating excess over virus such that nearly all the HA*-Cy3/Cy5 molecules were bound to GD1a in the liposomes. Prior to imaging, the surface was washed with Imaging Buffer at the desired pH to remove unbound liposomes. All smFRET imaging in the presence of liposomes was conducted at room temperature.
MD simulation
Atomic coordinates were taken from a crystallographic structure of the ectodomain of pre-fusion VN04 HA (PDB accession 2FK0) (Stevens et al., 2006). A post-fusion model was generated by threading the VN04 HA2 sequence into the crystallographic structure of an H1 HA (PDB accession 1HTM (Bullough et al., 1994b)) using the Phyre2 server. Coordinates for the VN04 HA2 amino acids proximal to the fusion peptide were generated by homology modeling using SWISS-MODEL. The fusion peptide-proximal residues were then attached to the post-fusion VN04 HA2 model in LEaP, and energy minimized as described below.
Atomic models of Cy3- and Cy5-tetrazine (Jena Bioscience) and the TCO* ncAA were constructed in PyMol (Schrödinger). The model geometries were initially optimized at the AM1 level of theory with the sqm program in the AmberTools software package. Geometries were further optimized, and the electrostatic potential (ESP) calculations were performed at the HF/6-31G(d) level of theory in Gaussian 9 (Gaussian, Inc.). Partial atomic charges were then derived by restrained electrostatic potential (RESP) fitting using antechamber in AmberTools. Atom types and bonded parameters from the Generalized Amber Force Field (GAFF2 (Wang et al., 2004)) were assigned using antechamber and parmchk2 in AmberTools. The fluorophores and linkers were attached at positions 17 and 127 in the atomic models of pre- and post-fusion VN04 HA in LEaP. The protein component of the system was parameterized with the Amber force field (ff14SB). The entire system was charge-neutralized and solvated in explicit water using the TIP3P model with periodic boundary conditions in LEaP. Simulations were run on the Bridges computer at the Pittsburgh Supercomputing Center using NAMD version 2.12, thanks to an XSEDE start-up allocation. The systems were energy minimized for 0.1 ns, followed by a 50 ns simulation run in the NPT ensemble, with temperature and pressure maintained at 300 K and 1 atm through the use of Langevin dynamics and the Nosé-Hoover Langevin piston method, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
smFRET data analysis
All smFRET data analysis was perform in Matlab (Mathworks) using the SPARTAN software package (Juette et al., 2016), with additional custom-written scripts. Fluorescence traces were extracted from the movies and corrected for bleedthrough of donor fluorescence onto the acceptor channel. The corrected fluorescence traces were used to calculate the apparent FRET efficiency according to FRET = IA/(γID+IA), where IA and ID are the fluorescence emission intensities of the acceptor and donor fluorescence, respectively, and γ is the empirically determined ratio of detection efficiencies on the acceptor and donor channels. smFRET trajectories were automatically identified according to several criteria: (1) donor and acceptor fluorescence trajectories displayed a single photobleaching event, which is indicative of a single FRETing fluorophore pair per virion; (2) FRET was detectable for minimally 15 frames before photobleaching; (3) the correlation coefficient of donor and acceptor fluorescence traces was less than 0.1; (4) the signal-to-noise ratio of the total fluorescence, defined as the ratio of the magnitude of the photobleaching event to the variance of the background signal, was greater than 8. All smFRET trajectories that met these criteria were fit to a hidden Markov model (HMM) consisting of four states, with FRET values of 0.00 ± 0.06, 0.20 ± 0.08, 0.53 ± 0.08, and 0.95 ± 0.08 (mean ± standard deviation) using the segmental k-means algorithm (Qin, 2004). All the observed FRET data points until the point of photobleaching (transition to 0 FRET) were compiled into histograms. Overlaid on the histograms are three Guassian distributions; the means and standard deviations of the Gaussian fits were constrained to reflect the results of the HMM analysis. This analysis identified the transitions between FRET states, which were used to construct TDPs (McKinney et al., 2006).
HMM analysis of the smFRET trajectories allowed identification of the sequence of dwell times in each FRET state. The dwell times for each FRET state were compiled into histograms and fit to exponential functions, A exp (−kt), where A is the amplitude and k is the rate of transition between every pair of states as defined in Figure 7. The results of this fitting are displayed in Table S3. Given the time resolution of imaging of 25 frames/s, according to the Shannon-Nyquist sampling theorem rates greater than 12.5 s−1 may not have been determined with high accuracy.
Supplementary Material
(A) Schematic of engineered influenza HA*, and sequence alignment of wild-type VN04 HA and HA*. The locations of TCO* residues at positions 17 and 127 are shown as asterisks. (B) Schematic outlining the steps involved in amber stop codon suppression and fluorescent labeling for VN04 HA pseudotyped on the HIV-1 core. See STAR Methods for details. (C) Western blot indicating the presence of wild-type VN04 HA0 and HA on the HIV-1 core. VN04 HA is efficiently cleaved by cellular furin. HA0, formed through mutation of the polybasic furin cleavage site, shows no cleavage. Lysate from cells transfected with VN04 HA0 and HA017TAG/127TAG plasmid. Generation of full length HA0* is dependent on the presence of NESPylRSAF/tRNAPyl and TCO* ncAA. Likewise, full-length VN04 HA0* is dependent on the presence of NESPylRSAF/tRNAPyl and TCO* ncAA. The antibody targeting HA1 was purchased from Thermo Fisher. (D) Pseudotyped virions containing wild-type HA or HA* were subjected to the fluorescent labeling reaction, surface immobilized, and imaged using TIRF microscopy as described in STAR Methods. Surface-localized fluorescence was only observed for HA* indicating specific attachment of the fluorophore. Shown here are representative fields of view.
(A) The fusogenicity of pseudotyped virions containing HA*-Cy3/Cy5 was evaluated using a Blam-based virus entry assay (Cavrois et al. 2002). As compared to virions with wild-type VN04 HA, virions containing HA* demonstrated approximately 80% fusogenicity with A549 target cells, and HA*-Cy3/Cy5 demonstrated approximately 76% fusogenicity. KB2 and CR9114 (1.5 μM) antibodies were incubated with the virions at 37°C for 30 minutes before passage to A549 target cells. VSV-G was used as a positive control for the virus-cell fusion assay. (B) A bulk fluorescence-based lipid mixing assay indicated dequenching of the lipophilic fluorophore (DiO) due to fusion of HA-pseudotyped virions to liposomes containing GD1a (Wessels et al., 2007). The kinetics of fusion for wild-type HA and HA*-Cy3/Cy5 were equivalent across a range of pHs. This indicates no loss of functionality or change in pH dependence for the modified HA. (C) ELISA assays performed with four HA stalk-binding antibodies (CR9114, FI6, KB2, and 6F12) showing equivalent binding to pseudovirions with HA*-Cy3/Cy5 and wild-type HA.
(A) Representative fluorescence (donor, green; acceptor, red) and FRET trajectories (blue) obtained from single HA*-Cy3/Cy5 protomers within HA trimers on the surface of viral particles at pH 7. At each time point FRET efficiency was calculated as the ratio of acceptor fluorescence intensity to total fluorescence intensity. Overlaid on the FRET trajectory in red is an idealized trace generated through HMM analysis. (B–E) The same data acquired at the indicated pH.
The mobility of the fluorophores on HA in the (blue) pre-fusion and (red) post-fusion coiled-coil conformations were evaluated by MD simulation (STAR Methods). (A) The average inter-fluorophore distances over the 50 ns simulations were 39 ± 3 Å and 105 ± 2 Å, for pre-fusion and post-fusion coiled-coil conformations, respectively. These distances are consistent with the observed 0.95-FRET state for the pre-fusion conformation, and the 0.20-FRET state for the post-fusion conformation. (B) Snap-shots of the structural models are shown with the centers of mass of the fluorophores from each frame in the simulation are indicated with green (Cy3) and red (Cy5) spheres.
The furin cleavage site was mutated (RKKR/QKQQ) to abrogate proteolytic processing. (A) (top) Population FRET contour plot and histogram of FRET traces with Gaussian fits overlaid acquired from HA0 at pH 7. (bottom) TDPs indicating relative frequencies of the observed transitions. Similarly, smFRET traces, contour plot, histogram and TDP plot for data acquired with HA0 at (B) pH 6.1 and (C) pH 5.3. HA0 maintains significant conformational dynamics across all pHs considered. (D) The G4E mutation in the fusion peptide of HA2 prevents stable formation of the high-FRET pre-fusion conformation. (top) FRET contour plot and histograms of FRET traces acquired from G4E HA at (D) pH 7 and (E) pH 5.6. (bottom) TDPs indicating relative frequencies of the observed transitions. The G4E HA undergoes significant fluctuations between the low-FRET state and the intermediate-FRET state, but never stably adopts the high-FRET pre-fusion state due to destabilization of the fusion peptide in the hydrophobic pocket. (F) HA stalk-targeting neutralizing antibodies prevent formation of stable low FRET. (top) FRET contour plot, histogram and TDPs (bottom) acquired for HA in the presence of stalk-targeting neutralizing antibody CR9114 at pH 6.1. (G) The same data acquired in the presence of antibody KB2 at pH 6.1. In all panels, data are displayed as in Figure 2.
(A–F) FRET contour plot and histogram displayed as in Figure 2 in the presence of the soluble LSTa oligosaccharide. Data were acquired at the indicated pH. (G–L) The same data acquired in the presence of biotinylated liposomes lacking sialic acid, which were bound to the virions containing a biotin-lipid in the viral membrane by way of a streptavidin bridge.
(A) (top) FRET contour plot, histograms and TDPs acquired in the presence of soluble LSTa immediately after exposure to pH 5.6. The same data were acquired after return to pH 7.5 following (middle) 5 min, or (bottom) 30 min. (B) The same data acquired in the presence of a target membrane lacking sialic acid. Data are displayed as in Figure 2.
HIGHLIGHTS.
Influenza HA spontaneously samples conformations related to membrane fusion.
Interaction with the receptor allosterically induces fusion peptide movement.
pH, receptor binding, and interaction with a target membrane regulate HA dynamics.
HA can return to the pre-fusion conformation after transient exposure to acidic pH.
Acknowledgments
The authors with to thank Dr. Peter Kwong for providing the VN04 HA expression plasmid, Drs. Gregory Melikian and Sergi Padilla-Parra for assistance with the virus entry assay, and Dr. Carol Weiss for assistance with production of viral particles containing VN04 HA and the HIV-1 core. This work was supported by NIH grants DP2 AI124384 and K22 AI116262, and the Gilead Sciences Research Scholars Program to J.B.M., and by the NIAID Centers for Excellence in Influenza Virus Research and Surveillance program (CEIRS, HHSN272201400008C) to F.K.
Footnotes
AUTHOR CONTRIBUTIONS
D.K.D. and J.B.M. designed the experiments. R.G., I.N.S., F.K., and E.A.L. provided resources. D.K.D. prepared the samples and conducted all the experiments. D.K.D. and J.B.M. analyzed and interpreted the data and wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aitken CE, Marshall RA, Puglisi JD. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher C, Ludwig K, Herrmann A, van Heel M, Stark H. Structure of influenza haemagglutinin at neutral and at fusogenic pH by electron cryo-microscopy. FEBS Lett. 1999;463:255–259. doi: 10.1016/s0014-5793(99)01475-1. [DOI] [PubMed] [Google Scholar]
- Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of Influenza Hemagglutinin at the Ph of Membrane-Fusion. Nature. 1994a;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Bullough PA, Hughson FM, Treharne AC, Ruigrok RW, Skehel JJ, Wiley DC. Crystals of a fragment of influenza haemagglutinin in the low pH induced conformation. J Mol Biol. 1994b;236:1262–1265. doi: 10.1016/0022-2836(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Carr CM, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 1998;95:409–417. doi: 10.1016/s0092-8674(00)81771-7. [DOI] [PubMed] [Google Scholar]
- Chen J, Skehel JJ, Wiley DC. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci USA. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampor F, BAYLEY PM, Nermut MV, Hirst EM, Sugrue RJ, HAY AJ. Evidence that the amantadine-induced, M2-mediated conversion of influenza A virus hemagglutinin to the low pH conformation occurs in an acidic trans Golgi compartment. Virology. 1992;188:14–24. doi: 10.1016/0042-6822(92)90730-d. [DOI] [PubMed] [Google Scholar]
- Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- Dave R, Terry DS, Munro JB, Blanchard SC. Mitigating unwanted photophysical processes for improved single-molecule fluorescence imaging. Biophys J. 2009;96:2371–2381. doi: 10.1016/j.bpj.2008.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, et al. Highly Conserved Protective Epitopes on Influenza B Viruses. Science. 2012;337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd DL, Ragains JR, Skehel JJ, Harrison SC, van Oijen AM. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci USA. 2008;105:15382–15387. doi: 10.1073/pnas.0807771105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J, Cardone G, Heymann JB, Winkler DC, Steven AC. Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J Virol. 2012;86:2919–2929. doi: 10.1128/JVI.06698-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia NK, Guttman M, Ebner JL, Lee KK. Dynamic changes during acid-induced activation of influenza hemagglutinin. Structure. 2015;23:665–676. doi: 10.1016/j.str.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething MJ, Doms RW, York D, White J. Studies on the Mechanism of Membrane-Fusion - Site-Specific Mutagenesis of the Hemagglutinin of Influenza-Virus. J Cell Biol. 1986;102:11–23. doi: 10.1083/jcb.102.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenke JA, Armstrong RT, Newcomb WW, Brown JC, White JM. New insights into the spring-loaded conformational change of influenza virus hemagglutinin. J Virol. 2002;76:4456–4466. doi: 10.1128/JVI.76.9.4456-4466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha Y, Stevens DJ, Skehel JJ, Wiley DC. X-ray structures of H5 avian and H9 swine influenza virus hemagglutinins bound to avian and human receptor analogs. Proc Natl Acad Sci USA. 2001;98:11181–11186. doi: 10.1073/pnas.201401198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai R, Krammer F, Tan GS, Pica N, Eggink D, Maamary J, Margine I, Albrecht RA, Palese P. Influenza Viruses Expressing Chimeric Hemagglutinins: Globular Head and Stalk Domains Derived from Different Subtypes. J Virol. 2012;86:5774–5781. doi: 10.1128/JVI.00137-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SC. Viral membrane fusion. Virology. 2015;479–480C:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic T, Choi JL, Whelan SP, van Oijen AM, Harrison SC. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife. 2013;2:e00333. doi: 10.7554/eLife.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Terry DS, Wasserman MR, Altman RB, Zhou Z, Zhao H, Blanchard SC. Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat Methods. 2016;13:341–344. doi: 10.1038/nmeth.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaoka Y, Webster RG. Sequence Requirements for Cleavage Activation of Influenza-Virus Hemagglutinin Expressed in Mammalian-Cells. Proc Natl Acad Sci USA. 1988;85:324–328. doi: 10.1073/pnas.85.2.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble GW, Bodian DL, Rose J, Wilson IA, White JM. Intermonomer Disulfide Bonds Impair the Fusion Activity of Influenza-Virus Hemagglutinin. J Virol. 1992;66:4940–4950. doi: 10.1128/jvi.66.8.4940-4950.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte T, Ludwig K, Krumbiegel M, Zirwer D, Damaschun G, Herrmann A. Transient changes of the conformation of hemagglutinin of influenza virus at low pH detected by time-resolved circular dichroism spectroscopy. J Biol Chem. 1997;272:9764–9770. doi: 10.1074/jbc.272.15.9764. [DOI] [PubMed] [Google Scholar]
- Krumbiegel M, Herrmann A, Blumenthal R. Kinetics of the Low Ph-Induced Conformational-Changes and Fusogenic Activity of Influenza Hemagglutinin. Biophys J. 1994;67:2355–2360. doi: 10.1016/S0006-3495(94)80721-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikina E, Ramos C, Markovic I, Zimmerberg J, Chernomordik LV. Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. Embo J. 2002;21:5701–5710. doi: 10.1093/emboj/cdf559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Eddy NR, Noel JK, Whitford PC, Wang Q, Ma J, Onuchic JN. Order and disorder control the functional rearrangement of influenza hemagglutinin. Proc Natl Acad Sci USA. 2014;111:12049–12054. doi: 10.1073/pnas.1412849111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci USA. 1998;95:6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic I, Leikina E, Zhukovsky M, Zimmerberg J, Chernomordik LV. Synchronized activation and refolding of influenza hemagglutinin in multimeric fusion machines. J Cell Biol. 2001;155:833–843. doi: 10.1083/jcb.200103005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay T, Patel M, Pickles RJ, Johnson LG, Olsen JC. Influenza M2 envelope protein augments avian influenza hemagglutinin pseudotyping of lentiviral vectors. Gene Ther. 2006;13:715–724. doi: 10.1038/sj.gt.3302715. [DOI] [PubMed] [Google Scholar]
- McKinney SA, Joo C, Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000;151:413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB, Kwong PD, et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science. 2014;346:759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikić I, Estrada Girona G, Kang JH, Paci G, Mikhaleva S, Koehler C, Shymanska NV, Ventura Santos C, Spitz D, Lemke EA. Debugging Eukaryotic Genetic Code Expansion for Site-Specific Click-PAINT Super-Resolution Microscopy. Angew Chem Int Ed Engl. 2016;55:16172–16176. doi: 10.1002/anie.201608284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikić I, Kang JH, Girona GE, Aramburu IV, Lemke EA. Labeling proteins on live mammalian cells using click chemistry. Nat Protoc. 2015;10:780–791. doi: 10.1038/nprot.2015.045. [DOI] [PubMed] [Google Scholar]
- Nikić I, Plass T, Schraidt O, Szymański J, Briggs JAG, Schultz C, Lemke EA. Minimal tags for rapid dual-color live-cell labeling and super-resolution microscopy. Angew Chem Int Ed Engl. 2014;53:2245–2249. doi: 10.1002/anie.201309847. [DOI] [PubMed] [Google Scholar]
- Qin F. Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys J. 2004;86:1488–1501. doi: 10.1016/S0006-3495(04)74217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T, Leser GP, Lamb RA. The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J Cell Biol. 1996;133:733–747. doi: 10.1083/jcb.133.4.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandrin V, Boson B, Salmon P, Gay W, Nègre D, Le Grand R, Trono D, Cosset FL. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood. 2002;100:823–832. doi: 10.1182/blood-2001-11-0042. [DOI] [PubMed] [Google Scholar]
- Stegmann T, White JM, Helenius A. Intermediates in Influenza Induced Membrane-Fusion. Embo J. 1990;9:4231–4241. doi: 10.1002/j.1460-2075.1990.tb07871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J, Blixt O, Tumpey TM, Taubenberger JK, Paulson JC, Wilson IA. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- Stieneke-Gröber A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, Klenk HD, Garten W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. Embo J. 1992;11:2407–2414. doi: 10.1002/j.1460-2075.1992.tb05305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Lamb RA. Influenza virus M2 protein ion channel activity stabilizes the native form of fowl plague virus hemagglutinin during intracellular transport. J Virol. 1994;68:911–919. doi: 10.1128/jvi.68.2.911-919.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan GS, Krammer F, Eggink D, Kongchanagul A, Moran TM, Palese P. A Pan-H1 Anti-Hemagglutinin Monoclonal Antibody with Potent Broad-Spectrum Efficacy In Vivo. J Virol. 2012;86:6179–6188. doi: 10.1128/JVI.00469-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian SA, Tamm LK. Reversible pH-dependent conformational change of reconstituted influenza hemagglutinin. J Mol Biol. 1996;260:312–316. doi: 10.1006/jmbi.1996.0402. [DOI] [PubMed] [Google Scholar]
- Tatulian SA, Hinterdorfer P, Baber G, Tamm LK. Influenza hemagglutinin assumes a tilted conformation during membrane fusion as determined by attenuated total reflection FTIR spectroscopy. Embo J. 1995;14:5514–5523. doi: 10.1002/j.1460-2075.1995.tb00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Wang W, Butler EN, Veguilla V, Vassell R, Thomas JT, Moos M, Ye Z, Hancock K, Weiss CD. Establishment of retroviral pseudotypes with influenza hemagglutinins from H1, H3, and H5 subtypes for sensitive and specific detection of neutralizing antibodies. J Virol Methods. 2008;153:111–119. doi: 10.1016/j.jviromet.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Weber T, Paesold G, Galli C, Mischler R, Semenza G, Brunner J. Evidence for H(+)-induced insertion of influenza hemagglutinin HA2 N-terminal segment into viral membrane. J Biol Chem. 1994;269:18353–18358. [PubMed] [Google Scholar]
- Wessels L, Elting MW, Scimeca D, Weninger K. Rapid membrane fusion of individual virus particles with supported lipid bilayers. Biophys J. 2007;93:526–538. doi: 10.1529/biophysj.106.097485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM, Wilson IA. Anti-Peptide Antibodies Detect Steps in a Protein Conformational Change - Low-Ph Activation of the Influenza-Virus Hemagglutinin. J Cell Biol. 1987;105:2887–2896. doi: 10.1083/jcb.105.6.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Xu R, Wilson IA. Structural Characterization of an Early Fusion Intermediate of Influenza Virus Hemagglutinin. J Virol. 2011;85:5172–5182. doi: 10.1128/JVI.02430-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW, Taylor A, Yellen A, Caton A, Gerhard W, Bachi T. Mutations in or Near the Fusion Peptide of the Influenza-Virus Hemagglutinin Affect an Antigenic Site in the Globular Region. J Virol. 1993;67:933–942. doi: 10.1128/jvi.67.2.933-942.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Viswanathan K, Raman R, Yu W, Sasisekharan R, Wilson IA. Structural Basis for a Switch in Receptor Binding Specificity of Two H5N1 Hemagglutinin Mutants. Cell Rep. 2015;13:1683–1691. doi: 10.1016/j.celrep.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic of engineered influenza HA*, and sequence alignment of wild-type VN04 HA and HA*. The locations of TCO* residues at positions 17 and 127 are shown as asterisks. (B) Schematic outlining the steps involved in amber stop codon suppression and fluorescent labeling for VN04 HA pseudotyped on the HIV-1 core. See STAR Methods for details. (C) Western blot indicating the presence of wild-type VN04 HA0 and HA on the HIV-1 core. VN04 HA is efficiently cleaved by cellular furin. HA0, formed through mutation of the polybasic furin cleavage site, shows no cleavage. Lysate from cells transfected with VN04 HA0 and HA017TAG/127TAG plasmid. Generation of full length HA0* is dependent on the presence of NESPylRSAF/tRNAPyl and TCO* ncAA. Likewise, full-length VN04 HA0* is dependent on the presence of NESPylRSAF/tRNAPyl and TCO* ncAA. The antibody targeting HA1 was purchased from Thermo Fisher. (D) Pseudotyped virions containing wild-type HA or HA* were subjected to the fluorescent labeling reaction, surface immobilized, and imaged using TIRF microscopy as described in STAR Methods. Surface-localized fluorescence was only observed for HA* indicating specific attachment of the fluorophore. Shown here are representative fields of view.
(A) The fusogenicity of pseudotyped virions containing HA*-Cy3/Cy5 was evaluated using a Blam-based virus entry assay (Cavrois et al. 2002). As compared to virions with wild-type VN04 HA, virions containing HA* demonstrated approximately 80% fusogenicity with A549 target cells, and HA*-Cy3/Cy5 demonstrated approximately 76% fusogenicity. KB2 and CR9114 (1.5 μM) antibodies were incubated with the virions at 37°C for 30 minutes before passage to A549 target cells. VSV-G was used as a positive control for the virus-cell fusion assay. (B) A bulk fluorescence-based lipid mixing assay indicated dequenching of the lipophilic fluorophore (DiO) due to fusion of HA-pseudotyped virions to liposomes containing GD1a (Wessels et al., 2007). The kinetics of fusion for wild-type HA and HA*-Cy3/Cy5 were equivalent across a range of pHs. This indicates no loss of functionality or change in pH dependence for the modified HA. (C) ELISA assays performed with four HA stalk-binding antibodies (CR9114, FI6, KB2, and 6F12) showing equivalent binding to pseudovirions with HA*-Cy3/Cy5 and wild-type HA.
(A) Representative fluorescence (donor, green; acceptor, red) and FRET trajectories (blue) obtained from single HA*-Cy3/Cy5 protomers within HA trimers on the surface of viral particles at pH 7. At each time point FRET efficiency was calculated as the ratio of acceptor fluorescence intensity to total fluorescence intensity. Overlaid on the FRET trajectory in red is an idealized trace generated through HMM analysis. (B–E) The same data acquired at the indicated pH.
The mobility of the fluorophores on HA in the (blue) pre-fusion and (red) post-fusion coiled-coil conformations were evaluated by MD simulation (STAR Methods). (A) The average inter-fluorophore distances over the 50 ns simulations were 39 ± 3 Å and 105 ± 2 Å, for pre-fusion and post-fusion coiled-coil conformations, respectively. These distances are consistent with the observed 0.95-FRET state for the pre-fusion conformation, and the 0.20-FRET state for the post-fusion conformation. (B) Snap-shots of the structural models are shown with the centers of mass of the fluorophores from each frame in the simulation are indicated with green (Cy3) and red (Cy5) spheres.
The furin cleavage site was mutated (RKKR/QKQQ) to abrogate proteolytic processing. (A) (top) Population FRET contour plot and histogram of FRET traces with Gaussian fits overlaid acquired from HA0 at pH 7. (bottom) TDPs indicating relative frequencies of the observed transitions. Similarly, smFRET traces, contour plot, histogram and TDP plot for data acquired with HA0 at (B) pH 6.1 and (C) pH 5.3. HA0 maintains significant conformational dynamics across all pHs considered. (D) The G4E mutation in the fusion peptide of HA2 prevents stable formation of the high-FRET pre-fusion conformation. (top) FRET contour plot and histograms of FRET traces acquired from G4E HA at (D) pH 7 and (E) pH 5.6. (bottom) TDPs indicating relative frequencies of the observed transitions. The G4E HA undergoes significant fluctuations between the low-FRET state and the intermediate-FRET state, but never stably adopts the high-FRET pre-fusion state due to destabilization of the fusion peptide in the hydrophobic pocket. (F) HA stalk-targeting neutralizing antibodies prevent formation of stable low FRET. (top) FRET contour plot, histogram and TDPs (bottom) acquired for HA in the presence of stalk-targeting neutralizing antibody CR9114 at pH 6.1. (G) The same data acquired in the presence of antibody KB2 at pH 6.1. In all panels, data are displayed as in Figure 2.
(A–F) FRET contour plot and histogram displayed as in Figure 2 in the presence of the soluble LSTa oligosaccharide. Data were acquired at the indicated pH. (G–L) The same data acquired in the presence of biotinylated liposomes lacking sialic acid, which were bound to the virions containing a biotin-lipid in the viral membrane by way of a streptavidin bridge.
(A) (top) FRET contour plot, histograms and TDPs acquired in the presence of soluble LSTa immediately after exposure to pH 5.6. The same data were acquired after return to pH 7.5 following (middle) 5 min, or (bottom) 30 min. (B) The same data acquired in the presence of a target membrane lacking sialic acid. Data are displayed as in Figure 2.