

Abstract
Background and Purpose
One of the hallmarks of ventral midbrain dopamine‐releasing neurons is membrane hyperpolarization in response to stimulation of somato‐dendritic D2 receptors. At early postnatal age, under sustained dopamine, this inhibitory response is followed by a slow recovery, resulting in dopamine inhibition reversal (DIR). In the present investigation, we aimed to get a better insight into the cellular mechanisms underlying DIR.
Experimental Approach
We performed single‐unit extracellular recordings with a multi‐electrode array device and conventional patch‐clamp recordings on midbrain mouse slices.
Key Results
While continuous dopamine (100 μM) perfusion gave rise to firing inhibition that recovered in 10 to 15 min, the same effect was not obtained with the D2 receptor agonist quinpirole (100 nM). Moreover, firing inhibition caused by the GABAB receptor agonist baclofen (300 nM) was reversed by dopamine (100 μM), albeit D2 receptors had been blocked by sulpiride (10 μM). Conversely, the block of the dopamine transporter (DAT) with cocaine (30 μM) prevented firing recovery by dopamine under GABAB receptor stimulation. Accordingly, in whole‐cell recordings from single cells, the baclofen‐induced outward current was counteracted by dopamine (100 μM) in the presence of sulpiride (10 μM), and this effect was prevented by the DAT antagonists cocaine (30 μM) and GBR12909 (2 μM).
Conclusions and Implications
Our results indicate that the DAT plays a major role in DIR, mediating it under conditions of sustained dopamine exposure, and point to DAT as an important target for pharmacological therapies leading to prolonged enhancement of the dopaminergic signal.
Abbreviations
- ACSF
artificial CSF
- CNQX
6‐cyano‐7‐nitroquinoxaline‐2,3‐dione
- DAT
dopamine transporter
- DIR
dopamine inhibition reversal
- DOPAC
3,4‐dihydroxyphenylacetic acid
- DOPAL
3,4‐dihydroxyphenylacetaldehyde
- GIRK (Kir3.1)
G‐protein‐coupled inwardly rectifying potassium conductance
- HVA
homovanillic acid
- l‐DOPA
l‐3,4‐dihydroxyphelylalanine
- MEA
multi‐electrode array
- PD
Parkinson's disease
- SNc
substantia nigra pars compacta
- VTA
ventral tegmental area
Introduction
The main source of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=940 in the CNS arises from a densely packed population of dopamine‐releasing (dopaminergic) neurons located in the ventral midbrain, arranged into two main subregions, the substantia nigra pars compacta (SNc) and the ventral tegmental area (VTA), projecting to striatal, as well as limbic and cortical regions (Van den Heuvel and Pasterkamp, 2008). In recent years, data have been put forward illustrating specific properties that differentiate these two subpopulations of dopaminergic neurons. However, there is a common consensus that, although to a variable extent, virtually all dopaminergic neurons of the ventral midbrain are inhibited by dopamine in response to stimulation of somato‐dendritic http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=215&familyId=20&familyType=GPCR, also called D2 autoreceptors, by interfering with http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=434) (Lacey et al., 1987; Lacey et al., 1988; Kim et al., 1995; Beckstead et al., 2004; Luscher and Slesinger, 2010; Krashia et al., 2017). Thus, through its somato‐dendritic release, dopamine acts on D2 receptors located on the dopaminergic neurons acting as a negative feedback system to modulate dopaminergic neurons' excitability (Beckstead et al., 2004; Ford, 2014; Evans et al., 2017). On this basis, a thorough understanding of the functional effects exerted by local extracellular dopamine is of considerable importance for the physiology of the dopaminergic system, as it affects the overall dopamine signal to specific target regions. Moreover, it provides more insights into the functional outcomes of a sustained increase in dopamine levels, like that achieved with drugs of abuse, or for the pharmacological treatment of pathologies linked to dysfunctions of the dopaminergic system, namely Parkinson's disease (PD) and schizophrenia. Indeed, the gold standard pharmacological therapy for the treatment of PD patients still relies on l‐3,4‐dihydroxyphelylalanine (l‐DOPA), in order to increase the dopamine content, thus inducing a steady enhancement of extracellular dopamine (Lipski et al., 2011).
With regard to the effects of high levels of extracellular dopamine, it has been shown that, while a transient exposure to dopamine results in the expected D2 receptor‐mediated hyperpolarization of midbrain dopaminergic neurons, this inhibitory action is not maintained when dopamine exposure is continued, giving rise to what has been defined as dopamine inhibition reversal (DIR) (Nimitvilai and Brodie, 2010). Therefore, the result of changes in extracellular dopamine levels cannot be predicted in a simplistic manner, according to the known acute D2 receptor‐mediated inhibitory effect. Further investigation on DIR has proposed a desensitizing process on D2 receptors, occurring in SNc dopaminergic neurons at an early postnatal age, and is progressively lost with the development of a calcium‐dependent interaction between D2 receptors and a neuronal calcium sensor (Dragicevic et al., 2014).
Another candidate that needs be taken into account when trying to understand how dopaminergic neurons respond to increased levels of extracellular dopamine is the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=927); its ability to remove dopamine is particularly increased in conditions of high extracellular dopamine (Mortensen and Amara, 2003; Benoit‐Marand et al., 2011; Vaughan and Foster, 2013; Ford, 2014).
In the present work, we investigated DIR by looking at the response of SNc dopaminergic neurons to sustained high levels of dopamine, in midbrain slices of mice at an early postnatal age, using both conventional patch‐clamp and multi‐electrode array (MEA) recordings, the latter in order to detect the single‐unit activity of a large population of neurons in a less invasive experimental condition (Berretta et al., 2010). By focusing our attention on D2 receptor desensitization processes and on DAT, we found that, although D2 receptor desensitization is present, it does not seem to play a significant role in the expression of DIR, while it is primarily DAT that promotes recovery from dopamine‐induced firing inhibition through the depolarizing current linked to its activity.
Methods
Slice preparation and ethical statement
Experiments were performed according to the national and international laws for laboratory animal welfare and experimentation (Leg. Decree no. 26, implementation of the UE directive 2010/63/UE, 4 March 2014) and were approved by the Italian Ministry of Health (authorization no. 91/2014 PR). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). C57BL6 mice (postnatal day 12–15) of either sex were housed with their mothers at a controlled temperature (24–25°C), with food and water available ad libitum and a 12 h light/dark cycle. Animals were randomly assigned to different treatments. For slice preparation, the mice were anaesthetized with halothane and killed by decapitation. The brain was removed from the skull, and horizontal midbrain slices (250–300 μm) were cut in cold (8–12°C) artificial CSF (ACSF) and left to recover at 34°C for at least 1 h. ACSF composition (in mM): NaCl 126, KCl 2.5, MgCl2 1.3, CaCl2 2.4, NaH2PO4 1.2, NaHCO3 24 and glucose 10, and saturated with 95% O2 and 5% CO2 (pH 7.4). The recording of data was not blinded to the operator, because each electrophysiological session required an awareness by the experimenter of the running protocol. Data analysis, although not blinded, was double checked by more than one researcher.
Multi‐electrode array recordings
A single horizontal slice of the ventral midbrain (300 μm) was placed over an 8 × 8 array of planar microelectrodes, each 20 × 20 μm in size, with an interpolar distance of 100 μm (MED‐P2105; Alpha MED Sciences, Kadoma, Japan). The slice was positioned under visual control through an upright microscope (Leica DM‐LFS; Leica Microsystems, Wetzlar, Germany), so that most of the planar electrodes were covered by the SNc area (Berretta et al., 2010). The slices were kept submerged in ACSF with a nylon mesh glued to a platinum ring, under a continuous flow of warm (34°C) oxygenated ACSF (5.5–6.0 mL·min−1).
Voltage signals were acquired using the MED64 System operated by Mobius software (Alpha MED Sciences), digitized at 20 kHz and filtered (0.1–10 kHz) with a 6071E Data Acquisition Card (National Instruments, Austin, TX, USA). The fast transients corresponding to spontaneous action potentials were captured offline using Spike2 6.0 software (Cambridge Electronic Design Ltd, Cambridge, UK), by means of an amplitude threshold adjusted by visual inspection for each individual active channel. When spontaneous action potentials arising from more than one neuron were detected from a single recording channel, spike sorting discrimination was achieved by generating spike templates with Spike2 6.0 (Cambridge Electronic Design Ltd), sorted with a Normal Mixtures algorithm on independent clusters obtained from principal component data.
Patch‐clamp recordings
Whole‐cell patch‐clamp recordings in horizontal slices (250 μm) of the ventral midbrain were obtained from neurons visualized under an upright microscope (BX51WI; Olympus, Segrate, Italy) using infrared differential interference contrast and recorded with glass electrodes (3–5 MΩ) filled with (in mM): 145 K‐gluconate, 0.1 CaCl2, 2 MgCl2, 10 HEPES, 0.75 EGTA, 4 ATP‐Mg2 and 0.3 GTP‐Na3; 10 phosphocreatine‐Na2 (pH 7.3 with KOH; 280 mOsm).
Signals were acquired with pClamp 10 software, using a Multiclamp 700A amplifier (Molecular Devices, San Jose, California, USA), digitized at 10 kHz and filtered at 3 kHz. Nigral dopaminergic neurons were identified as tightly packed cells located close to the medial terminal nucleus of the accessory optic tract. Moreover, at the beginning of each patch‐clamp experimental session, the neurons were tested in current‐clamp mode, in order to verify the presence of normal spontaneous firing (<5 Hz) and a depolarizing ‘sag’ in response to hyperpolarizing current steps, paralleled in voltage‐clamp mode by the presence of a prominent I h (hyperpolarization‐activated cation current) in response to hyperpolarizing voltage commands (Krashia et al., 2017).
I/V relationships were obtained in slices perfused in ACSF with added CsCl2 (1 mM), in order to block the I h current and using voltage ramp waveforms (from −120 to −60 mV, 80 mV·s−1) preceded by a 700 ms period at −120 mV.
Data analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). In all experiments, summary data are expressed as mean ± SEM. The estimate of sample size for each experimental paradigm was performed using software PS Power and Sample Size Calculation (version 3.1.2) and calculated in order to achieve a power of 0.9, keeping a minimum of five slices per experimental session.
For the experiments on single units with MEA, the relative change in the firing rate was calculated in each neuron over periods of 5 min at various time points from drug perfusion, with respect to a 3 min control period before drug perfusion. These values, taken from all neurons of the same slice, were used to obtain a median value relative to each time point that was then averaged across the corresponding median values of all other slices exposed to the same treatment. Such analytical protocol, instead of using a general mean value from all neurons recorded in all slices, allowed us to escape the bias of results obtained from slices particularly rich in active neurons; moreover, the use of the median, rather than the mean value, allowed us to circumvent the bias due to the largely skewed distribution of the relative change of firing inhibition, due to those neurons whose firing was completely stopped by the treatment. Thus, for each experimental set, we refer to the sample size (n) as the number of slices used. The data were then normalized and expressed as % of baseline, in order to reduce variability of the data, due to differences in absolute firing rate, which may display a high degree of variability (Berretta et al., 2010).
In patch‐clamp experiments, the degree of reversal in the outward current induced by dopamine or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1084 was calculated as ratio (in %) between the Δ current at a specific time point relative to the peak current and the Δ current at the peak relative to baseline, with 100 and 0%, thereby indicating complete or no reversal respectively.
The data were compared with one‐way ANOVA followed by post hoc comparisons assessed with Bonferroni's test when the F statistic was significant. Alternatively, when only two groups of data were compared, Student's two‐tailed t‐test was used. P < 0.05 was considered as minimum level for statistical significance under both statistical comparisons.
Drugs
All drugs were dissolved in ACSF and applied in the bath at the desired final concentration through a three‐way tap. Drugs used were dopamine, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7262, homovanillic acid (HVA), 3,4‐dihydroxyphenylacetic acid (DOPAC), 3,4‐dihydroxyphenylacetaldehyde (DOPAL), benomyl, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=502, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=564, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2286 (from Sigma‐Aldrich, Milan, Italy), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5501, baclofen, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=938), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5475; 1‐(2‐[bis(4‐fluorophenyl)methoxy]ethyl)‐4‐(3‐phenylpropyl)piperazine dihydrochloride (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9983) (from Tocris Bioscience, Bristol, UK). In the experiments with pargyline, the slices were incubated at the desired concentration for at least 1 h before being placed into the recording chamber in the continuous presence of the same drug.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Results
Firing inhibition under prolonged dopamine exposure in SNc neurons
The spontaneous firing of SNc neurons in slices was detected with an MEA device as extracellular single‐unit spikes, as previously reported (Berretta et al., 2010). The slice was exposed to 100 μM dopamine, and a total of 58 active neurons (six slices, from three animals) responded with inhibition of their firing rate (from 1.93 ± 0.20 to 0.24 ± 0.10 Hz) that reached a maximal effect within 30 s to 1 min, as expected from typical dopaminergic neurons, due to activation of their D2 autoreceptors (Lacey et al., 1987; Kim et al., 1995; Krashia et al., 2017). We thus focused our investigation on this presumed dopaminergic neuronal population. With prolonged exposure to 100 μM dopamine for 15 min, the same neurons did not remain inhibited, but partially, or even totally, recovered their firing in the presence of dopamine, giving rise to DIR. Overall, the firing rate of the dopaminergic neurons was initially inhibited to 4.70 ± 1.59% of control by 100 μM dopamine, while it recovered during continuous dopamine perfusion to a level of firing inhibition (48.18 ± 16.65% of control after 15 min) that was significantly different from the initial maximal effect (P < 0.05; F (2, 15) = 5.50). A total recovery was then observed at dopamine washout 98.44 ± 9.19% (Figure 1).
Figure 1.
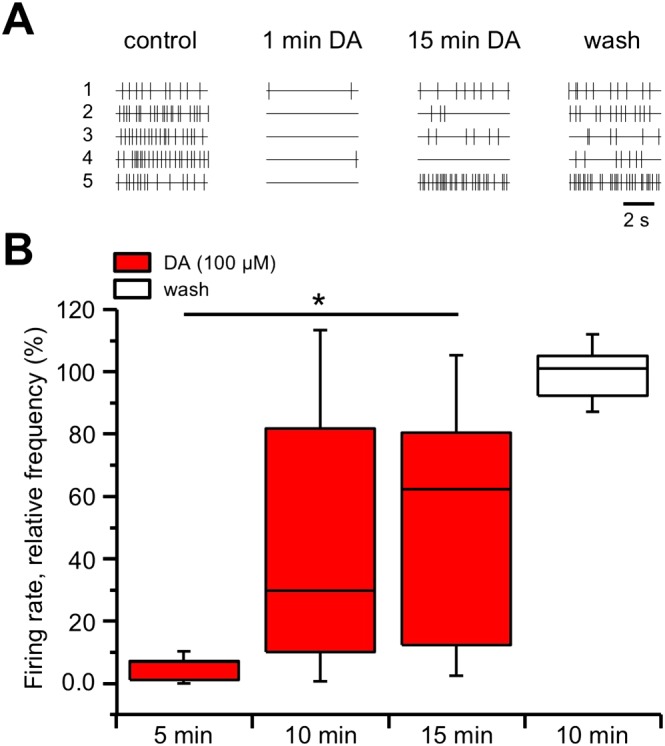
DIR in SNc neurons recorded with MEA. (A) Raster plots of single units of five sample neurons recorded from the same slice. Upon dopamine (DA, 100 μM) perfusion, a rapid inhibition of action potential firing was observed in all neurons (1 min dopamine) that was followed by a partial or full recovery of the firing during the course of the 15 min dopamine challenge (neurons 1–3 and 5). Not all neurons restored their firing in the presence of dopamine (neuron 4), although a full recovery was attained at dopamine washout. (B) Box‐and‐whisker plots of the median values of the firing rate of all the neurons recorded in each slice with MEA, normalized (in %) to control firing rate and measured at the times indicated at the bottom. The centre lines denote medians, edges are upper and lower quartiles and whiskers show minimum and maximum values. The relative firing rate was strongly inhibited at 5 min dopamine perfusion but slowly recovered at 10 and 15 min, such that the relative firing rates at 5 and 15 min were significantly different (* P < 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test, n = 6 slices). A full recovery was obtained at 10 min dopamine washout.
DIR requires the presence of dopamine but not through its continuous stimulation of D2 receptors
We thus examined whether an analogous reversal of firing inhibition could be obtained in the dopaminergic neurons by D2 receptor stimulation with the selective agonist quinpirole, rather than dopamine. Using the MEA device we recorded the firing rate of 71 neurons (in five slices, from two animals) whose firing was inhibited by 100 nM quinpirole. Differently from the previous results with dopamine, the firing of the dopaminergic neurons attained a maximal inhibition to 1.75 ± 0.70% of control that was maintained throughout the 15 min period of continuous quinpirole perfusion. Interestingly though, if 100 μM dopamine was subsequently added, still in the continuous presence of 100 nM quinpirole, the firing gradually recovered, reaching 62.26 ± 11.01% of control. The level of firing inhibition after 15 min in 100 μM dopamine and 100 nM quinpirole was significantly lower than that in quinpirole alone (P < 0.05; F (2, 12) = 36.25; Figure 2A).
Figure 2.
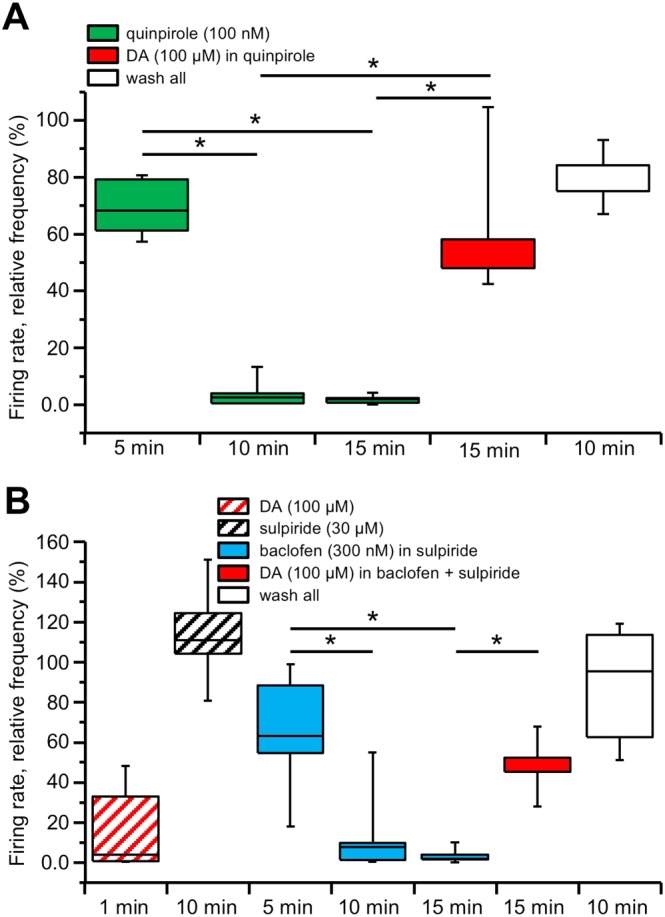
DIR relies on the presence of dopamine (DA) but not through D2 receptor stimulation. Box‐and‐whisker plots in both panels represent the median values of the firing rate of all the neurons recorded in each slice with MEA, normalized (in %) to control firing rate and measured at the times indicated at the bottom. The centre lines denote medians, edges are upper and lower quartiles and whiskers show minimum and maximum values. (A) Perfusion with the D2 receptor agonist quinpirole (100 nM) induced a pronounced inhibition of the firing rate that reached a plateau within 10 min and remained inhibited at 15 min. Conversely, the firing rate was largely restored in response to a 15 min perfusion with dopamine (100 μM), in the continuous presence of 100 nM quinpirole, such that the relative firing rates at 15 min in quinpirole and at 15 min in quinpirole and dopamine were significantly different (* P < 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test, n = 5 slices). Further recovery was attained 10 min after washout of all applied drugs. (B) An initial challenge with a brief 1–2 min perfusion with dopamine (100 μM) inhibited the firing rate of the recorded neurons, confirming their dopaminergic identity. Following this brief dopamine challenge, the slice was perfused with the D2 receptor antagonist sulpiride (30 μM) and kept in the medium thereafter. Perfusion with baclofen (300 nM) induced a pronounced inhibition of the firing rate, reaching a plateau at 15 min. The firing rate was largely restored in response to a 15 min perfusion with dopamine (100 μM), in the continuous presence of 300 nM baclofen, such that the relative firing rates at 15 min in baclofen and at 15 min in baclofen and dopamine were significantly different (* P < 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test, n = 5 slices). Further recovery was attained 10 min after washout of all applied drugs.
This result strongly suggests that a prolonged perfusion with dopamine reverses its autoinhibition by a mechanism other than just D2 receptor stimulation and that dopamine as such is required for DIR expression. This hypothesis was confirmed in experiments in which we completely bypassed D2 receptor stimulation, taking advantage of the converging ionic mechanism underlying D2 receptor and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=26 activation in dopaminergic neurons, through opening of a Kir3.1 channel (GIRK)‐mediated conductance (Lacey et al., 1988; Beckstead et al., 2004; Cruz et al., 2004).
Using the MEA device we recorded the firing rate of 38 neurons (in five slices, from three animals), previously identified as dopaminergic by the transient inhibition of their firing rate in response to a brief (1–2 min) challenge with 100 μM dopamine. The slices were then treated with the specific D2 receptor antagonist sulpiride (10 μM) and kept in the medium thereafter. Following perfusion with the GABAB receptor agonist baclofen (300 nM), dopaminergic neurons firing remained inhibited, attaining 3.52 ± 1.76% of control after 15 min of continuous baclofen perfusion. However, when we added 100 μM dopamine, in the continuous presence of baclofen and sulpiride, a gradual recovery of the firing was observed, reaching 49.00 ± 6.44% of control. The level of firing inhibition after 15 min in 100 μM dopamine in baclofen was significantly lower than that in baclofen alone (P < 0.05; F (3, 16) = 10.94; Figure 2B).
These results demonstrate that dopamine is necessary for DIR but through a mechanism that does not rely on continuous D2 receptor stimulation.
Dopamine does not act on excitatory dopamine‐sensitive receptors to induce DIR
Dopamine may cause a delayed increase in the firing rate through stimulation of excitatory receptors, which parallels and eventually overcomes D2 receptor‐mediated firing inhibition. We ruled out this possibility with a series of control experiments on theoretical candidates of excitatory side effects of dopamine exposure. In particular, we focused our attention on a possible cross‐stimulation on adrenoceptors (Cornil et al., 2002; Swaminath et al., 2004; Zhang et al., 2004; Cucchiaroni et al., 2011) or α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) glutamate receptors (Guatteo et al., 2013).
We found that DIR was still present following perfusion of 100 μM dopamine in phentolamine (30 μM) and propanolol (30 μM), α‐ and β‐adrenoceptor antagonists respectively (P < 0.05; F (2, 12) = 9.74, n = 5 slices, from three animals, with total 62 neurons; Figure 3A). Similar results were obtained with the AMPA glutamate receptor antagonist CNQX (10 μM). Thus, a significant recovery from firing inhibition was still observed (P < 0.05; F (2, 12) = 15.34, n = 5 slices, from two animals, with total 39 neurons; Figure 3B).
Figure 3.
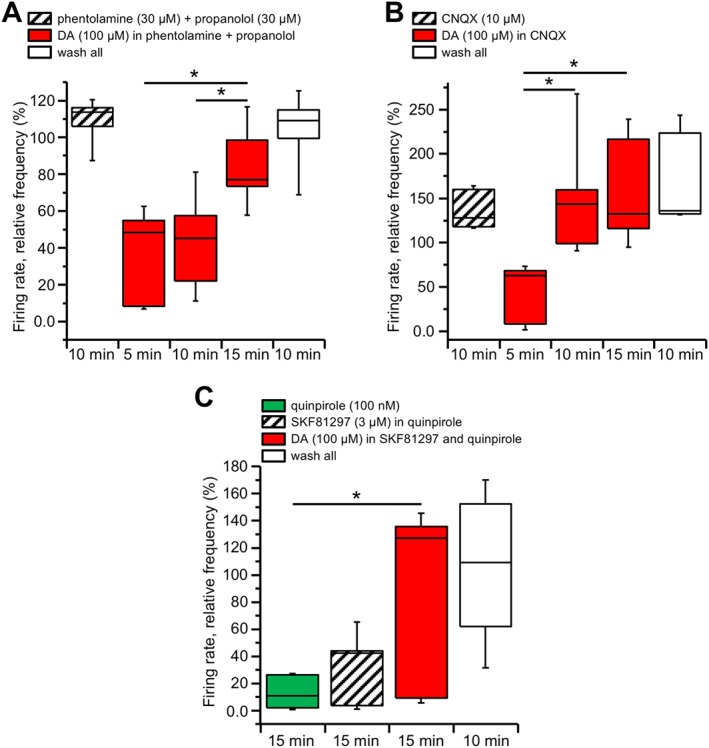
DIR is not secondary to adrenoceptor, AMPA or D1 receptor stimulation. Box‐and‐whisker plots in all panels represent the median values of the firing rate of all the neurons recorded in each slice with MEA, normalized (in %) to control firing rate and measured at the times indicated at the bottom. The centre lines denote medians, edges are upper and lower quartiles and whiskers show minimum and maximum values. (A) The slices were perfused with the α‐ and β‐adrenoceptor antagonists phentolamine (30 μM) and propranolol (30 μM) and kept in the medium thereafter. Perfusion with dopamine (100 μM) still induced an initial firing rate inhibition that slowly recovered at 10 and 15 min (* P < 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test, n = 5 slices), with full recovery at washout. (B) Similar experiments were performed under the continuous presence of the AMPA receptor antagonist CNQX (30 μM). Again, dopamine (100 μM) induced an initial firing rate inhibition that slowly recovered after 10 to 15 min (* P < 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test, n = 5 slices), with full recovery at washout. (C) Perfusion with the D2 receptor agonist quinpirole (100 nM) induced a pronounced and long‐lasting firing rate inhibition. The firing rate was not restored in response to the D1 receptor agonist SKF81297 (3 μM), while a significant recovery was achieved when dopamine (100 μM) was added in the medium (* P < 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test, n = 6 slices). Further recovery was attained 10 min after washout of all applied drugs.
Finally, we also considered the possible involvement of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=214. The D1 receptor agonist SKF81297 (3 μM), differently from dopamine, did not produce any recovery from firing inhibition by 100 nM quinpirole (11.76 ± 4.97% of control after 15 min quinpirole and 26.84 ± 11.02% after additional 15 min in quinpirole and SKF81297). However, when 100 μM dopamine was added in the same slices, in the continuous presence of quinpirole and SKF81297, a significant recovery from firing inhibition was obtained (73.81 ± 28.00% of control; P < 0.05; F (2, 15) = 6.95, n = 6 slices, from two animals, with total of 58 neurons; Figure 3C).
DIR is prevented by DAT inhibition
With the aim of understanding why dopamine is required for DIR induction, albeit independently from its interaction with D2 receptors, we considered a possible role played by the DAT, activated by extracellular dopamine and responsible for its reuptake by the dopaminergic neurons.
The pharmacological block of DAT induces an increase in endogenous dopamine in the extracellular space, thereby causing firing inhibition (Mercuri et al., 1992) and preventing further estimate of the effects of exogenous dopamine. For this reason, we evaluated the effect of prolonged dopamine exposure using the previously shown protocol of recovery from GABAB‐mediated firing inhibition under D2 receptor block. Using the MEA device we recorded the firing rate of 82 neurons (in five slices, from three animals), previously identified as dopaminergic by the transient inhibition of their firing rate in response to a brief (1–2 min) challenge with 100 μM dopamine. The slices were then treated with the specific D2 receptor antagonist sulpiride (10 μM) and the DAT inhibitor cocaine (30 μM), both kept in the medium thereafter. Following perfusion with baclofen (300 nM), the dopaminergic neurons firing was inhibited to 22.79 ± 16.29% of control (P < 0.05; F (2, 12) = 44.90). However, in these experimental conditions, when 100 μM dopamine was added, in the continuous presence of baclofen, cocaine and sulpiride, the firing remained inhibited, and no significant recovery of firing was observed (25.15 ± 11.61% after 15 min in dopamine; Figure 4). These results demonstrate that DAT activation is necessary for DIR expression.
Figure 4.
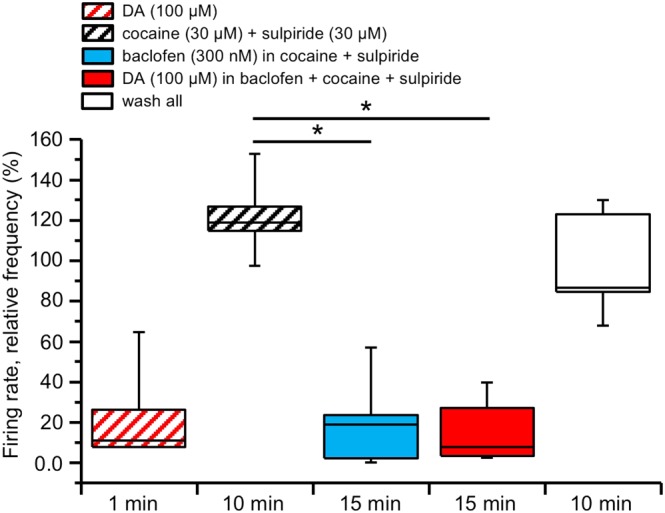
DIR is prevented under DAT inhibition. Box‐and‐whisker plots in both panels represent the median values of the firing rate of all the neurons recorded in each slice with MEA, normalized (in %) to control firing rate and measured at the times indicated at the bottom. The centre lines denote medians, edges are upper and lower quartiles and whiskers show minimum and maximum values. An initial challenge with a brief 1–2 min perfusion with dopamine (DA, 100 μM) inhibited the firing rate of the recorded neurons, confirming their dopaminergic identity. Following this brief dopamine challenge, the slice was perfused with the D2 receptor antagonist sulpiride (30 μM) and the DAT antagonist cocaine (30 μM) and kept in the medium thereafter. Perfusion with baclofen (300 nM) induced a pronounced inhibition of the firing rate, reaching a plateau at 15 min (P < 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test, n = 5 slices). In these conditions, dopamine (100 μM) did not restor the firing rate, while a full recovery was achieved at washout.
DIR requires MAO activity but not their main enzymatic products
Since DAT activation is required for DIR, we next addressed the possibility that DIR may be caused by dopamine down‐products, due to the intracellular enzymatic degradation that follows its reuptake. To this aim, we first investigated whether DIR could be prevented by inhibition of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=766, the main enzymatic pathway in the mesencephalic dopaminergic system, responsible of dopamine inactivation through its oxidation into a number of downstream metabolites (Kopin, 1985; Shih et al., 1999; Eisenhofer et al., 2004).
Using the MEA device we recorded the firing rate of 117 neurons from seven slices (three animals), previously incubated for at least 1 h in the presence of the MAO inhibitor pargyline (10 μM) and kept in the continuous presence of the same drug during recording. In these conditions, perfusion of 100 μM dopamine inhibited the spontaneous firing rate, and no significant recovery was observed during the 15 min of dopamine perfusion (P > 0.05, F (2, 18) = 2.21). Interestingly, recovery of the firing at drug washout was very much delayed and could only be accelerated by a bolus of 10 μM sulpiride (Figure 5A).
Figure 5.
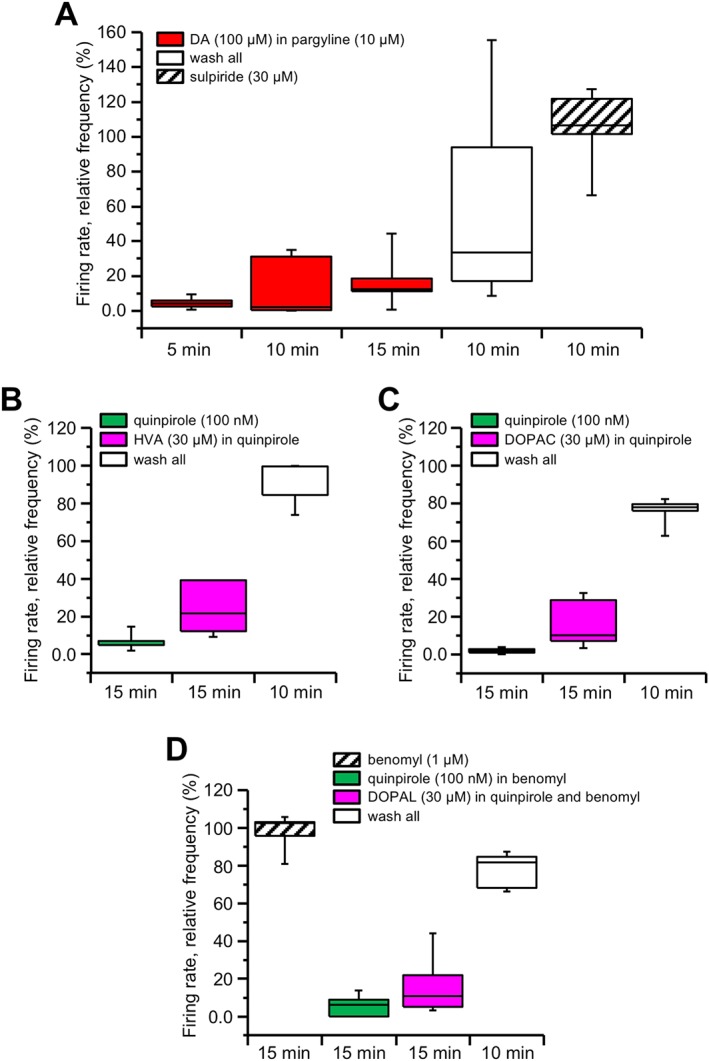
DIR is not present under MAO inhibition but does not rely on its enzymatic products. Box‐and‐whisker plots in all panels represent the median values of the firing rate of all the neurons recorded in each slice with MEA, normalized (in %) to control firing rate and measured at the times indicated at the bottom. The centre lines denote medians, edges are upper and lower quartiles and whiskers show minimum and maximum values. (A) Recordings obtained from slices previously incubated in the MAO inhibitor pargyline (10 μM) and kept in the medium for all the time of recording. Dopamine (DA, 100 μM) inhibited the relative firing rate, which did not significantly recover after 10 or 15 min perfusion (P > 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test, n = 7 slices). A partial recovery was obtained at 10 min dopamine washout, while full recovery could be accelerated by the D2 receptor antagonist sulpiride (30 μM). (B) Perfusion with the D2 receptor agonist quinpirole (100 nM) induced a pronounced and long‐lasting firing rate inhibition. The firing rate was not restored in response to 30 μM HVA (P > 0.05, paired t‐test, n = 5 slices). The firing rate then recovered at drugs washout. (C) Similar results were obtained when attempting to restore quinpirole‐mediated firing inhibition with 30 μM DOPAC (P > 0.05, paired t‐test, n = 5 slices) or (D) 30 μM DOPAL (P > 0.05, paired t‐test, n = 5 slices), with the latter being applied after previous exposure in 1 μM benomyl.
Based on the above results, we postulated that products of dopamine degradation through MAO activity, rather than dopamine proper, may participate in the recovery from firing inhibition under prolonged dopamine exposure. Therefore, we tested the main metabolites of this enzymatic pathway, to see if they promoted recovery from inhibition of the firing rate in response to prolonged exposure to 100 nM quinpirole. In particular, we tested HVA, DOPAC and DOPAL. However, none of these metabolites was effective, as just a small but no significant recovery from firing inhibition by quinpirole was observed at 15 min of 30 μM HVA (P > 0.05, n = 5 slices, from two animals, with total of 82 neurons; Figure 5B) or of 30 μM DOPAC (P > 0.05, n = 5 slices, from two animals, with total of 60 neurons; Figure 5C) or of 30 μM DOPAL (P > 0.05, n = 5 slices, from two animals, with total of 48 neurons; Figure 5D), with the latter being applied in the presence of the aldehyde dehydrogenase inhibitor benomyl (1 μM), in order to prevent its conversion into DOPAC. Thus, the degree of firing recovery was significantly different from that produced by 100 μM dopamine in 100 nM quinpirole (P < 0.05, dopamine vs. HVA; P < 0.05, dopamine vs. DOPAC; P < 0.05, dopamine vs. DOPAL, unpaired t‐test).
These experiments indicate that, although MAO activity is required for firing rate recovery during prolonged dopamine exposure, this effect is not due to the participation of metabolites produced by dopamine enzymatic degradation in DIR expression.
Dopamine‐induced DAT current counteracts the Kir3.1channel‐mediated outward current
In order to investigate DIR mechanisms at single‐cell level, we measured specific membrane currents by means of patch‐clamp whole‐cell experiments on single SNc dopaminergic neurons.
Bath perfusion of 100 μM dopamine produced an outward current that reached a peak level in 1 to 2 min (n = 26 cells, from 10 animals). Upon continued dopamine perfusion, the outward current steadily decayed, going beyond the initial holding level after 10 min, giving rise to a delayed inward current (Figure 6A). Prolonged 100 μM dopamine exposure was also capable to inhibit the Kir3.1 (GIRK) channel‐mediated outward current induced by baclofen (2 μM) in the presence of 10 μM sulpiride (64.02 ± 10.20% of maximal outward current after 10 min in baclofen and dopamine; n = 13 cells, from four animals; Figure 6B).
Figure 6.
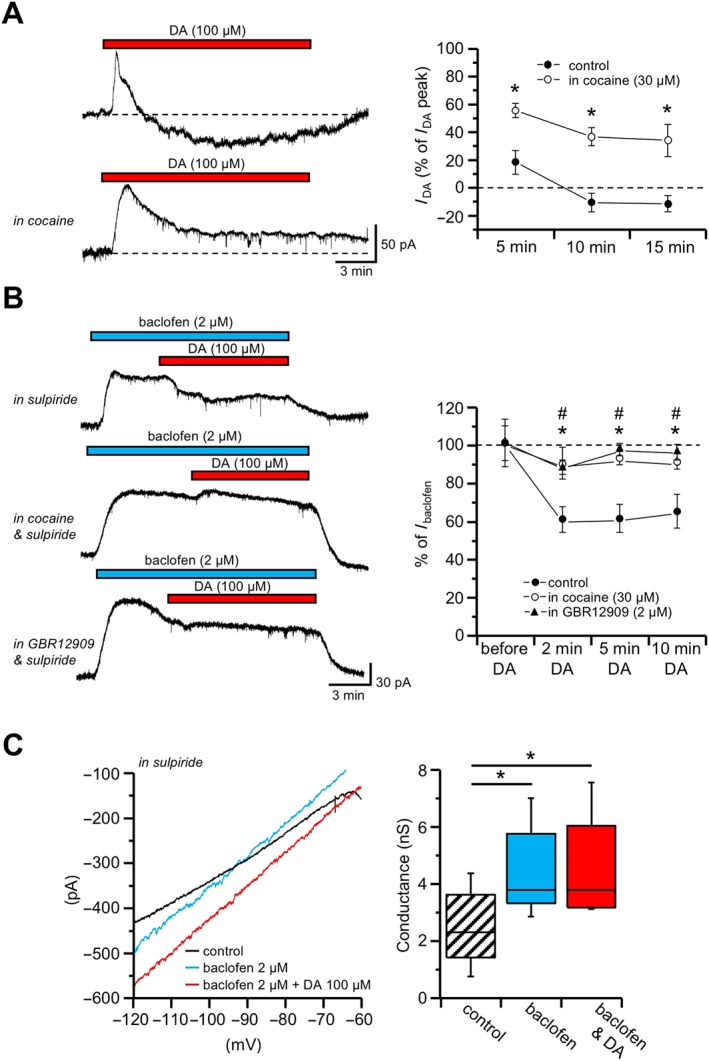
A cocaine‐sensitive inward current counteracts D2 and GABAB receptor‐mediated outward currents. Whole‐cell patch‐clamp recordings from SNc dopaminergic neurons recorded in voltage‐clamp mode. (A) In control conditions, dopamine (DA, 100 μM) induced an outward current that slowly decayed upon continuous exposure to dopamine, giving rise to an inward current at later stages (top trace). In the presence of the DAT antagonist cocaine (30 μM), the decay of the outward current induced by dopamine (100 μM) was strongly attenuated (bottom trace). The plot on the right indicates the current induced by dopamine as percentage (±SEM) of the peak outward current, at various times from onset of drug perfusion, in control conditions (n = 26) and in cocaine (n = 9; * P < 0.05, unpaired t‐test). (B) The outward current induced by the GABAB receptor agonist baclofen (2 μM) was attenuated by dopamine (100 μM), in the presence of 10 μM sulpiride (top trace). This effect of dopamine was not observed when 30 μM cocaine was added in the bath (middle trace). Similarly, the dopamine‐mediated inward current was prevented in the presence of the DAT inhibitor GBR12909 (2 μM; bottom trace). The plot on the right indicates the current induced by dopamine as a percentage (±SEM) of the baclofen‐induced outward current at steady state, at various times from onset of dopamine perfusion, in control conditions (n = 13), in cocaine (n = 13) and in GBR12909 (n = 6). * P < 0.05, unpaired t‐test control versus cocaine; # P < 0.05, unpaired t‐test control versus GBR12909. (C) I–V relationships obtained by means of voltage ramps (from −120 to −60 mV, 80 mV·s−1) in the continuous presence of 10 μM sulpiride, in control conditions, following exposure in 2 μM baclofen and during maximal effect of 100 μM dopamine over the baclofen‐mediated outward current. Note the change in slope induced by baclofen, as opposed to the rightward shift induced by dopamine. The box‐and‐whisker plots on the right show the membrane slope conductance measured in the same neurons (n = 6) in control conditions, in 2 μM baclofen and in 2 μM baclofen plus 100 μM dopamine. The centre lines denote medians, edges are upper and lower quartiles and whiskers show minimum and maximum values. * P < 0.05, one‐way repeated measures ANOVA with Bonferroni's post hoc test.
These observations are consistent with the previously shown data, obtained with MEA recordings, on the transient effect of dopamine on the neuronal firing, as well as the recovery from GABAB‐mediated firing inhibition by dopamine under D2 receptor block.
Based on the results obtained with MEA on the role played by DAT, we tested whether the dopamine‐induced inward current could be abolished by the pharmacological block of DAT. Indeed, the decay of the outward current in response to prolonged exposure to 100 μM dopamine was strongly attenuated in the presence of the DAT antagonist cocaine (30 μM; Figure 6A), such that a significantly smaller reduction was achieved from the initial peak, compared with control conditions without DAT inhibition (P < 0.05 after 15 min dopamine, unpaired t‐test, n = 9 cells, from two animals; Figure 6A). Moreover, the outward current produced by 2 μM baclofen, in the continuous presence of 10 μM sulpiride, was not counteracted by 100 μM dopamine when the experiments were conducted in 30 μM cocaine (89.84 ± 5.90% of maximal outward current after 10 min in baclofen and dopamine; P < 0.05 after 10 min dopamine, unpaired t‐test, n = 13 cells, from five animals; Figure 6B). Similar results were obtained using the DAT antagonists GBR12909 (2 μM; 94.24 ± 2.91% of maximal outward current after 10 min in baclofen and dopamine; P < 0.05 after 10 min dopamine, n = 6 cells, from two animals; Figure 6B).
With the aim to investigate the I–V relationship of the ionic current underlying the effect of prolonged dopamine exposure, we measured the membrane slope conductance with voltage ramps (from −120 to −60 mV, 80 mV·s−1) applied in control conditions, during 2 μM baclofen and during baclofen and 100 μM dopamine, in the continuous presence of 10 μM sulpiride.
The outward current produced in response to baclofen was associated with an increase of membrane conductance (from 2.37 ± 0.56 nS in control to 4.38 ± 0.66 nS in baclofen; P < 0.05, F (2, 15) = 61.23, n = 6 cells, from four animals), having a reversal potential close to the K+ equilibrium potential (approximately −95 mV), as expected from Kir3.1channel opening in response to GABAB receptor stimulation (Lacey et al., 1988). When the same voltage ramp protocol was applied during the maximal dopamine effect on the baclofen‐induced outward current, no additional change in the slope conductance was detected within the voltage range tested (from 4.38 ± 0.66 nS in baclofen to 4.48 ± 0.76 nS in baclofen and dopamine; Figure 6C).
These results indicate that dopamine counteracts the Kir3.1 channel‐mediated outward current promoting the development of an inward current, rather than by acting on the previously opened K+ conductance, and are in agreement with a role played by the depolarizing current elicited in response to DAT activation (Ingram et al., 2002).
Discussion
We have used a MEA device to investigate the effect of prolonged dopamine exposure in slices of the SNc, in order to record the firing of a large population of neurons, in a condition that is unperturbed by physical or chemical interference of any sort, as with conventional recording electrodes, and thus possibly closer to physiological conditions (Berretta et al., 2010). We selected from each recorded slice all active neurons within the SNc area, whose firing was inhibited by dopamine and hence identified as dopaminergic based on the well‐characterized D2 receptor‐mediated autoinhibition, typical of this SNc neuronal population (Lacey et al., 1987; Kim et al., 1995; Krashia et al., 2017). In these experimental conditions, we were able to describe an initial profound dopamine‐mediated firing inhibition that reached a plateau in 2 to 5 min, followed by a progressive re‐emergence of the firing in spite of persisting dopamine exposure, giving rise to DIR.
DIR has been ascribed to a D2 receptor desensitization process that is present at early postnatal life, while being progressively lost towards adulthood, through development of a calcium‐dependent process involving L‐type calcium channels (Dragicevic et al., 2014). According to our results, D2 receptor desensitization is indeed present, as indicated by the partial recovery of the dopamine‐mediated outward current in the presence of cocaine (see Figure 6A); however, in our experimental conditions, it seems to be a minor factor in determining firing recovery under persistent dopamine exposure. This conclusion is based on the lack of firing recovery under continuous exposure to quinpirole, while it was restored by addition of dopamine. Accordingly, recent data of Robinson et al. (2017) reported a relatively small desensitization of D2 autoreceptors by quinpirole or dopamine that was largely independent of their internalization. Furthermore, when dopaminergic neurons' firing had been inhibited by the opening of Kir3.1 (GIRK) channels through GABAB receptor stimulation, instead of D2 receptors, dopamine was still able to restore neuronal firing even in the presence of the D2 receptor antagonist sulpiride, thus demonstrating a dopamine‐mediated mechanism occurring independent of D2 receptors.
Rather, our results strongly indicate a key role played by DAT activation, because the pharmacological inhibition of DAT prevented firing recovery by dopamine in MEA recordings, and accordingly, it strongly reduced the delayed fading of the dopamine‐mediated outward current, as well as that mediated by GABAB receptors, in response to dopamine. DAT plays a major role in the clearance of dopamine from the extracellular space, carrying this monoamine inside the cell against its concentration gradient, using the driving force of 2 Na+ and 1 Cl−, co‐transported with dopamine, as energy source (Kilty et al., 1991; Gu et al., 1994; Mortensen and Amara, 2003; Vaughan and Foster, 2013). Additionally, this transport has been shown to cause a channel‐dependent current (Carvelli et al., 2004; De Felice, 2017) such that DAT activation generates an overall depolarizing current, capable to increase the firing rate of the dopaminergic neurons in cultures (Ingram et al., 2002) and slices (Branch and Beckstead, 2012). We suggest that DAT activation, due to accumulation of extracellular dopamine, behaves like an ionotropic receptor by inducing a depolarizing current in the dopaminergic neurons, which is large enough to bring the membrane potential back to threshold for action potential generation, thereby restoring the firing while still in the presence of exogenously applied dopamine. This hypothesis is corroborated in our results, not just by the pharmacological evidence using DAT antagonists and also by the observation that, while the GABAB receptor‐mediated outward current was associated to an expected increase of slope conductance reverting close to K+ equilibrium potential, the dopamine‐mediated recovery of the same current was not paralleled by an opposite effect on the same conductance (Ingram et al., 2002; Swant et al., 2011).
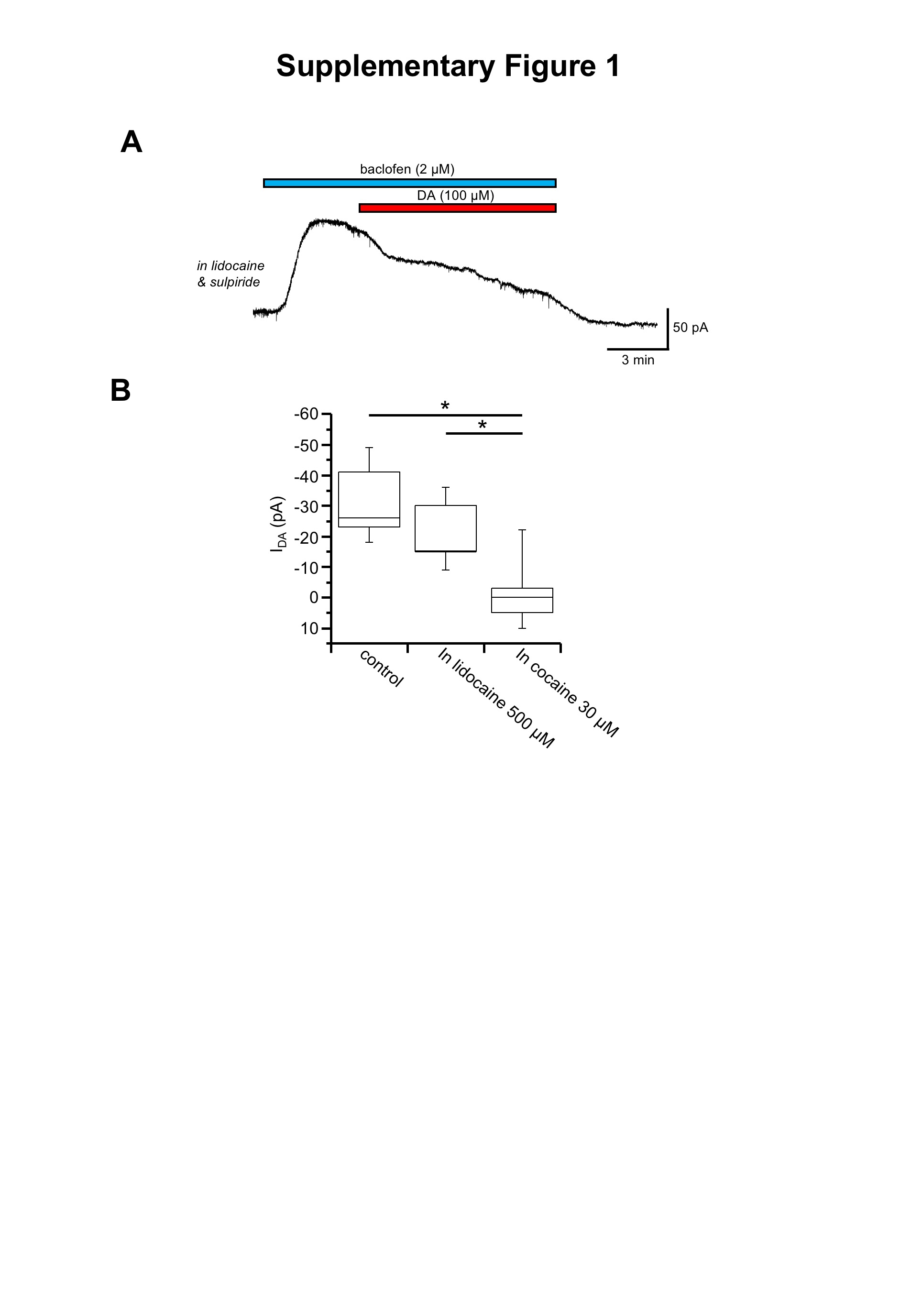
A possible criticism of this hypothesis arises from the high concentration of cocaine (30 μM) needed to prevent DIR and the dopamine‐induced current, arguing for possible side effects due to its anaesthetic action, known to occur at high dose (Yasuda et al., 1984). Indeed, we found that a lower concentration of cocaine (5 and 10 μM; not shown) was ineffective; however, two pieces of evidence lead us to exclude this interpretation: (i) inconsistently with the hypothesis that cocaine acts by exerting an anaesthetic action, we found that the fading of the baclofen‐induced current by 100 μM dopamine was not prevented by the local anaesthetic lidocaine (500 μM; Supporting information Figure S1) and (ii) similarly to cocaine, the dopamine‐induced inward current was blocked by the DAT inhibitor GBR12909 at a highly selective concentration (2 μM). Indeed, GBR12909 has previously been shown to be at least 50 times more potent than cocaine as DAT inhibitor (Izenwasser et al., 1990; Masserano et al., 1994), suggesting that a high cocaine dose is needed in order to overcome the high concentration of dopamine used. On this regard, the need of a complete DAT block in order to prevent DIR may explain the seemingly divergent results of Nimitvilai et al. (2012a), who found that cocaine at low concentration facilitates the ability of dopamine to induce DIR. Possibly, the DAT‐mediated current causing DIR requires the transporter to be completely blocked, while when it is only partially inhibited, the prevailing effect is that of increased extracellular dopamine levels.
The need of concomitant D1 and D2 receptor stimulation in order to induce recovery from dopamine‐mediated firing inhibition has been previously reported in VTA dopaminergic neurons (Nimitvilai and Brodie, 2010). According to our experimental evidence, the co‐application of their respective agonists, SKF81297 and quinpirole, was not able to mimic dopamine effects on firing recovery, thus suggesting a different mechanism of action.
These discrepancies from previously proposed mechanisms may be the result of differences in the species used (mice vs. rats) or in the precise area under investigation (SNc vs. VTA), or the age of the animal, as the DIR reported by Nimitvilai et al. (2012a) became evident only after 15 postnatal day, as opposed to 12–15 in the present work. However, we should also consider the possibility that more than one mechanism might be implicated in DIR, including D2 receptor desensitization (Dragicevic et al., 2014) or a D1 receptor‐dependent phosphorylation mechanism (Nimitvilai et al., 2012a; Nimitvilai et al., 2013), with varying relative weight of contribution according to particular experimental conditions. In any case, our experimental evidences bring forward the electrogenic DAT current as a major actor of DIR expression in SNc dopaminergic neurons.
Interestingly, we found that, when MAO enzymes were inhibited with pargyline, firing recovery from prolonged dopamine exposure was abolished. This effect was not due to the block in the release of metabolic down‐products of dopamine degradation, as neither HVA nor DOPAC nor DOPAL were capable to restore quinpirole‐mediated firing inhibition. This result is reminiscent of a previous report from our own laboratories, showing that MAO inhibition transforms the typical transient firing inhibition due to brief dopamine exposure, into one that lasts for several tens of minutes (Mercuri et al., 1997). Similarly, we now found that, in the presence of the MAO inhibitor pargyline, not only was DIR absent but also firing recovery at dopamine washout was even delayed, such that a pharmacological block of D2 receptors with sulpiride was needed in order to accelerate a full recovery (see Figure 5A). Possibly, when dopamine degradation is blocked, extracellular dopamine available for D2 receptor stimulation becomes high enough to overcome the counteracting effect of the DAT current. Moreover, whatever is the cellular mechanism responsible for the abnormally sustained dopamine inhibition caused by MAO inhibition, its stimulation by the previous exposure in pargyline might have similarly compromised the recovery from inhibition under prolonged dopamine. In addition to these possible explanations, we cannot rule out an indirect effect on DAT due to MAO inhibition. For instance, it has been reported that stimulation of Rho family GTPases mediates an internalization of DAT. This effect occurs in response to amphetamine entering the dopaminergic neurons, and also, intracellular dopamine appears to be capable to stimulate the same process (Wheeler et al., 2015). On this basis, we hypothesize that the rise of intracellular dopamine due to MAO inhibition may cause DAT internalization, thus preventing firing recovery. This result strengthens the importance of MAO activity in controlling the dopaminergic signal, and it is for this reason that inhibition of this enzymatic process is often combined with l‐DOPA treatment in order to boost its therapeutic action in PD patients (McCormack, 2014; Finberg and Rabey, 2016; Cereda et al., 2017).
It is known that stimulation of D2 autoreceptors by extracellular dopamine not only alters dopaminergic neurons' basal excitability but also affects action potential backpropagation and calcium current dynamics and hence dopamine transmission to target areas (Cardozo and Bean, 1995; Gentet and Williams, 2007; Ford, 2014; Evans et al., 2017). On this basis, our results suggest that extracellular dopamine modulates the local processing of active information by means of an interplay between D2 receptor‐mediated hyperpolarization and an opposing current generated by DAT activity. Interestingly, extracellular dopamine also dampens GABAB receptor‐mediated inhibition. A similar interplay has previously been reported in VTA dopaminergic neurons, occurring through a cross‐desensitization process (Nimitvilai et al., 2012b), while our results point to a DAT‐dependent mechanism, as it is prevented by cocaine. In any case, these results confirm a much more complex and heterogeneous action exerted by extracellular dopamine in the ventral midbrain, than a simple self‐inhibitory role through D2 receptor stimulation.
A greater awareness of the significant role played by the DAT current in shaping SNc dopaminergic neurons firing can be of considerable importance also in relation to the vulnerability of this neuronal population in PD. It should be noted that, while DIR in control mice is normally present only at early postnatal age (Dragicevic et al., 2014), in the PARK7 gene knockout mouse model of PD, in adults, there is also a strong degree of recovery from dopamine‐mediated firing inhibition by sustained extracellular dopamine in SNc dopaminergic neurons (Goldberg et al., 2005), as well as a higher level of oxidative stress linked to their pacemaking action potential firing (Guzman et al., 2010). Interestingly, DAT‐overexpressing mice also display histopathological and behavioural features of PD (Masoud et al., 2015). This effect can indeed be ascribed to the accumulation of cytosolic dopamine (Lohr et al., 2017); however, we may also hypothesize a significant role played by the DAT current that keeps action potential firing at higher levels, thereby exacerbating neurodegenerative processes in SNc dopaminergic neurons because of an increased metabolic demand.
Another fundamental issue raised by our experimental results is the need to optimize l‐DOPA‐based pharmacological therapies in PD patients, taking into account the adverse side effects of DAT stimulation in response to the increased levels of exogenous dopamine. An aggravating factor in this opposing interplay between D2 receptors and DAT is the finding that DAT activity and expression are increased by D2 receptor stimulation in response to high extracellular dopamine levels, as those that can be achieved under l‐DOPA treatment (Cass and Gerhardt, 1994; Mayfield and Zahniser, 2001; Benoit‐Marand et al., 2011; Ford, 2014). Thus, adding to possible oxidative damage due to dopamine accumulation (Lipski et al., 2011), l‐DOPA treatment may be limited in its efficacy by the concomitant DAT stimulation and cause in the meantime a higher metabolic demand in the surviving population of dopaminergic neurons.
Author contributions
D.A. performed the MEA recordings and analysed the data; A.M. performed the whole‐cell recordings and analysed the data; N.B., N.B.M, E.G. and A.P. designed and supervised the study and interpreted the data; N.B. drafted the manuscript; and all authors took part in correcting the proofs and approved the final manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 The presence of the anaesthetic drug lidocaine did not prevent the dopamine‐induced inward current. While still in cell‐attached, before gaining a whole‐cell access, the cell was exposed to lidocaine 500 μM, in order to ensure the disappearance of spontaneous extracellularly recorded action potentials, thereby confirming the efficacy of the drug. (A) The outward current induced by the GABAB receptor agonist baclofen (2 μM), in the presence of 10 μM sulpiride and 500 μM lidocaine, was attenuated by dopamine (100 μM; n = 9, from 3 animals). (B) Box‐and‐whisker plots of the dopamine‐induced inward current in control conditions, in lidocaine and in cocaine. The center lines denote medians, edges are upper and lower quartiles, whiskers show minimum and maximum values. As shown by the plots, the dopamine‐induced current was significantly reduced in the presence of cocaine, while not in lidocaine. *P < 0.05, F (2,32) = 16.86, One‐Way ANOVA with Boferroni's post hoc test.
{kind=link}
Data S1 Supporting info item
Acknowledgements
This work has been financially supported by ‘Ricerca Corrente’ of the Italian Ministry of Health. D.A. and A.M. were supported by postgraduate studentships provided by the Italian Ministry of University and Research.
Aversa, D. , Martini, A. , Guatteo, E. , Pisani, A. , Mercuri, N. B. , and Berretta, N. (2018) Reversal of dopamine‐mediated firing inhibition through activation of the dopamine transporter in substantia nigra pars compacta neurons. British Journal of Pharmacology, 175: 3534–3547. 10.1111/bph.14422.
Contributor Information
Nicola Biagio Mercuri, Email: mercurin@med.uniroma2.it.
Nicola Berretta, Email: n.berretta@hsantalucia.it.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide To PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide To PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide To PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide To PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT (2004). Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 42: 939–946. [DOI] [PubMed] [Google Scholar]
- Benoit‐Marand M, Ballion B, Borrelli E, Boraud T, Gonon F (2011). Inhibition of dopamine uptake by D2 antagonists: an in vivo study. J Neurochem 116: 449–458. [DOI] [PubMed] [Google Scholar]
- Berretta N, Bernardi G, Mercuri NB (2010). Firing properties and functional connectivity of substantia nigra pars compacta neurones recorded with a multi‐electrode array in vitro . J Physiol 588: 1719–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch SY, Beckstead MJ (2012). Methamphetamine produces bidirectional, concentration‐dependent effects on dopamine neuron excitability and dopamine‐mediated synaptic currents. J Neurophysiol 108: 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo DL, Bean BP (1995). Voltage‐dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol 74: 1137–1148. [DOI] [PubMed] [Google Scholar]
- Carvelli L, McDonald PW, Blakely RD, DeFelice LJ (2004). Dopamine transporters depolarize neurons by a channel mechanism. Proc Natl Acad Sci U S A 101: 16046–16051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass WA, Gerhardt GA (1994). Direct in vivo evidence that D2 dopamine receptors can modulate dopamine uptake. Neurosci Lett 176: 259–263. [DOI] [PubMed] [Google Scholar]
- Cereda E, Cilia R, Canesi M, Tesei S, Mariani CB, Zecchinelli AL et al (2017). Efficacy of rasagiline and selegiline in Parkinson's disease: a head‐to‐head 3‐year retrospective case–control study. J Neurol 264: 1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornil CA, Balthazart J, Motte P, Massotte L, Seutin V (2002). Dopamine activates noradrenergic receptors in the preoptic area. J Neurosci 22: 9320–9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Lüscher C (2004). Bi‐directional effects of GABAB receptor agonists on the mesolimbic dopamine system. Nat Neurosci 7: 153–159. [DOI] [PubMed] [Google Scholar]
- Cucchiaroni ML, Freestone PS, Berretta N, Viscomi MT, Bisicchia E, Okano H et al (2011). Properties of dopaminergic neurons in organotypic mesencephalic‐striatal co‐cultures – evidence for a facilitatory effect of dopamine on the glutamatergic input mediated by α‐1 adrenergic receptors. Eur J Neurosci 33: 1622–1636. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice LJ (2017). Monoamine transporters as ionotropic receptors. Trends Neurosci 40: 195–196. [DOI] [PubMed] [Google Scholar]
- Dragicevic E, Poetschke C, Duda J, Schlaudraff F, Lammel S, Schiemann J et al (2014). Cav1.3 channels control D2‐autoreceptor responses via NCS‐1 in substantia nigra dopamine neurons. Brain 137: 2287–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS (2004). Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 56: 331–349. [DOI] [PubMed] [Google Scholar]
- Evans RC, Zhu M, Khaliq ZM (2017). Dopamine inhibition differentially controls excitability of substantia nigra dopamine neuron subpopulations through T‐type calcium channels. J Neurosci 37: 3704–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finberg JP, Rabey JM (2016). Inhibitors of MAO‐A and MAO‐B in psychiatry and neurology. Front Pharmacol 7: 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP (2014). The role of D2‐autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 282: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet LJ, Williams SR (2007). Dopamine gates action potential backpropagation in midbrain dopaminergic neurons. J Neurosci 27: 1892–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C et al (2005). Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism‐linked gene DJ‐1. Neuron 45: 489–496. [DOI] [PubMed] [Google Scholar]
- Guzman JN, Sanchez‐Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT et al (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ‐1. Nature 468: 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Wall SC, Rudnick G (1994). Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem 269: 7124–7130. [PubMed] [Google Scholar]
- Guatteo E, Yee A, McKearney J, Cucchiaroni ML, Armogida M, Berretta N et al (2013). Dual effects of l‐DOPA on nigral dopaminergic neurons. Exp Neurol 247: 582–594. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Prasad BM, Amara SG (2002). Dopamine transporter‐mediated conductances increase excitability of midbrain dopamine neurons. Nat Neurosci 5: 971–978. [DOI] [PubMed] [Google Scholar]
- Izenwasser S, Werling LL, Cox BM (1990). Comparison of the effects of cocaine and other inhibitors of dopamine uptake in rat striatum, nucleus accumbens, olfactory tubercle, and medial prefrontal cortex. Brain Res 520: 303–309. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilty JE, Lorang D, Amara SG (1991). Cloning and expression of a cocaine‐sensitive rat dopamine transporter. Science 254: 578–579. [DOI] [PubMed] [Google Scholar]
- Kim KM, Nakajima Y, Nakajima S (1995). G protein‐coupled inward rectifier modulated by dopamine agonists in cultured substantia nigra neurons. Neuroscience 69: 1145–1158. [DOI] [PubMed] [Google Scholar]
- Kopin IJ (1985). Catecholamine metabolism: basic aspects and clinical significance. Pharmacol Rev 37: 333–364. [PubMed] [Google Scholar]
- Krashia P, Martini A, Nobili A, Aversa D, D'Amelio M, Berretta N et al (2017). On the properties of identified dopaminergic neurons in the mouse substantia nigra and ventral tegmental area. Eur J Neurosci 45: 92–105. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA (1987). Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol 392: 397–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA (1988). On the potassium conductance increase activated by GABAB and dopamine D2 receptors in rat substantia nigra neurones. J Physiol 401: 437–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipski J, Nistico R, Berretta N, Guatteo E, Bernardi G, Mercuri NB (2011). l‐DOPA: a scapegoat for accelerated neurodegeneration in Parkinson's disease? Prog Neurobiol 94: 389–407. [DOI] [PubMed] [Google Scholar]
- Lohr KM, Masoud ST, Salahpour A, Miller GW (2017). Membrane transporters as mediators of synaptic dopamine dynamics: implications for disease. Eur J Neurosci 45: 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Slesinger PA (2010). Emerging roles for G protein‐gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11: 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoud ST, Vecchio LM, Bergeron Y, Hossain MM, Nguyen LT, Bermejo MK et al (2015). Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and l‐DOPA reversible motor deficits. Neurobiol Dis 74: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masserano JM, Venable D, Wyatt RJ (1994). Effects of chronic cocaine administration on [3H] dopamine uptake in the nucleus accumbens, striatum and frontal cortex of rats. J Pharmacol Exp Ther 270: 133–141. [PubMed] [Google Scholar]
- Mayfield RD, Zahniser NR (2001). Dopamine D2 receptor regulation of the dopamine transporter expressed in Xenopus laevis oocytes is voltage‐independent. Mol Pharmacol 59: 113–121. [DOI] [PubMed] [Google Scholar]
- McCormack PL (2014). Rasagiline: a review of its use in the treatment of idiopathic Parkinson's disease. CNS Drugs 28: 1083–1097. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Scarponi M, Bonci A, Siniscalchi A, Bernardi G (1997). Monoamine oxidase inhibition causes a long‐term prolongation of the dopamine‐induced responses in rat midbrain dopaminergic cells. J Neurosci 17: 2267–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Calabresi P, Bernardi G (1992). The electrophysiological actions of dopamine and dopaminergic drugs on neurons of the substantia nigra pars compacta and ventral tegmental area. Life Sci 51: 711–718. [DOI] [PubMed] [Google Scholar]
- Mortensen OV, Amara SG (2003). Dynamic regulation of the dopamine transporter. Eur J Pharmacol 479: 159–170. [DOI] [PubMed] [Google Scholar]
- Nimitvilai S, Brodie MS (2010). Reversal of prolonged dopamine inhibition of dopaminergic neurons of the ventral tegmental area. J Pharmacol Exp Ther 333: 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimitvilai S, Arora DS, Brodie MS (2012a). Reversal of dopamine inhibition of dopaminergic neurons of the ventral tegmental area is mediated by protein kinase C. Neuropsychopharmacology 37: 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimitvilai S, Arora DS, McElvain MA, Brodie MS (2012b). Reversal of inhibition of putative dopaminergic neurons of the ventral tegmental area: interaction of GABAB and D2 receptors. Neuroscience 226: 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimitvilai S, McElvain MA, Brodie MS (2013). Reversal of dopamine D2 agonist‐induced inhibition of ventral tegmental area neurons by Gq‐linked neurotransmitters is dependent on protein kinase C, G protein‐coupled receptor kinase, and dynamin. J Pharmacol Exp Ther 344: 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BG, Bunzow JR, Grimm JB, Lavis LD, Dudman JT, Brown J et al (2017). Desensitized D2 autoreceptors are resistant to trafficking. Sci Rep 7: 4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JC, Chen K, Ridd MJ (1999). Monoamine oxidase: from genes to behavior. Ann Rev Neurosci 22: 197–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK (2004). Sequential binding of agonists to the β2 adrenoceptor. Kinetic evidence for intermediate conformational states. J Biol Chem 279: 686–691. [DOI] [PubMed] [Google Scholar]
- Swant J, Goodwin JS, North A, Ali AA, Gamble‐George J, Chirwa S et al (2011). α‐Synuclein stimulates a dopamine transporter‐dependent chloride current and modulates the activity of the transporter. J Biol Chem 286: 43933–43943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Heuvel DM, Pasterkamp RJ (2008). Getting connected in the dopamine system. Prog Neurobiol 85: 75–93. [DOI] [PubMed] [Google Scholar]
- Vaughan RA, Foster JD (2013). Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol Sci 34: 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DS, Underhill SM, Stolz DB, Murdoch GH, Thiels E, Romero G et al (2015). Amphetamine activates Rho GTPase signaling to mediate dopamine transporter internalization and acute behavioral effects of amphetamine. Proc Natl Acad Sci U S A 112: E7138–E7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda RP, Zahniser NR, Dunwiddie TV (1984). Electrophysiological effects of cocaine in the rat hippocampus in vitro. Neurosci Lett 45: 199–204. [DOI] [PubMed] [Google Scholar]
- Zhang WP, Ouyang M, Thomas SA (2004). Potency of catecholamines and other l‐tyrosine derivatives at the cloned mouse adrenergic receptors. Neuropharmacology 47: 438–449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The presence of the anaesthetic drug lidocaine did not prevent the dopamine‐induced inward current. While still in cell‐attached, before gaining a whole‐cell access, the cell was exposed to lidocaine 500 μM, in order to ensure the disappearance of spontaneous extracellularly recorded action potentials, thereby confirming the efficacy of the drug. (A) The outward current induced by the GABAB receptor agonist baclofen (2 μM), in the presence of 10 μM sulpiride and 500 μM lidocaine, was attenuated by dopamine (100 μM; n = 9, from 3 animals). (B) Box‐and‐whisker plots of the dopamine‐induced inward current in control conditions, in lidocaine and in cocaine. The center lines denote medians, edges are upper and lower quartiles, whiskers show minimum and maximum values. As shown by the plots, the dopamine‐induced current was significantly reduced in the presence of cocaine, while not in lidocaine. *P < 0.05, F (2,32) = 16.86, One‐Way ANOVA with Boferroni's post hoc test.
Data S1 Supporting info item