

Abstract
Rickets and hyperparathyroidism caused by a defective vitamin D receptor (VDR) can be prevented in humans and animals by high calcium intake, suggesting that intestinal calcium absorption is critical for 1,25(OH)2 vitamin D [1,25(OH)2D3] action on calcium homeostasis. We assessed the rate of serum 45Ca accumulation within 10 min of oral gavage in two strains of VDR-knockout (KO) mice (Leuven and Tokyo KO) and observed a 3-fold lower area under the curve in both KO strains. Moreover, we evaluated the expression of intestinal candidate genes involved in transcellular calcium transport. The calcium transport protein1 (CaT1) was more abundantly expressed at mRNA level than the epithelial calcium channel (ECaC) in duodenum, but both were considerably reduced (CaT1>90%, ECaC>60%) in the two VDR-KO strains on a normal calcium diet. Calbindin-D9K expression was decreased only in the Tokyo KO, whereas plasma membrane calcium ATPase (PMCA1b) expression was normal in both VDR-KOs. In Leuven wild-type mice, a high calcium diet inhibited (>90%) and 1,25(OH)2D3 injection or low calcium diet induced (6-fold) duodenal CaT1 expression and, to a lesser degree, ECaC and calbindin-D9K expression. In Leuven KO mice, however, high or low calcium intake decreased calbindin-D9K and PMCA1b expression, whereas CaT1 and ECaC expression remained consistently low on any diet. These results suggest that the expression of the novel duodenal epithelial calcium channels (in particular CaT1) is strongly vitamin D-dependent, and that calcium influx, probably interacting with calbindin-D9K, should be considered as a rate-limiting step in the process of vitamin D-dependent active calcium absorption.
It is commonly accepted that 1,25(OH)2 vitamin D [1,25(OH)2D3] is one of the main hormones for calcium homeostasis and bone mineralization (1). The genomic effects underlying part of these processes are mediated by the interaction of 1,25(OH)2D3 with the nuclear vitamin D receptor (VDR) in a ligand-dependent manner (2).
Hereditary hypocalcemic vitamin D-resistant rickets (HVDDR), an autosomal recessive disorder, is caused by a defect in the human VDR gene, resulting in target tissue insensitivity to 1,25(OH)2D3 (2). The concomitant bone pathology is cured by frequent i.v. calcium infusions (3) and prevented by high oral doses of calcium (4). VDR-knockout (KO) mice display a phenotype similar to HVDDR: rickets, hypocalcemia, hypophosphatemia, elevated serum 1,25(OH)2D3, hyperparathyroidism, and alopecia (5, 6). Consistent with the standard therapy for HVDDR, a high calcium diet prevented rickets and hyperparathyroidism in VDR-KO mice (7, 8).
On the basis of these data, it is obvious that the small intestine plays an indispensable role in 1,25(OH)2D3 action on calcium homeostasis and bone mineralization (2). Possible targets involved in vitamin D-dependent active duodenal calcium absorption are: (i) calcium influx, which was elusive until the epithelial calcium channel (ECaC) (9–13) and the calcium transport protein type 1 (CaT1) (14) were described as possible gatekeepers for calcium entering the cell; (ii) intracellular calcium transfer by calbindin-D9K; (iii) calcium extrusion by the plasma membrane calcium ATPase (PMCA1b) (15). The calbindin-D9K mRNA level is decreased in duodenum of the two known VDR-KO strains, compared with wild-type (WT) mice (5, 16), suggesting a critical role for calbindin-D9K in vitamin D-dependent calcium absorption.
However, no data on the regulation of the other proteins or assessment of active calcium absorption in VDR-KO mice are available. We used two strains of VDR-KO mice to evaluate calcium absorption in vivo and to measure the expression of the intestinal candidate genes possibly involved in transcellular calcium transport.
Materials and Methods
Animals.
Leuven VDR-KO mice.
The VDR targeting vector (Fig. 1A) contained a 5′ flanking 5.5-kb BamHI-XhoI and a 3′ flanking 5.5-kb XhoI fragment encompassing exons 1 and 2, respectively. A “floxed” cassette containing the neomycin phosphotransferase (neo) gene was inserted at the XhoI site upstream of exon 2, and a third lox site was introduced downstream of exon 2 at the inactivated EcoRV site. After electroporation of this vector, R1 embryonic stem (ES) cells were selected in G418 (200 μg/ml) and gancyclovir (2 μM) and assessed by Southern analysis. Targeted ES cell clones were then transiently electroporated with a Cre-recombinase expression cassette, and Southern analysis demonstrated correct excision of the “floxed” neo cassette. Chimeric mice were generated by morula aggregation and bred for germline transmission with Swiss mice in an open animal facility (Flanders Interuniversity Institute for Biotechnology). VDR-KO mice were obtained by crossing the VDRlox mice with phosphoglycerate kinase-cre mice (17).
Figure 1.

Targeting of the VDR gene. (A) Modification of the VDR gene. 1, Targeting vector pNT.VDR and VDRWT allele; 2, Homologously recombined VDR allele (VDRneo); 3, Modified VDR allele after Cre-excision of the floxed neo cassette; 4, VDRmin allele after Cre-excision of the floxed exon 2. exons  ; intron sequences
; intron sequences  ; D external (1.5-kb XhoI fragment) and C internal (1-kb EcoRV-SacI fragment) hybridization probe. The observed size of the diagnostic restriction fragments, used to distinguish the WT and mutant alleles, corresponds to their expected size. (B) Southern analysis of EcoRV-digested genomic DNA from VDRWT and VDRneo embryonic stem cell clones by using the external 3′ probe D, which generates a 8.5-kb VDRWT and a 10.5-kb VDRneo fragment. (C) Southern analysis of EcoRV-digested genomic DNA from VDRWT and VDR+/lox mice by using the external 3′ probe D, which generates a 8.5-kb VDRWT and a 16-kb VDRlox fragment. (D) Southern analysis of SacI-digested genomic DNA from VDRWT, VDR+/− and VDRKO mice by using the internal probe C. (E) RT-PCR analysis of RNA isolated from duodenum, kidney, and femur of VDRWT and VDRKO mice for VDR and HPRT RNA level expression.
; D external (1.5-kb XhoI fragment) and C internal (1-kb EcoRV-SacI fragment) hybridization probe. The observed size of the diagnostic restriction fragments, used to distinguish the WT and mutant alleles, corresponds to their expected size. (B) Southern analysis of EcoRV-digested genomic DNA from VDRWT and VDRneo embryonic stem cell clones by using the external 3′ probe D, which generates a 8.5-kb VDRWT and a 10.5-kb VDRneo fragment. (C) Southern analysis of EcoRV-digested genomic DNA from VDRWT and VDR+/lox mice by using the external 3′ probe D, which generates a 8.5-kb VDRWT and a 16-kb VDRlox fragment. (D) Southern analysis of SacI-digested genomic DNA from VDRWT, VDR+/− and VDRKO mice by using the internal probe C. (E) RT-PCR analysis of RNA isolated from duodenum, kidney, and femur of VDRWT and VDRKO mice for VDR and HPRT RNA level expression.
Tokyo VDR-KO mice.
Tokyo VDR-KO mice were generated by targeted ablation of exon 2. They were kindly provided by S. Kato (University of Tokyo) (5).
The animals, bred in our animal housing facilities [Proefdierencentrum, Katholieke Universiteit Leuven (KUL)], received a normal diet containing 1.1% calcium, 0.8% phosphorus, 0% lactose (Standard, Carfil, Oud-Turnhout, Belgium). In addition, the Leuven strain was studied on either a low calcium diet (0.02% calcium/1% phosphorus/0% lactose: 4403.03 Purified Diet, Hope Farms, Woerden, The Netherlands) or high calcium (rescue) diet (2% calcium/1.25% phosphorus/20% lactose: TD94112, Teklad, Madison, WI). The diets were started immediately after weaning. Animals were killed at around 10 weeks of age. A group of 8-week-old WT mice on a normal diet were injected with 2 μg of 1,25(OH)2D3/kg body weight (BW) (kindly given by J. P. Vandevelde, Solvay Pharmaceuticals BV, Weesp, The Netherlands) or vehicle (peanut oil) 6 h before euthanasia. The Animal Ethics Board of the KUL approved all experimental procedures.
Functional Calcium Absorption Assay.
To validate the method, two control groups of Swiss mice were used: a first group was injected with 2 μg of 1,25(OH)2D3/kg BW or vehicle (peanut oil) 6 h before the assay; the second group received either a normal, low, or high calcium diet during 1 week before the experiment. The Leuven strain was studied after 1 week of low calcium diet. The Tokyo strain was fed a normal diet throughout, because the Tokyo KO did not tolerate calcium deprivation.
Mice were fasted 12 h before the test. Animals were hemodynamically stable under anesthesia (urethane 1.4 mg/g BW) during the entire experiment. The test solution contained 0.1 mM CaCl2, 125 mM NaCl, 17 mM Tris, 1.8 g/liter fructose, and was enriched with 20 μCi 45CaCl2/ml (18 Ci/g, New England Nuclear) for oral tests (Leuven and Tokyo mice) or with 6.5 μCi 45CaCl2/ml for i.v. tests (Leuven mice). For oral tests, 15 μl/g of BW was administered by gavage (p.o.) and 5 μl/g of BW for i.v. tests. Blood samples were taken at indicated time intervals. Serum (10 μl) was analyzed by liquid scintillation counting. The change in the plasma calcium concentration (ΔμMol) was calculated from the 45Ca content of the plasma samples and the specific activity of the administered calcium. Results are also expressed as area under the curve between 0 or 1 and 10 min (AUC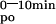 , AUC
, AUC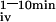 ).
).
RNA Isolation and Quantitative Reverse Transcription–PCR (RT-PCR).
Mice were exsanguinated under anesthesia. Distal duodenum and kidney were homogenized in TriZol (GIBCO/BRL). Total RNA was isolated as specified by the manufacturer. RNA (5 μg) was reverse transcribed by using Superscript II RT (GIBCO/BRL). Quantitative RT-PCR reactions (qRT-PCR) were performed by using an ABI-prism 7700 sequence detector (Gene-Amp PCR system 9600, PE Biosystems, Foster City, CA). Sequences of PCR primers and fluorescent probes for ECaC, CaT1, calbindin-D9K, calbindin-D28K, PMCA1b, Na+/Ca2+ exchanger (NCX), VDR, 25(OH)D3-1α-hydroxylase (Cyp27B1), and hypoxanthine–guanine phosphoribosyl transferase (HPRT) are published as supporting information on the PNAS web site (Table 4, www.pnas.org). The obtained amplicons were verified by sequencing. The relative expression levels of the target genes were calculated as a ratio to the HPRT gene.
Biochemical Assays.
Serum analysis.
Serum calcium, 1,25(OH)2D3, and parathyroid hormone (PTH) levels were determined by microcolorimetric assay (Sigma), [125I]1,25(OH)2D3 RIA kit (Diasorin, Stillwater, MN), and mouse-PTH Elisa kit (Immutopics, San Clemente, CA), respectively.
Bone calcium content.
Femur dry weight was obtained by burning the bones for 24 h at 100°C, followed by 24 h at 600°C to obtain ash weight. Calcium was measured by microcolorimetry in HCl-dissolved ash dilutions and reported corrected for the dry weight.
Calbindin-D9K protein measurements.
Proximal duodenum was dissected as described for RNA extraction. Calbindin-D9K protein content was measured by RIA (18) [a gift from M. Thomasset (Institut National de la Santé et de la Recherche Médicale, U.120, Le Vésinet, France)]. Calbindin-D9K purified from mouse duodenum was used as reference standard.
Statistical Analysis.
Statistical analysis was performed by using the software program ncss (NCSS, Kaysville, UT). The results are expressed as mean ± SEM. One- (genotype) and two-way (genotype vs. diet or genotype vs. time) ANOVA was carried out to detect intergroup differences, followed by Bonferroni (Versus Control) Multiple-Comparison Test. Differences were considered significant at P < 0.05.
Results
Phenotype of the Leuven VDR-KO Mice.
After mating of heterozygotes, Leuven VDR-KO, -heterozygote, and WT mice were obtained in the expected Mendelian ratio. Homozygosity for the inactivated allele was demonstrated by Southern blot (Fig. 1D). The lack of functional VDR was demonstrated by the absence of VDR mRNA on RT-PCR analysis of total RNA from duodenum, kidney, and bone of 4-week-old mice (Fig. 1E). The Leuven KO exhibited the same phenotype as the two other VDR-KO strains (5, 6): hypocalcemia, hyperparathyroidism, elevated serum 1,25(OH)2D3 levels, decreased calcium content in bone (Table 1), rickets, and a progressive alopecia after weaning. Similar to the phenotypic rescue of the Boston KO (7, 8), we found normalization of serum calcium, calcium content in bone, and serum PTH on the rescue diet. Consequently, renal expression of 1α-hydroxylase and serum 1,25(OH)2D3 levels dropped (Table 1). The low calcium diet resulted in mild (WT animals) to severe hypocalcemia (KO mice). Calcium content in bone showed the same pattern as serum calcium. Although serum 1,25(OH)2D3 and PTH levels in WT mice on the low calcium tended to be higher than in WT animals on the normal diet, these differences did not prove to be significant (Table 1).
Table 1.
Phenotypic characterization of Leuven mice raised on different diets and of Tokyo mice raised on a normal diet
| Leuven: low calcium diet
|
Leuven: normal diet
|
Leuven: rescue diet
|
Tokyo: normal diet
|
|||||
|---|---|---|---|---|---|---|---|---|
| WT (n = 12) | KO (n = 12) | WT (n = 12) | KO (n = 12) | WT (n = 12) | KO (n = 12) | WT (n = 6) | KO (n = 6) | |
| Femoral calcium (mg/g of dry weight) | 15.5 ± 0.7* | 11.5 ± 0.5*† | 20.6 ± 0.4 | 17.3 ± 0.5* | 20.0 ± 0.4 | 20.1 ± 0.5† | 19.2 ± 0.7 | 13.3 ± 0.6* |
| Serum calcium (mg/dl) | 7.4 ± 0.4* | 4.2 ± 0.1*† | 10.1 ± 0.3 | 7.3 ± 0.3* | 10.2 ± 0.2 | 10.1 ± 0.4† | 8.6 ± 0.2 | 6.0 ± 0.1* |
| Serum PTH (pg/ml) | 41 ± 14 | 345 ± 50*† | 17 ± 3 | 182 ± 34* | 16 ± 1 | 15 ± 1.1† | ||
| Serum 1.25(OH)2D3 (pg/ml) | 226 ± 28 | 1843 ± 261*† | 57 ± 7 | 3634 ± 268* | 19 ± 4 | 9 ± 1.9† | ||
| Kidney Cyp27B1 RNA level | 57 ± 8.9 | 144 ± 21* | 2 ± 0.3 | 276 ± 49* | 2 ± 0.6 | 12 ± 10† | 1 ± 0.2 | 177 ± 18* |
Cyp27B1 RNA levels, assessed by qRT-PCR, are expressed as a ratio to the HPRT RNA level.
, P < 0.001 vs. WT on normal diet;
, P < 0.001 vs. KO on normal diet.
Calcium Absorption Assay.
We assessed calcium absorption by measuring serum 45Ca at early time points after oral gavage: 45Ca was already detectable in serum within 2 min after administration. Administration of 1,25(OH)2D3 to control mice, induced significantly higher values at each time point (Fig. 2A), resulting in a 1.8-fold higher AUC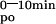 (Table 2), compared with vehicle-treated littermates. In the second validation experiment, 1 week of low calcium diet doubled, whereas 1 week of a high calcium diet halved, the AUC
(Table 2), compared with vehicle-treated littermates. In the second validation experiment, 1 week of low calcium diet doubled, whereas 1 week of a high calcium diet halved, the AUC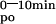 (Fig. 2B; Table 2) when compared with values of Swiss mice on a normal diet.
(Fig. 2B; Table 2) when compared with values of Swiss mice on a normal diet.
Figure 2.

Intestinal calcium absorption assay. Changes in serum calcium (ΔμMol) within 10 min after administration of 45Ca: (A) orally in vehicle (■) and 1,25(OH)2D3 (▴) treated Swiss mice; (B) orally in Swiss mice after 1 week of normal (□), low calcium (▵), or rescue diet (◊); (C) orally (WT ● and KO ▾) and intravenously (WT ○ and KO ▿) in the Leuven strain; (D) orally (WT ● and KO ▾) in the Tokyo strain. ΔμMol is obtained by the equation: (cpm 10 μl serum/cpm 10 μl stock solution) × 102 μM. *, P < 0.001 vs. vehicle at same time point; †, P < 0.05 vs. normal diet at same time point; ‡, P < 0.001 vs. WT at same time point.
Table 2.
The effect of 1,25(OH)2D3-treatment, dietary intervention and VDR inactivation on serum calcium kinetics after oral or intravenous administration (Leuven mice) of 45Ca
| Mouse strain | Swiss (oral 45Ca)
|
Swiss (oral 45Ca)
|
Leuven (oral 45Ca)
|
Leuven (i.v. 45Ca)
|
Tokyo (oral 45Ca)
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Vehicle | 1,25(OH)2D3 | Low Ca diet | Normal diet | High Ca diet | WT | KO | WT | KO | WT | KO | |
| Number of animals | 17 | 13 | 15 | 15 | 15 | 20 | 20 | 11 | 10 | 23 | 13 |
| Age, days | 67 ± 1 | 69 ± 1 | 60 ± 0.3 | 60 ± 0.3 | 60 ± 0 | 65 ± 1 | 67 ± 1 | 64 ± 1 | 63 ± 0.3 | 88 ± 2 | 86 ± 3 |
| Weight, g | 22 ± 0.4 | 22 ± 0.3 | 20 ± 0.4 | 20 ± 0.3 | 19 ± 0.3 | 22 ± 1 | 20 ± 1 | 20 ± 1 | 18 ± 1§ | 19 ± 1 | 13 ± 0.6‡ |
| Serum calcium, mg/dl | ND | ND | ND | ND | ND | 8.5 ± 0.2 | 6.9 ± 0.3‡ | 8.6 ± 0.3 | 6.6 ± 0.2‡ | 8.7 ± 0.2 | 4.8 ± 0.3‡ |
AUC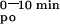 (ΔμMol*min) (ΔμMol*min) |
8.5 ± 0.6 | 15.1 ± 1.2* | 16.8 ± 1.0† | 8.3 ± 1.2 | 4.3 ± 0.8† | 13.3 ± 0.9 | 4.4 ± 0.4‡ | 7.0 ± 0.4 | 2.3 ± 0.3‡ | ||
AUC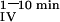 (ΔMol*min) (ΔMol*min) |
10.4 ± 0.6 | 9.2 ± 0.4 | |||||||||
, P < 0.001 vs. vehicle;
, P < 0.001 vs. normal diet;
, P < 0.001 and
, P < 0.05 vs. WT.
Decreased active calcium absorption is claimed to contribute to the hypocalcemia of VDR-KO mice. The Leuven strain was fed a low calcium diet for 1 week, which favors active over passive calcium transport and which resulted in lower serum calcium levels in both WT and KO mice, compared with levels on a normal diet (Tables 1 and 2). The time curve of calcium absorption of the KO mice was significantly lower at each time point, resulting in a 3-fold lower AUC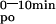 compared with the WT littermates (Fig. 2C; Table 2). Calcium kinetics during the first 10 min after i.v. bolus in the KO animals were indistinguishable from kinetics in WT (Fig. 2C; Table 2). Hence, low serum 45Ca concentrations in the KO were not because of an enhanced clearance from the blood compartment. This reduced calcium absorption in VDR-KO mice was confirmed in the Tokyo strain (Fig. 2D; Table 2). The older age of these animals, the normal calcium diet, and strain differences are possible explanations for the lower absolute values of calcium absorption, compared with the Leuven strain (Fig. 2; Table 2).
compared with the WT littermates (Fig. 2C; Table 2). Calcium kinetics during the first 10 min after i.v. bolus in the KO animals were indistinguishable from kinetics in WT (Fig. 2C; Table 2). Hence, low serum 45Ca concentrations in the KO were not because of an enhanced clearance from the blood compartment. This reduced calcium absorption in VDR-KO mice was confirmed in the Tokyo strain (Fig. 2D; Table 2). The older age of these animals, the normal calcium diet, and strain differences are possible explanations for the lower absolute values of calcium absorption, compared with the Leuven strain (Fig. 2; Table 2).
Effects on Gene Expression in Duodenum.
In a next step, we investigated whether expression of genes involved in transcellular calcium absorption was altered in duodenum of VDR-KO mice on a normal diet. A 3-fold decrease of calbindin-D9K RNA (P < 0.01) and protein (P < 0.001) level was found in the duodenum of the Tokyo KO (Fig. 3 A and B) (5). The Leuven KO presented with a small decrease in calbindin-D9K mRNA level (38% reduction, P < 0.001), which did not affect calbindin-D9K protein content (Fig. 3 A and B). PMCA1b gene expression was not significantly altered in any strain. In contrast, duodenal CaT1 expression was significantly down-regulated in Leuven (P < 0.001) and in Tokyo (P < 0.01) KO mice to less than 10% of WT animals (Fig. 3). ECaC gene expression was also significantly lower in duodenum of KO mice (P < 0.01). However, detection of CaT1 RNA in duodenum was markedly easier because of higher relative mRNA levels (±200-fold) than ECaC RNA, which suggests that CaT1 is the dominant duodenal calcium channel at RNA level in these mice (Table 3).
Figure 3.

Gene expression patterns in duodenum and kidney of Leuven mice on different diets (n = 12/group) and of Tokyo mice on normal diet (n = 6/group). (A) Duodenal gene expression of CaT1 , calbindin-D9K , and PMCA  , assessed by qRT-PCR analysis, is calculated as a ratio to the HPRT RNA level and is expressed relatively to levels of WT mice on normal diet. (B) Calbindin-D9K protein content in duodenum, measured by RIA, is expressed as % (mg/mg) of total duodenal protein. (C) Renal gene expression of ECaC
, assessed by qRT-PCR analysis, is calculated as a ratio to the HPRT RNA level and is expressed relatively to levels of WT mice on normal diet. (B) Calbindin-D9K protein content in duodenum, measured by RIA, is expressed as % (mg/mg) of total duodenal protein. (C) Renal gene expression of ECaC  , calbindin-D9K
, calbindin-D9K  , calbindin-D28K
, calbindin-D28K  , PMCA
, PMCA  , and NCX , assessed by qRT-PCR analysis, is calculated as a ratio to the HPRT RNA level and expressed relatively to levels of WT mice on normal diet. *, P < 0.001, †, P < 0.01, and ‡, P < 0.05 vs. WT on normal diet.
, and NCX , assessed by qRT-PCR analysis, is calculated as a ratio to the HPRT RNA level and expressed relatively to levels of WT mice on normal diet. *, P < 0.001, †, P < 0.01, and ‡, P < 0.05 vs. WT on normal diet.
Table 3.
Duodenal and renal ECaC and CaT1 gene expression in Leuven mice raised on different diets and in Tokyo mice raised on a normal diet
| Leuven: low calcium diet
|
Leuven: normal diet
|
Leuven: rescue diet
|
Tokyo: normal diet
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| WT (n = 12) | KO (n = 12) | WT (n = 12) | KO (n = 12) | WT (n = 12) | KO (n = 12) | WT (n = 6) | KO (n = 6) | ||
| Duodenum | ECaC | 8.2 ± 1.1* | 0.4 ± 0.1* | 4.4 ± 0.8 | 1.6 ± 0.4† | 0.9 ± 0.2* | 0.4 ± 0.2* | 12 ± 3 | 1.0 ± 0.4† |
| CaT1 | 18,061 ± 1158* | 40 ± 11* | 3,020 ± 552 | 283 ± 74* | 64 ± 15* | 18 ± 8* | 2,940 ± 636 | 97 ± 26† | |
| Kidney | ECaC | 2,608 ± 216 | 1,726 ± 124 | 2,927 ± 554 | 3,618 ± 826 | 2,627 ± 286 | 4,703 ± 1010 | 3,684 ± 572 | 4,484 ± 420 |
| CaT1 | 21 ± 2 | 27 ± 3 | 22 ± 4 | 30 ± 4 | 26 ± 4 | 45 ± 8* | 31 ± 6 | 64 ± 5† | |
ECaC and CaT1 RNA levels, assessed by qRT-PCR, are expressed as a ratio to the HPRT RNA level.
, P < 0.001 and
, P < 0.01 vs. WT on normal diet.
These observations led us to study the influence of dietary calcium in the Leuven strain on expression levels of these proteins (Fig. 3 A and B). First, in WT animals, qRT-PCR analysis of calbindin-D9K RNA levels revealed no significant difference between normal and low calcium diet, but a 2.5-fold increase of calbindin-D9K protein content was found in WT mice on the low calcium diet (P < 0.001). Conversely, a more than 70% decrease in calbindin-D9K RNA (P < 0.001) and protein level (P < 0.05) was found in WT animals consuming the rescue diet. In the KO animals on a high and on a low calcium diet, calbindin-D9K RNA level decreased to less than 20% of KO on a normal diet (P < 0.001). This resulted in a reduction of calbindin-D9K protein content of 70% on the rescue diet and 40% on the low calcium diet, compared with levels of KO mice on a normal diet. Second, PMCA1b gene expression was decreased two to three times (P < 0.001) in WT on the low calcium diet and in KO on both low and high calcium diet. Third, CaT1 expression varied considerably in WT mice: a 6-fold increase with calcium restriction (P < 0.001) and a 90% reduction with calcium abundance (P < 0.001), compared with levels of WT mice on normal diet. In KO animals, CaT1 expression was severely impaired, regardless of calcium intake (P < 0.001). Despite less pronounced differences and lower detection levels, a similar pattern as for CaT1 gene expression was found for ECaC RNA levels in the different conditions (Table 3).
As a different approach to elicit vitamin D genomic responses, a single dose of 2 μg of 1,25(OH)2D3/kg BW was administered to Leuven WT animals (see supporting information Table 5, www.pnas.org). RNA levels of genes involved in each step of transcellular calcium absorption were up-regulated: CAT1 6.5-fold (P < 0.001), ECaC 1.6-fold (P < 0.05), calbindin-D9K 2.2-fold (P < 0.01), and PMCA1b 1.8-fold (P < 0.05).
Changes in Gene Expression in Kidney.
Although the role of the kidney in vitamin D-regulated calcium homeostasis is considered less important, similar mechanisms as in intestinal calcium absorption are involved in renal calcium reabsorption (19–21). On a normal diet, renal calbindin-D9K RNA levels were significantly reduced in kidneys of both KOs to less than 10% of WT (Leuven: P < 0.001; Tokyo: P < 0.05) (Fig. 3C). Gene expression of the second renal calcium-binding protein calbindin-D28K was not influenced by the absence of VDR, neither was the expression of PMCA1b. NCX RNA levels were unaffected in the Leuven KO and halved in the Tokyo KO (P < 0.05). ECaC was abundantly expressed in kidney, in contrast to CaT1 (Table 3), suggesting a major role for ECaC in renal calcium reabsorption. Moreover, ECaC and CaT1 expression in kidney of KO mice appeared to be normal, or slightly above normal, as was detected for CaT1 expression in the Tokyo KO on normal diet (P < 0.01) (Fig. 3C and Table 3).
During low calcium intake, calbindin-D9K RNA level in kidneys of Leuven WT doubled (P < 0.001), whereas on high calcium intake, it dropped to 25% (P < 0.001) (Fig. 3C). Its expression in the KO mice was impaired on all diets (P < 0.001). Calbindin-D28K as well as NCX RNA levels were decreased to less than half in both WT and KO on the low calcium diet (P < 0.001). In addition, calbindin-D28K gene expression was also slightly reduced in WT and KO animals on the rescue diet (P < 0.05). ECaC, CaT1, and PMCA1b RNA levels remained stable regardless of dietary calcium, except for an increase of CaT1 RNA level in the KO on a rescue diet (P < 0.001); (Fig. 3C and Table 3). Finally, after 1,25(OH)2D3 treatment, ECaC (2.8-fold, P < 0.001), CaT1 (2.2-fold, P < 0.01), and calbindin-D9K (1.6-fold, P < 0.05) RNA levels were increased (see supporting information Table 5, www.pnas.org). Calbindin-D28K and PMCA1b gene expression were not significantly changed by 1,25(OH)2D3 injection, whereas NCX expression was reduced to one-third (P < 0.05).
Discussion
We analyzed active intestinal calcium absorption at a functional and molecular level in two different VDR-KO strains. The phenotypic rescue of these mice by a high calcium diet (7, 8) identified the intestine as the primary target tissue for vitamin D action. Moreover, the recent reports of two epithelial calcium channels in the small intestine (9, 14) justified the evaluation of the regulation and importance of the known duodenal calcium handling proteins for active calcium transport.
Intestinal calcium absorption capacities of VDR-KO mice appeared to be reduced, as reflected by the 3-fold lower AUC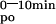 , without enhanced clearance of calcium from the blood compartment during this time frame. The setting of this experiment did not allow us to calculate absolute active calcium absorption. Nevertheless, serum 45Ca accumulation is vitamin D-mediated and regulated by dietary calcium: the area under curve (AUC)
, without enhanced clearance of calcium from the blood compartment during this time frame. The setting of this experiment did not allow us to calculate absolute active calcium absorption. Nevertheless, serum 45Ca accumulation is vitamin D-mediated and regulated by dietary calcium: the area under curve (AUC)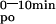 doubled after a single dose of 1,25(OH)2D3 or 1 week of low calcium diet and conversely was halved on a high calcium diet. Moreover, low CaCl2 concentration in the stock solution, early time points, and 1 week of dietary calcium deprivation in the Leuven strain were chosen as conditions in which active duodenal calcium absorption processes should play a dominant role (22–24). The results of the VDR-KO mice agree with an estimated 20–60% reduction of intestinal calcium absorption in HVDDR patients (3). They are also in the same range as the observed difference in duodenal calcium absorption between vitamin D-deficient (±11% within 20 min) and vitamin D-repleted (±74%) chickens, estimated by the ligated duodenal loop technique (24). Calcium absorption in Tokyo mice was lower than in the Leuven strain, which, besides a different calcium intake, may be because of strain differences.
doubled after a single dose of 1,25(OH)2D3 or 1 week of low calcium diet and conversely was halved on a high calcium diet. Moreover, low CaCl2 concentration in the stock solution, early time points, and 1 week of dietary calcium deprivation in the Leuven strain were chosen as conditions in which active duodenal calcium absorption processes should play a dominant role (22–24). The results of the VDR-KO mice agree with an estimated 20–60% reduction of intestinal calcium absorption in HVDDR patients (3). They are also in the same range as the observed difference in duodenal calcium absorption between vitamin D-deficient (±11% within 20 min) and vitamin D-repleted (±74%) chickens, estimated by the ligated duodenal loop technique (24). Calcium absorption in Tokyo mice was lower than in the Leuven strain, which, besides a different calcium intake, may be because of strain differences.
The second part of our study focused on possible molecular mechanisms of impaired calcium absorption in both VDR-KO strains. No defect in PMCA1b expression in either of the KOs could be established, although former studies of 1,25(OH)2D3 repletion in chickens and rats and 1,25(OH)2D3 injection in our WT mice resulted in an increased PMCA1b expression (15, 25–27). Calcium absorption in vivo and calbindin-D9K protein levels correlated fairly well in the Tokyo strain on a normal diet and in the Leuven strain on the low calcium diet: a 3-fold reduction in the KO animals, when compared with their WT littermates. These findings agree with previous reports ascribing the defective calcium homeostasis of the Tokyo (5) and Boston KO (16) mice to low calbindin-D9K RNA levels. They are also supported by experimental evidence that regulation of calbindin expression is rate-limiting for vitamin D-dependent active calcium absorption (15, 19, 28–31), despite a certain lack of correlation between the early rise in calcium transport and the later increase in calbindin content after 1,25(OH)2D3 injection (32, 33). The normal duodenal calbindin-D9K protein content, however, could not explain the HVDDR phenotype of the Leuven KO on a normal diet. By contrast, duodenal CaT1 and, to a lesser degree, ECaC RNA levels were dramatically and consistently down-regulated in the Tokyo and the Leuven KOs. Until recently, the unidentified calcium influx mechanism was not regarded as primordial, as calcium entered into the enterocyte even in the absence of vitamin D (15, 24, 34). Surprisingly, the present duodenal gene expression pattern indicates that, among the candidate calcium-transporting genes, only CaT1 and ECaC are severely impaired in VDR-KO mice. This may imply a regulatory role for the epithelial calcium channels in active calcium absorption or a more complex regulation driven by the interaction between calcium influx and calbindin-D9K. The permanent activation of these channels may indeed require the immediate intracellular uptake of each calcium molecule delivered through the channel by calbindin-D9K, with subsequent mobilization of calcium toward the basolateral membrane (24, 34, 35). In this view, the major down-regulation of CaT1 and ECaC expression combined with a reduced calbindin-D9K expression may contribute to the more profound hypocalcemia and reduced calcium in bone of the Tokyo KO on the normal diet. Moreover, an increased calcium transfer into the enterocyte was seen on ion microscopic images in vitamin D-repleted chickens 2.5 min after calcium administration, although calcium also concentrated, to a lesser extent, in the duodenal brush border region of vitamin D-deficient chickens (24). Additional investigation will be needed to verify (i) whether variations of ECaC and CaT1 are reflected by similar changes in duodenal calcium entry in vitamin D-repleted animals, which was formerly explained by nongenomic 1,25(OH)2D3 effects on microvillar membrane fluidity, and (ii) whether ECaC or CaT1 are directly involved in calcium entry in vitamin D deficiency.
Subsequently, studies with 1,25(OH)2D3 administration and low or high calcium intake were performed in the Leuven strain, in view of a more thorough evaluation of the calcium and vitamin D dependency of these calcium transporting proteins. A single injection of 1,25(OH)2D3 resulted in a significant increase of calbindin-D9K (19, 28), ECaC, and especially of CaT1 expression in WT mice. 1,25(OH)2D3-responsive ECaC expression was previously demonstrated in rat kidney (13), and four putative vitamin D3-responsive elements were described in the promoter region of the human ECaC gene (12). However, in the duodenum of rats, Northern blot did not reveal vitamin D-dependent regulation of CaT1 expression (14), and no significant relationship was shown between human CaT1 homologue (officially approved HGMW-gene symbol: ECaC2) expression in duodenal samples and serum 1,25(OH)2D3 levels of healthy volunteers (36). At present, no conclusive explanation for this differential regulation in different species is available. On the other hand, the alterations in CaT1 and ECaC gene expression in our WT mice on the different diets reflected changes in serum 1,25(OH)2D3 levels and were in concordance with the need for active calcium uptake from the intestine: the low calcium diet induced, and the rescue diet inhibited, their duodenal expression. Calbindin-D9K protein content in WT mice showed a similar but less pronounced pattern (19), although calbindin-D9K RNA level was not significantly altered on the low calcium diet, suggesting posttranscriptional modifications (31). Finally, PMCA1b gene expression was affected in the opposite way: reduction on the low calcium diet, but not on the rescue diet. Obviously, the gene expression pattern we observed in the duodenum of VDR-KO mice on a normal diet is complementary to changes in gene expression induced by 1,25(OH)2D3 injection or dietary calcium alteration in WT mice. In the duodenum of Leuven VDR-KO mice, not only was CaT1 and ECaC expression impaired on any diet, but calbindin-D9K and PMCA1b expression was substantially reduced either in calcium deficiency or abundance as well. The exact VDR independent gene repressor mechanism of this latter observation is elusive: the two remaining calciotropic hormones varied inversely in both dietary conditions, and we found no significant difference in duodenal expression of the calcium sensing receptor (unpublished data) on any diet (37). Interestingly, the inhibition of intestinal calbindin synthesis in vitamin D-replete chickens on a high calcium diet was also attributed to kidney 1α-hydroxylase-independent mechanisms (38).
As renal calcium reabsorption resembles intestinal calcium absorption (19, 21), we evaluated VDR-KO mice for possible defects of known factors involved in this process. Comparable to the stimulatory effect of 1,25(OH)2D3 repletion on renal ECaC RNA and protein expression in rats (13), expression of both channels (although CaT1 was less abundantly present) more than doubled in kidney of our WT mice after a 1,25(OH)2D3 injection. In contrast, expression of neither ECaC nor CaT1 was impaired in the KO mice nor was it influenced by changes in dietary calcium. In fact, only the RNA levels of calbindin-D9K changed considerably and with an almost similar pattern as in duodenum (except for the KO on a normal diet): reduction in the KO on any diet (16) and in the WT on the rescue diet; enhanced expression in WT animals on low calcium diet and after 1,25(OH)2D3 injection (16, 28). Nevertheless, it remains to be clarified whether these changes or the smaller changes in calbindin-D28K (16) and NCX expression have any influence on calcium reabsorption. Renal calcium handling in the VDR-KO and the contribution to its phenotype have not yet been characterized.
Further analysis and quantification of ECaC and CaT1 protein expression are eagerly anticipated to (i) verify whether reduced RNA levels result in decreased protein levels in duodenum of VDR-KO mice and (ii) substantiate the notion of a tissue-specific distribution of these channels, bearing on opposite ratio of RNA expression levels in duodenum (CaT1>ECaC) versus kidney (ECaC>CaT1) in our analysis and in former reports (14, 36).
In conclusion, we provide arguments for an impaired calcium absorption in VDR-KO mice. The two recently described intestinal calcium channels ECaC and CaT1 are expressed in duodenum, but CaT1 RNA levels are severalfold higher than ECaC RNA levels. The expression of both channels is strongly VDR-dependent and consistently down-regulated in two VDR-KO strains. This expression fluctuates in WT animals according to dietary calcium driven variations in 1,25(OH)2D3. Consequently, among the presently known active calcium absorption mechanisms, calcium influx, probably interacting with intracellular calcium transfer mechanisms, should be considered as a rate-limiting step in the process of vitamin D-dependent duodenal calcium absorption.
Supplementary Material
Acknowledgments
S.J.V.C. is a Research Assistant of the Fonds voor Wetenschappelyk Onderzoek–Vlaanderen (FWO) Table 1. J. G. J Hoenderop was supported by a long-term European Molecular Biology fellowship (ALTF 160-2000). This work was supported by a grant from FWO, Belgium (Grant G.0225.00).
Abbreviations
- 1,25(OH)2D3
1,25(OH)2 vitamin D
- VDR
vitamin D receptor
- KO
knockout
- WT
wild type
- CaT1
calcium transport protein type 1
- ECaC
epithelial calcium channel
- PMCA1b
plasma membrane calcium ATPase
- AUC
area under the curve
- NCX
Na+/Ca2+ exchanger
- HPRT
hypoxanthine–guanine phosphoribosyl transferase
- HVDDR
hypocalcemic vitamin D-resistant rickets
- BW
body weight
- p.o.
orally
- PTH
parathyroid hormone
- RT-PCR
reverse transcription–PCR
- qRT-PCR
quantitative RT-PCR
References
- 1.Reichel H, Koeffler H P, Norman A W. N Engl J Med. 1989;320:980–991. doi: 10.1056/NEJM198904133201506. [DOI] [PubMed] [Google Scholar]
- 2.Haussler M R, Whitfield G K, Haussler C A, Hsieh J C, Thompson P D, Selznick S H, Dominguez C E, Jurutka P W. J Bone Miner Res. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 3.Balsan S, Garabedian M, Larchet M, Gorski A M, Cournot G, Tau C, Bourdeau A, Silve C, Ricour C. J Clin Invest. 1986;77:1661–1667. doi: 10.1172/JCI112483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hochberg Z, Tiosano D, Even L. J Pediatr. 1992;121:803–808. doi: 10.1016/s0022-3476(05)81919-5. [DOI] [PubMed] [Google Scholar]
- 5.Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, Kawakami T, Arioka K, Sato H, Uchiyama Y, et al. Nat Genet. 1997;16:391–396. doi: 10.1038/ng0897-391. [DOI] [PubMed] [Google Scholar]
- 6.Li Y C, Pirro A E, Amling M, Delling G, Baron R, Bronson R, Demay M B. Proc Natl Acad Sci USA. 1997;94:9831–9835. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y C, Amling M, Pirro A E, Priemel M, Meuse J, Baron R, Delling G, Demay M B. Endocrinology. 1998;139:4391–4396. doi: 10.1210/endo.139.10.6262. [DOI] [PubMed] [Google Scholar]
- 8.Amling M, Priemel M, Holzmann T, Chapin K, Rueger J M, Baron R, Demay M B. Endocrinology. 1999;140:4982–4987. doi: 10.1210/endo.140.11.7110. [DOI] [PubMed] [Google Scholar]
- 9.Hoenderop J G, van der Kemp A W, Hartog A, van de Graaf S F, van Os C H, Willems P H, Bindels R J. J Biol Chem. 1999;274:8375–8378. doi: 10.1074/jbc.274.13.8375. [DOI] [PubMed] [Google Scholar]
- 10.Hoenderop J G, Hartog A, Stuiver M, Doucet A, Willems P H, Bindels R J. J Am Soc Nephrol. 2000;11:1171–1178. doi: 10.1681/ASN.V1171171. [DOI] [PubMed] [Google Scholar]
- 11.Muller D, Hoenderop J G, Meij I C, van den Heuvel L P, Knoers N V, den Hollander A I, Eggert P, Garcia-Nieto V, Claverie-Martin F, Bindels R J. Genomics. 2000;67:48–53. doi: 10.1006/geno.2000.6203. [DOI] [PubMed] [Google Scholar]
- 12.Muller D, Hoenderop J G, Merkx G F, van Os C H, Bindels R J. Biochem Biophys Res Commun. 2000;275:47–52. doi: 10.1006/bbrc.2000.3227. [DOI] [PubMed] [Google Scholar]
- 13.Hoenderop J G, Muller D, Kemp A W, Hartog A, Suzuki M, Ishibashi K, Imai M, Sweep F, Willems P H, Os C H, et al. J Am Soc Nephrol. 2001;12:1342–1349. doi: 10.1681/ASN.V1271342. [DOI] [PubMed] [Google Scholar]
- 14.Peng J B, Chen X Z, Berger U V, Vassilev P M, Tsukaguchi H, Brown E M, Hediger M A. J Biol Chem. 1999;274:22739–22746. doi: 10.1074/jbc.274.32.22739. [DOI] [PubMed] [Google Scholar]
- 15.Wasserman R H. In: Vitamin D. Feldman D, Glorieux F H, Pike J W, editors. New York: Academic; 1997. pp. 259–273. [Google Scholar]
- 16.Li Y C, Pirro A E, Demay M B. Endocrinology. 1998;139:847–851. doi: 10.1210/endo.139.3.5803. [DOI] [PubMed] [Google Scholar]
- 17.Lallemand Y, Luria V, Haffner-Krausz R, Lonai P. Transgenic Res. 1998;7:105–112. doi: 10.1023/a:1008868325009. [DOI] [PubMed] [Google Scholar]
- 18.Thomasset M, Parkes C O, Cuisinier-Gleizes P. Am J Physiol. 1982;243:E483–E488. doi: 10.1152/ajpendo.1982.243.6.E483. [DOI] [PubMed] [Google Scholar]
- 19.Matkovits T, Christakos S. Endocrinology. 1995;136:3971–3982. doi: 10.1210/endo.136.9.7649106. [DOI] [PubMed] [Google Scholar]
- 20.Friedman P A, Gesek F A. Physiol Rev. 1995;75:429–471. doi: 10.1152/physrev.1995.75.3.429. [DOI] [PubMed] [Google Scholar]
- 21.Sooy K, Kohut J, Christakos S. Curr Opin Nephrol Hypertens. 2000;9:341–347. doi: 10.1097/00041552-200007000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Bronner F. J Nutr. 1998;128:917–920. doi: 10.1093/jn/128.5.917. [DOI] [PubMed] [Google Scholar]
- 23.Wood R J, Fleet J C, Cashman K, Bruns M E, Deluca H F. Endocrinology. 1998;139:3843–3848. doi: 10.1210/endo.139.9.6176. [DOI] [PubMed] [Google Scholar]
- 24.Chandra S, Fullmer C S, Smith C A, Wasserman R H, Morrison G H. Proc Natl Acad Sci USA. 1990;87:5715–5719. doi: 10.1073/pnas.87.15.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai Q, Chandler J S, Wasserman R H, Kumar R, Penniston J T. Proc Natl Acad Sci USA. 1993;90:1345–1349. doi: 10.1073/pnas.90.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pannabecker T L, Chandler J S, Wasserman R H. Biochem Biophys Res Commun. 1995;213:499–505. doi: 10.1006/bbrc.1995.2159. [DOI] [PubMed] [Google Scholar]
- 27.Zelinski J M, Sykes D E, Weiser M M. Biochem Biophys Res Commun. 1991;179:749–755. doi: 10.1016/0006-291x(91)91880-l. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Christakos S. Endocrinology. 1991;128:2844–2852. doi: 10.1210/endo-128-6-2844. [DOI] [PubMed] [Google Scholar]
- 29.Christakos S, Gabrielides C, Rhoten W B. Endocr Rev. 1989;10:3–26. doi: 10.1210/edrv-10-1-3. [DOI] [PubMed] [Google Scholar]
- 30.Wasserman R H, Fullmer C S. Adv Exp Med Biol. 1989;249:45–65. doi: 10.1007/978-1-4684-9111-1_5. [DOI] [PubMed] [Google Scholar]
- 31.Dupret J M, Brun P, Perret C, Lomri N, Thomasset M, Cuisinier-Gleizes P. J Biol Chem. 1987;262:16553–16557. [PubMed] [Google Scholar]
- 32.Thomasset M. In: Vitamin D. Feldman D, Glorieux F H, Pike J W, editors. New York: Academic; 1997. pp. 223–232. [Google Scholar]
- 33.Bishop C W, Kendrick N C, Deluca H F. J Biol Chem. 1983;258:1305–1310. [PubMed] [Google Scholar]
- 34.Fullmer C S, Chandra S, Smith C A, Morrison G H, Wasserman R H. Histochem Cell Biol. 1996;106:215–222. doi: 10.1007/BF02484403. [DOI] [PubMed] [Google Scholar]
- 35.Vennekens R, Hoenderop J G, Prenen J, Stuiver M, Willems P H, Droogmans G, Nilius B, Bindels R J. J Biol Chem. 2000;275:3963–3969. doi: 10.1074/jbc.275.6.3963. [DOI] [PubMed] [Google Scholar]
- 36.Barley N F, Howard A, O'Callaghan D, Legon S, Walters J R. Am J Physiol Gastrointest Liver Physiol. 2001;280:G285–G290. doi: 10.1152/ajpgi.2001.280.2.G285. [DOI] [PubMed] [Google Scholar]
- 37.Chattopadhyay N, Cheng I, Rogers K, Riccardi D, Hall A, Diaz R, Hebert S C, Soybel D I, Brown E M. Am J Physiol. 1998;274:G122–G130. doi: 10.1152/ajpgi.1998.274.1.G122. [DOI] [PubMed] [Google Scholar]
- 38.Bar A, Shani M, Fullmer C S, Brindak M E, Striem S. Mol Cell Endocrinol. 1990;72:23–31. doi: 10.1016/0303-7207(90)90236-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.